
Human Radiosensitivity and Radiosusceptibility: What Are the Differences?

 , ,
, ,
Abstract
:1. Introduction
1.1. Historical Features
1.2. A Current Confusion
1.3. The Evidence of a Molecular Difference
1.4. Univocal Definitions
- the “genomic approach” which consists of inventorying all the genes involved by their expression or polymorphisms in the high throughput studies of radiosensitivity, in order to establish causal links with clinical features [21]. The major advantage of this approach is to get a large number of candidate genes. The major inconvenience is to consider gene expression as a major feature of the response to radiation, while some cases of radiosensitivity are not necessarily linked to a higher or lower gene expression but to protein dysfunction [8];
- the “clinical approach” which consists of defining the major clinical features of the response to radiation, and thereafter to identify genes in each category. The major advantage of this approach is to gather all the different types of radiation-induced events observed by clinicians. The major inconvenience is to omit some genes that may be involved in the response to radiation while their mutations lead to non-viability and therefore are not associated with any described syndrome [8].
- The radiosensitivity is the proneness to the adverse tissue events that are considered as non-cancer radiation-induced effects and attributable to cell death. Radiosensitivity is generally correlated with unrepaired DNA damage [12];
- The radiosusceptibility is the proneness to the radiation-induced cancers that are non-toxic radiation-induced effects attributable to cell transformation and genomic instability. Radiosusceptibility is generally correlated with misrepaired DNA damage [22]. As IR is considered as a carcinogenic agent, radiosusceptibility is strongly linked to susceptibility to spontaneous cancers. The term “radiosusceptibility” was proposed through its similarities with “cancer susceptibility”, extensively used in the ICRP reports, and as it introduces the notions of stochastic events [8];
- The radiodegeneration responses are non-cancer effects attributable to mechanisms related to accelerated aging. Radiodegeneration should be correlated with unrepaired DNA damage that is tolerated by and can cumulate in cells [8]. Radiodegeneration responses cannot be considered similar to radiosensitivity responses as defined above, as their incidence rates, the types of cellular death, and the genes involved are different.
2. The Different Features of Human Radiosensitivity
2.1. What do We Learn from the Quantification of Human Radiosensitivity?
2.2. Genetic Syndromes Associated with Radiosensitivity from the Clinical, Cellular and Molecular Criteria
- AT patients show a very high risk of leukemia/lymphoma, and were treated by radiotherapy (total body irradiations) in the 1970s [16,34,35,36]. The severity of their post-radiotherapy reactions (nearly all fatal) and their extreme sensitivity to other DSB-inducing drugs have imposed a particular care for treating them with radiomimetic drugs [16,37,38,39]. AT is caused by homozygous or compound heterozygous mutations of the ATM gene [40,41];
- LIG4 syndrome was first described from an acute lymphoblastic leukemia patient who overresponded to radiation therapy and died following radiation morbidity, without showing any clinical features in common with AT. The radiobiological characterization of his cells revealed homozygous mutation of Ligase IV [42,43,44,45];
- a case of a Xeroderma Pigmentosum C patient who suffered from an angiosarcoma and showed a fatal reaction to radiotherapy was reported [50]. However, the radiosensitivity of this case has been shown to be complemented by the transfer of a wild-type chromosome 8, suggesting that this XPC variant patient may hold other gene mutation responsible of his abnormal response to radiation [51,52].
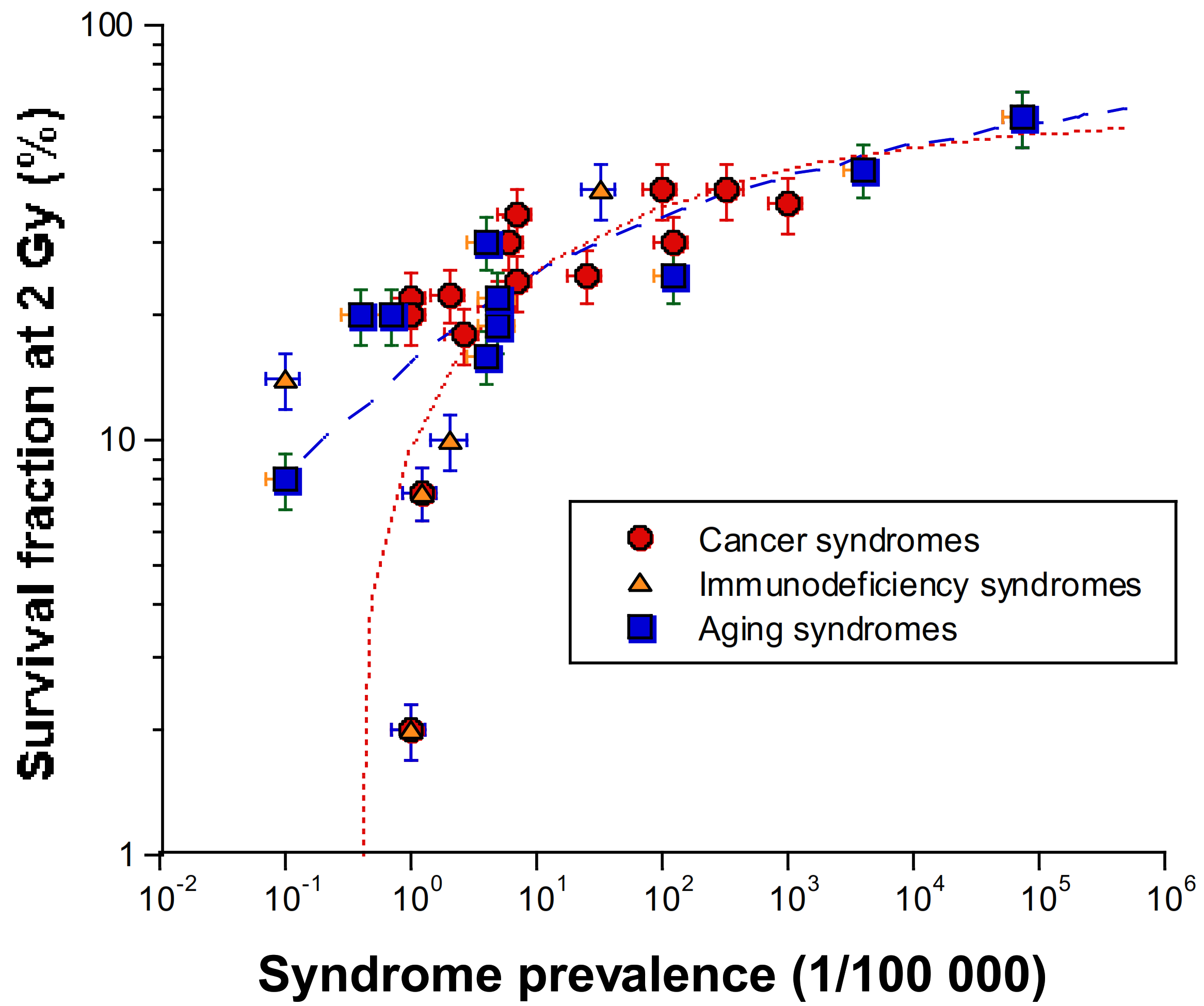
- as SF2 increases, the syndromes caused by homozygous mutations (leading to loss of functions) are progressively replaced by syndromes caused by heterozygous mutations (leading to “leaky” functions) (Table 1). Syndromes caused by heterozygous mutations being more frequent than those caused by homozygous mutations, SF2 increases with prevalence (Figure 2).
- homozygous mutations of ATM, LIG4, and NBS1, involved in the DSB repair and signalling pathways, cause the most hyper-radiosensitive syndromes; this hyper-radiosensitivity has been observed both clinically and in vitro;
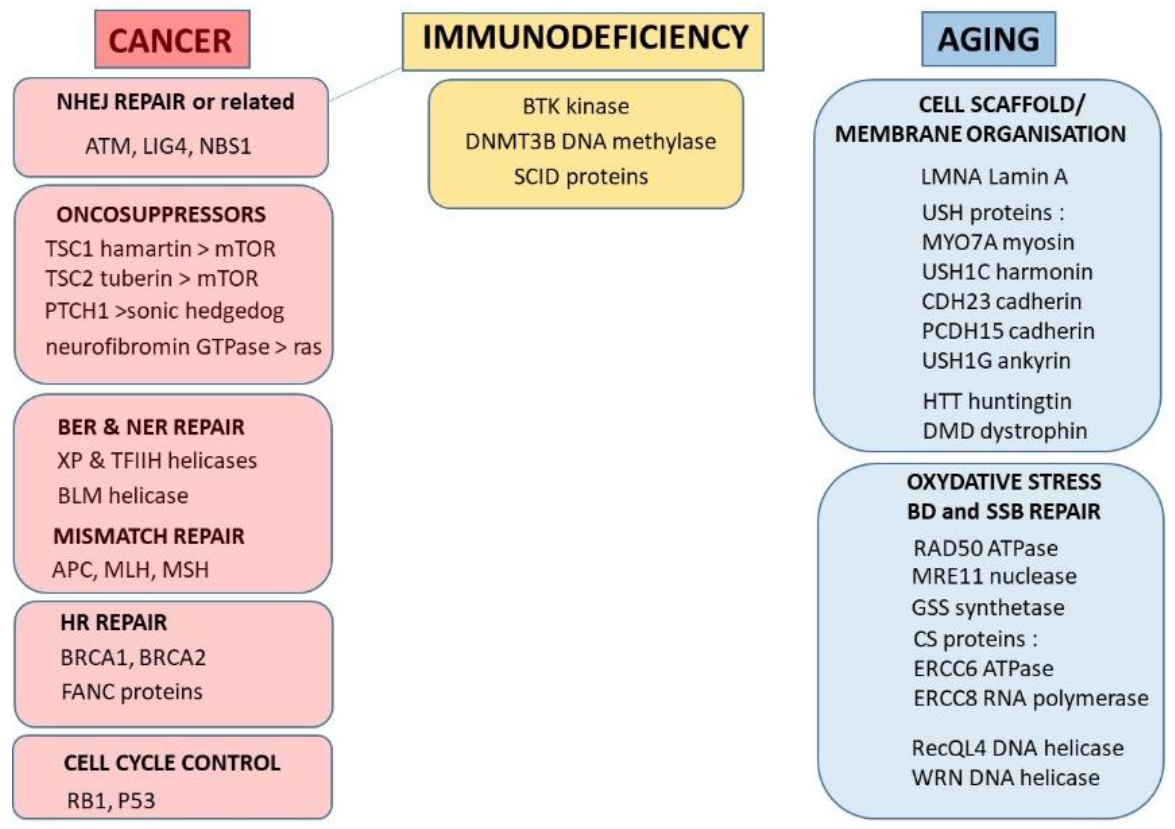
- there is a continuum of SF2 values form 1% (ataxia telangiectasia) to about 60% (average radioresistance). Surprisingly, to the notable exceptions of the three precited syndromes, there are few syndromes caused by mutations of DSB repair proteins, probably as DSB is a key-DNA damage that impacts on each step of embryogenesis. About 50% of radiosensitive syndromes are caused by genes involved in the repair of radiation-induced DNA damage (other than DSB) that may affect cell survival after irradiation. The remaining 50% are caused by genes involved in the cell scaffold and the nuclear membrane, and whose encoded proteins are cytoplasmic (Figure 1).
3. The Different Features of Human Radiosusceptibility
3.1. What do We Learn from the Quantification of Human Radiosusceptibility?
- these cohorts/databases, derived from epidemiological data, do not highlight any individual predispositions to specific malignancies, but concern a whole population of individuals considered as equally radioresistant. The dose-effect curve shape may vary according to the type of radiation-induced cancer;
- there is no clinical equivalent of CTCAE/RTOG scales grading the different steps of carcinogenesis. Consequently, the relative risk (RR) or the excess of relative risk (ERR) are the only parameters to express cancer incidence as a function of dose. It is noteworthy that these parameters are calculated from epidemiological data [8];
- there is no consensual mathematical model that describes (similar to the LQ model for cell survival) the cancer incidence, or its risk as a function dose. Indeed, the radiation-induced cancer incidence curves are generally described as either linear with no threshold (LNT) or non-linear with a threshold (NLT). The LNT/NLT controversies have long reflected a societal issue, raising the question of the existence of a dose-threshold below which there are no significant association between malignancies and exposures to ionizing radiation [77,78]. From Hiroshima survivors data, the threshold doses have been found to be 100 mGy for radiation-induced leukemia and 200 mGy for radiation-induced solid cancers [75]. However, these dose thresholds are relevant only for high dose-rate (flash) exposures to radiation. The corresponding dose thresholds for low dose-rate exposures are still unknown [79].
3.2. Genetic Syndromes Associated with Radiosusceptibility from the Clinical, Cellular, and Molecular Criteria
- homozygous mutations of ATM, LIG4, and NBS1 genes are associated with high risks of leukaemia/lymphoma;
- there is no consensual parameter to quantify radiosusceptibility, notably as the intrinsic mechanisms of carcinogenesis are still unknown;
- the radiosensitive syndromes that are associated with radiosusceptibility may be associated with a large spectrum of malignancies for a single gene mutation;
- the radiosusceptible syndromes are caused by mutations of genes related to proto-oncogenes, to radiation-induced misrepaired DNA damage, or else to cell cycle checkpoints (Figure 1). Again, among these syndromes, some are caused by mutated cytoplasmic proteins.
4. Toward a Unified Model for Radiosensitivity and Radiosusceptibility
4.1. Biological Function of Proteins as Proteins or as Substrates?
4.2. The Nucleo-Shuttling of ATM as a Primum Movens of the Molecular Response to Radiation
- the “dosimetry step”: after irradiation, the production of reactive oxygen species (ROS) is dose-dependent. Under the effect of the radiation-induced oxidative stress, some cytoplasmic dimeric forms of ATM become monomeric in a dose-dependent manner;
- the “diffusion step”: the ATM monomers diffuse to the nucleus; however, during their course from the cytoplasm to the nucleus, they can meet some cytoplasmic ATM substrates with which they can form multiprotein complexes that prevents the nucleo-shutting;
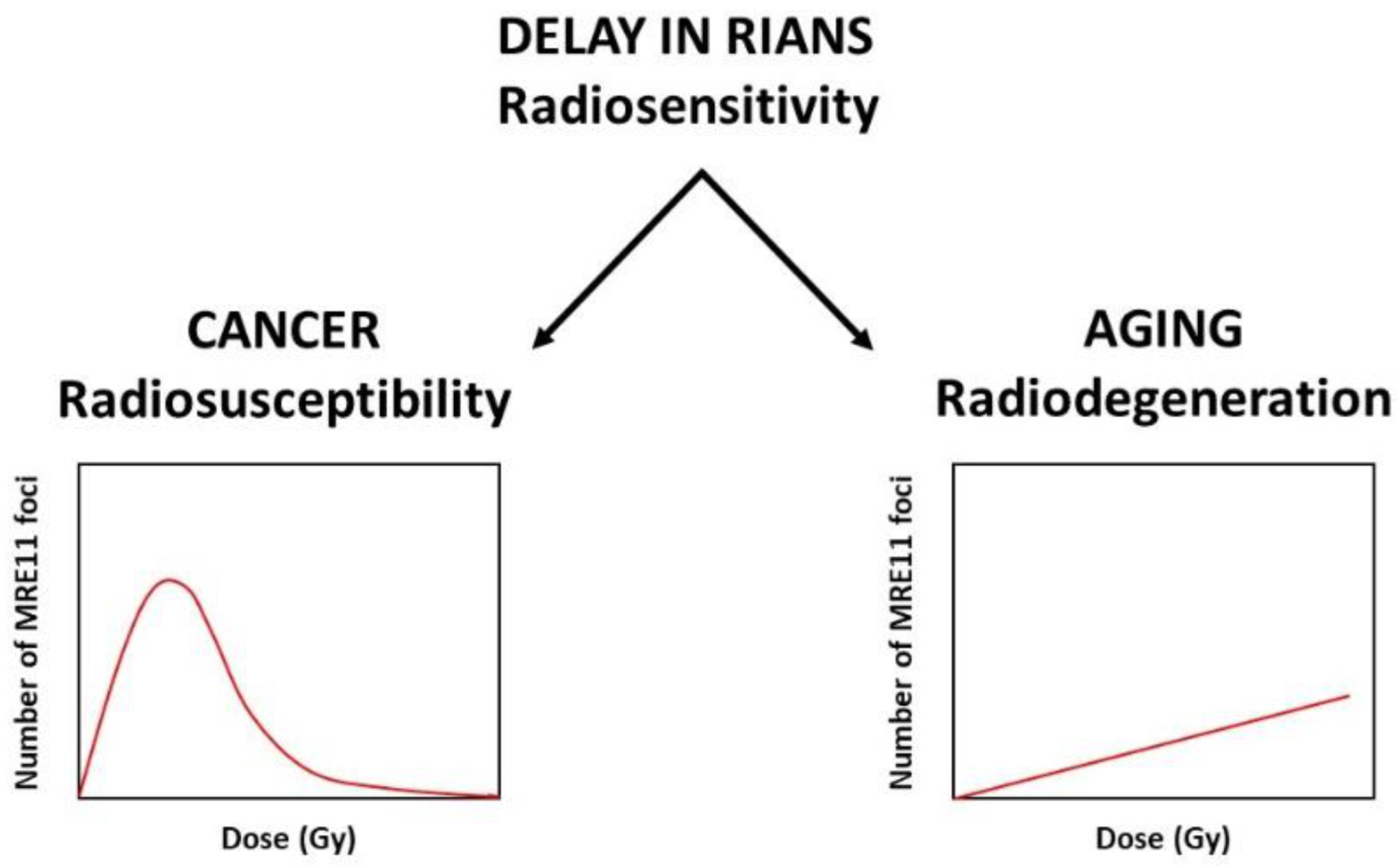
- the “recognition step”: the remaining free ATM monomers diffuse to the nucleus and phosphorylate H2AX molecules at DSB sites, which activates NHEJ. The ATM monomers will re-dimerize during the DSB repair process and can be easily quantifiable as nuclear foci by immunofluorescence [61].
- DSB are not repaired, whatever the repair pathway: these DSB become lethal and lead to radiosensitivity. Less than two unrepaired DSB are sufficient to cause cell death in humans;
- DSB are not recognized by NHEJ, but they are managed by a rapid but illegitimate hyper-recombination process: these DSB become misrepaired and lead to radiosusceptibility; they can be accompanied by additional DNA strand breaks due to hyper-recombination early after irradiation;
- DSB are tolerated (i.e., non-lethal immediately, likely as a longer cellular death process such as senescence rather than mitotic death or apoptosis). Progressively with time, the number of these DSB and SSB cumulate in cells to give a late subset of additional DNA damage.
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Britel, M.; Bourguignon, M.; Foray, N. Radiosensitivity: A term with various meanings at the origin of numerous confusions. A semantic analysis. Int. J. Radiat. Biol. 2018, 94, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Becquerel, H. Sur les radiations émises par phosphorescence. C. R. Acad. Sci. 1896, 122, 420–421. [Google Scholar]
- Regaud, C. Notice Sur les Travaux Scientifiques Publiés de 1893 à 1935; Presses Universitaires de France: Paris, France, 1935. [Google Scholar]
- Albers-Schönberg, H. Über die Benadlung des Lupus und des chronischen Ekzems mit Röntgenstrahlen. Fortschr. Rôntgenstr. 1898, 2, 20–29. [Google Scholar]
- Bouchacourt, L. Sur la différence de sensibilité aux rayons de Roentgen de la peau des différents sujets, et, sur le même sujet des différents régions du corps. In Proceedings of the Comptes-Rendus des Sessions de l’Association Française pour l’Avancement des Sciences, 40ème Congrès, Dijon, France, 1911; pp. 942–947. [Google Scholar]
- Frieben, A. Cancroid des rechten Handrückens. Dtsch. Med. Wochenschr. 1902, 28, 335. [Google Scholar]
- Gunderman, R.B.; Gonda, A.S. Radium Girls. Radiology 2015, 274, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Foray, N.; Bourguignon, M.; Hamada, N. Individual response to ionizing radiation. Mutat. Res. Mutat. Res. 2016, 770, 369–386. [Google Scholar] [CrossRef] [PubMed]
- ICRP. Radiation and Your Patient—A Guide for Medical Practitioners; Supporting Guidance Annals of ICRP; ICRP: Stockholm, Sweden, 2001; p. 31. [Google Scholar]
- Sinnott, B.; Ron, E.; Schneider, A.B. Exposing the Thyroid to Radiation: A Review of Its Current Extent, Risks, and Implications. Endocr. Rev. 2010, 31, 756–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.P. The lens is more sensitive to radiation than we had believed. Br. J. Ophthalmol. 1997, 81, 257. [Google Scholar] [CrossRef] [Green Version]
- Granzotto, A.; Benadjaoud, M.A.; Vogin, G.; Devic, C.; Ferlazzo, M.L.; Bodgi, L.; Pereira, S.; Sonzogni, L.; Forcheron, F.; Viau, M.; et al. Influence of Nucleoshuttling of the ATM Protein in the Healthy Tissues Response to Radiation Therapy: Toward a Molecular Classification of Human Radiosensitivity. Int. J. Radiat. Oncol. 2016, 94, 450–460. [Google Scholar] [CrossRef]
- Foray, N.; Bourguignon, M. Comment on ‘Considerations on the use of the terms radiosensitivity and radiosusceptibility’ by Wojcik et al. J. Radiol. Prot. 2019, 39, 309–313. [Google Scholar] [CrossRef]
- Le, A.N.; Harton, J.; Desai, H.; Powers, J.; Zelley, K.; Bradbury, A.R.; Nathanson, K.L.; Shah, P.D.; Doucette, A.; Freedman, G.M.; et al. Frequency of radiation-induced malignancies post-adjuvant radiotherapy for breast cancer in patients with Li-Fraumeni syndrome. Breast Cancer Res. Treat. 2020, 181, 181–188. [Google Scholar] [CrossRef]
- Amirifar, P.; Ranjouri, M.R.; Lavin, M.; Abolhassani, H.; Yazdani, R.; Aghamohammadi, A. Ataxia-telangiectasia: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Expert Rev. Clin. Immunol. 2020, 16, 859–871. [Google Scholar] [CrossRef]
- Schoenaker, M.; Suarez, F.; Szczepanski, T.; Mahlaoui, N.; Loeffen, J. Treatment of acute leukemia in children with ataxia telangiectasia (A–T). Eur. J. Med. Genet. 2016, 59, 641–646. [Google Scholar] [CrossRef]
- Vessoni, A.T.; Guerra, C.C.C.; Kajitani, G.S.; Nascimento, L.L.S.; Garcia, C.C.M. Cockayne Syndrome: The many challenges and approaches to understand a multifaceted disease. Genet. Mol. Biol. 2020, 43 (Suppl. S1), e20190085. [Google Scholar] [CrossRef]
- Lehmann, A.R. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 2003, 85, 1101–1111. [Google Scholar] [CrossRef]
- Larizza, L.; Roversi, G.; Volpi, L. Rothmund-Thomson syndrome. Orphanet J. Rare Dis. 2010, 5, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Halliday, D.; Parry, A.; Evans, D.G. Neurofibromatosis type 2 and related disorders. Curr. Opin. Oncol. 2019, 31, 562–567. [Google Scholar] [CrossRef]
- Pavlopoulou, A.; Bagos, P.G.; Koutsandrea, V.; Georgakilas, A.G. Molecular determinants of radiosensitivity in normal and tumor tissue: A bioinformatic approach. Cancer Lett. 2017, 403, 37–47. [Google Scholar] [CrossRef]
- Joubert, A.; Zimmerman, K.M.; Bencokova, Z.; Gastaldo, J.; Rénier, W.; Chavaudra, N.; Favaudon, V.; Arlett, C.F.; Foray, N. DNA double-strand break repair defects in syndromes associated with acute radiation response: At least two different assays to predict intrinsic radiosensitivity? Int. J. Radiat. Biol. 2008, 84, 107–125. [Google Scholar] [CrossRef]
- Morère, J.-F.; Mornex, F.; Soulières, D. Thérapeutique du Cancer; Springer: Paris, France, 2011. [Google Scholar]
- Trotti, A.; Colevas, A.; Setser, A.; Rusch, V.; Jaques, D.; Budach, V.; Langer, C.; Murphy, B.; Cumberlin, R.; Coleman, C.N.; et al. CTCAE v3.0: Development of a comprehensive grading system for the adverse effects of cancer treatment. Semin. Radiat. Oncol. 2003, 13, 176–181. [Google Scholar] [CrossRef]
- Cox, J.D.; Stetz, J.; Pajak, T.F. Toxicity criteria of the Radiation Therapy Oncology Group (RTOG) and the European organization for research and treatment of cancer (EORTC). Int. J. Radiat. Oncol. 1995, 31, 1341–1346. [Google Scholar] [CrossRef]
- Puck, T.T.; Marcus, P.I. Action of x-rays on mammalian cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef]
- Kellerer, A.M.; Rossi, H.H. The theory of dual radiation action. Curr. Top. Radiat. Res. 1972, 8, 85–158. [Google Scholar]
- Chadwick, K.H.; Leenhouts, H.P. A molecular theory of cell survival. Phys. Med. Biol. 1973, 18, 78–87. [Google Scholar] [CrossRef]
- Bodgi, L.; Canet, A.; Pujo-Menjouet, L.; Lesne, A.; Victor, J.-M.; Foray, N. Mathematical models of radiation action on living cells: From the target theory to the modern approaches. A historical and critical review. J. Theor. Biol. 2016, 394, 93–101. [Google Scholar] [CrossRef]
- Fertil, B.; Malaise, E.-P. Inherent cellular radiosensitivity as a basic concept for human tumor radiotherapy. Int. J. Radiat. Oncol. 1981, 7, 621–629. [Google Scholar] [CrossRef]
- Grote, S.; Joshi, G.; Revell, S.; Shaw, C. Observations of Radiation-induced Chromosome Fragment Loss in Live Mammalian Cells in Culture, and Its Effect on Colony-forming Ability. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1981, 39, 395–408. [Google Scholar] [CrossRef]
- Darroudi, F.; Fomina, J.; Meijers, M.; Natarajan, A. Kinetics of the formation of chromosome aberrations in X-irradiated human lymphocytes, using PCC and FISH. Mutat. Res. Mol. Mech. Mutagen. 1998, 404, 55–65. [Google Scholar] [CrossRef]
- Cornforth, M.N.; Bedford, J.S. A quantitative comparison of potentially lethal damage repair and the rejoining of interphase chromosome breaks in low passage normal human fibroblasts. Radiat. Res. 1987, 111, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Metcalfe, J.A.; Thick, J.; Mak, Y.F. Leukemia and lymphoma in ataxia telangiectasia. Blood 1996, 87, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.R.; Harnden, D.G.; Arlett, C.F.; Harcourt, S.A.; Lehmann, A.R.; Stevens, S.; Bridges, B.A. Ataxia telangiectasia: A human mutation with abnormal radiation sensitivity. Nature 1975, 258, 427–429. [Google Scholar] [CrossRef]
- Morgan, J.L.; Holcomb, T.M.; Morrissey, R.W. Radiation reaction in ataxia telangiectasia. Am. J. Dis. Childr. 1968, 116, 557–558. [Google Scholar] [CrossRef]
- Tamminga, R.Y.J.; Dolsma, W.V.; Leeuw, J.A.; Kampinga, H.H. Chemo- and radiosensitivity testing in a patient with ataxia telangiectasia and hodgkin disease. Pediatr. Hematol. Oncol. 2002, 19, 163–171. [Google Scholar] [CrossRef]
- Sandoval, C.; Swift, M. Hodgkin disease in ataxia-telangiectasia patients with poor outcomes. Med. Pediatr. Oncol. 2003, 40, 162–166. [Google Scholar] [CrossRef]
- Pietrucha, B.M.; Heropolitanska-Pliszka, E.; Wakulinska, A.; Skopczynska, H.; Gatti, R.A.; Bernatowska, E. Ataxia-telangiectasia with hyper-IgM and Wilms tumor: Fatal reaction to irradiation. J. Pediatr. Hematol. Oncol. 2010, 32, e28–e30. [Google Scholar] [CrossRef]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; A Tagle, D.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Savitsky, K.; Sfez, S.; Tagle, D.A.; Ziv, Y.; Sartiel, A.; Collins, F.S.; Shiloh, Y.; Rotman, G. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Mol. Genet. 1995, 4, 2025–2032. [Google Scholar] [CrossRef]
- Badie, C.; Iliakis, G.; Foray, N.; Alsbeih, G.; E Pantellias, G.; Okayasu, R.; Cheong, N.; Russell, N.S.; Begg, A.C.; Arlett, C.F. Defective repair of DNA double-strand breaks and chromosome damage in fibroblasts from a radiosensitive leukemia patient. Cancer Res. 1995, 55, 1232–1234. [Google Scholar]
- Badie, C.; Goodhardt, M.; Waugh, A.; Doyen, N.; Foray, N.; Calsou, P.; Singleton, B.; Gell, D.; Salles, B.; Jeggo, P.; et al. A DNA double-strand break defective fibroblast cell line (180BR) derived from a radiosensitive patient represents a new mutant phenotype. Cancer Res. 1997, 57, 4600–4607. [Google Scholar]
- Riballo, E.; Critchlow, S.; Teo, S.-H.; Doherty, A.; Priestley, A.; Broughton, B.; Kysela, B.; Beamish, H.; Plowman, N.; Arlett, C.; et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol. 1999, 9, 699–S2. [Google Scholar] [CrossRef] [Green Version]
- Altmann, T.; Gennery, A.R. DNA ligase IV syndrome: A review. Orphanet J. Rare Dis. 2016, 11, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Distel, L.; Neubauer, S.; Varon, R.; Holter, W.; Grabenbauer, G. Fatal toxicity following radio- and chemotherapy of medulloblastoma in a child with unrecognized Nijmegen Breakage Syndrome. Med. Pediatr. Oncol. 2003, 41, 44–48. [Google Scholar] [CrossRef]
- Chrzanowska, K.H.; Gregorek, H.; Dembowska-Bagińska, B.; A Kalina, M.; Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 2012, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Dembowska-Baginska, B.; Perek, D.; Brozyna, A.; Wakulinska, A.; Olczak-Kowalczyk, D.; Gladkowska-Dura, M.; Grajkowska, W.; Chrzanowska, K.H. Non-Hodgkin lymphoma (NHL) in children with Nijmegen Breakage syndrome (NBS). Pediatr. Blood Cancer 2009, 52, 186–190. [Google Scholar] [CrossRef]
- Matsuura, S.; Weemaes, C.; Smeets, D.; Takami, H.; Kondo, N.; Sakamoto, S.; Yano, N.; Nakamura, A.; Tauchi, H.; Endo, S.; et al. Genetic Mapping Using Microcell-Mediated Chromosome Transfer Suggests a Locus for Nijmegen Breakage Syndrome at Chromosome 8q21-24. Am. J. Hum. Genet. 1997, 60, 1487–1494. [Google Scholar] [CrossRef] [Green Version]
- Rogers, P.B.; Plowman, P.N.; Harris, S.J.; Arlett, C.F. Four radiation hypersensitivity cases and their implications for clinical radiotherapy. Radiother. Oncol. 2000, 57, 143–154. [Google Scholar] [CrossRef]
- Abbaszadeh, F.; Clingen, P.H.; Arlett, C.F.; Plowman, P.N.; Bourton, E.C.; Themis, M.; Makarov, E.; Newbold, R.F.; Green, M.H.L.; Parris, C.N. A novel splice variant of the DNA-PKcs gene is associated with clinical and cellular radiosensitivity in a patient with xeroderma pigmentosum. J. Med. Genet. 2009, 47, 176–181. [Google Scholar] [CrossRef]
- Arlett, C.F.; Plowman, P.N.; Rogers, P.B.; Parris, C.N.; Abbaszadeh, F.; Green, M.H.L.; McMillan, T.J.; Bush, C.; Foray, N.; Lehmann, A.R. Clinical and cellular ionizing radiation sensitivity in a patient with xeroderma pigmentosum. Br. J. Radiol. 2006, 79, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Corry, J.; Rischin, D.; Leong, T.; Peters, L. Combined modality treatment for locally advanced squamous-cell carcinoma of the oropharynx in a woman with Bloom’s syndrome: A case report and review of the literature. Ann. Oncol. 2001, 12, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Radiosensitivity in Fanconi’s anemia patients. Radiother. Oncol. 2002, 62, 345–347. [Google Scholar] [CrossRef]
- Arlett, C.F.; A Harcourt, S. Survey of radiosensitivity in a variety of human cell strains. Cancer Res. 1980, 40, 926–932. [Google Scholar]
- Little, J.B.; Nichols, W.W.; Troilo, P.; Nagasawa, H.; Strong, L.C. Radiation sensitivity of cell strains from families with genetic disorders predisposing to radiation-induced cancer. Cancer Res. 1989, 49, 4705–4714. [Google Scholar]
- Deschavanne, P.J.; Fertil, B. A review of human cell radiosensitivity in vitro. Int. J. Radiat. Oncol. 1996, 34, 251–266. [Google Scholar] [CrossRef]
- Varela, I.; Pereira, S.; Ugalde, A.P.; Navarro, C.; Suárez, M.F.; Cau, P.; Cadiñanos, J.; Osorio, F.G.; Foray, N.; Cobo, J.; et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 2008, 14, 767–772. [Google Scholar] [CrossRef]
- Ozgenc, A.; Loeb, L. Werner Syndrome, Aging and Cancer. Genome Dyn. 2006, 1, 206–217. [Google Scholar] [CrossRef]
- Kremer, H.; Van Wijk, E.; Märker, T.; Wolfrum, U.; Roepman, R. Usher syndrome: Molecular links of pathogenesis, proteins and pathways. Hum. Mol. Genet. 2006, 15, R262–R270. [Google Scholar] [CrossRef] [Green Version]
- Berthel, E.; Foray, N.; Ferlazzo, M.L. The Nucleoshuttling of the ATM Protein: A Unified Model to Describe the Individual Response to High- and Low-Dose of Radiation? Cancers 2019, 11, 905. [Google Scholar] [CrossRef] [Green Version]
- Pastwa, E.; Blasiak, J. Non-homologous DNA end joining. Acta Biochim. Pol. 2003, 50, 891–908. [Google Scholar] [CrossRef] [Green Version]
- Dudáš, A.; Chovanec, M. DNA double-strand break repair by homologous recombination. Mutat. Res. Mutat. Res. 2004, 566, 131–167. [Google Scholar] [CrossRef]
- Şimşek, D.; Brunet, E.; Wong, S.Y.-W.; Katyal, S.; Gao, Y.; McKinnon, P.J.; Lou, J.; Zhang, L.; Li, J.; Rebar, E.J.; et al. DNA Ligase III Promotes Alternative Nonhomologous End-Joining during Chromosomal Translocation Formation. PLoS Genet. 2011, 7, e1002080. [Google Scholar] [CrossRef] [Green Version]
- De Villartay, J.P. V(D)J recombination deficiencies. Adv. Exp. Med. Biol. 2009, 650, 46–58. [Google Scholar]
- Woodbine, L.; Gennery, A.R.; Jeggo, P.A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA repair 2014, 16C, 84–96. [Google Scholar] [CrossRef]
- Epstein, J.; Williams, J.R.; Little, J.B. Deficient DNA Repair in Human Progeroid Cells. Proc. Natl. Acad. Sci. USA 1973, 70, 977–981. [Google Scholar] [CrossRef] [Green Version]
- Navarro, C.; Cau, P.; Lévy, N. Molecular bases of progeroid syndromes. Hum. Mol. Genet. 2006, 15, R151–R161. [Google Scholar] [CrossRef] [Green Version]
- Nove, J.; Tarone, R.E.; Little, J.B.; Robbins, J.H. Radiation sensitivity of fibroblast strains from patients with Usher’s syndrome, Duchenne muscular dystrophy, and Huntington’s disease. Mutat. Res. 1987, 184, 29–38. [Google Scholar] [CrossRef]
- Huo, Y.K.; Wang, Z.; Hong, J.H.; Chessa, L.; McBride, W.H.; Perlman, S.L.; A Gatti, R. Radiosensitivity of ataxia-telangiectasia, X-linked agammaglobulinemia, and related syndromes using a modified colony survival assay. Cancer Res. 1994, 54, 2544–2547. [Google Scholar]
- Mattsson, P.T.; Vihinen, M.; Smith, C.I.E. X-linked agammaglobulinemia (XLA): A genetic tyrosine kinase (Btk) disease. Bioessays 1996, 18, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Arlett, C.F.; Muriel, W.J. Radiosensitivity in Huntington’s disease. Heredity 1979, 42, 276. [Google Scholar]
- Ferlazzo, M.L.; Foray, N. Huntington Disease: A Disease of DNA Methylation or DNA Breaks? Am. J. Pathol. 2016, 186, 1750–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlazzo, M.L.; Sonzogni, L.; Granzotto, A.; Bodgi, L.; Lartin, O.; Devic, C.; Vogin, G.; Pereira, S.; Foray, N. Mutations of the Huntington’s Disease Protein Impact on the ATM-Dependent Signaling and Repair Pathways of the Radiation-Induced DNA Double-Strand Breaks: Corrective Effect of Statins and Bisphosphonates. Mol. Neurobiol. 2013, 49, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Preston, D.L.; Shimizu, Y.; A Pierce, D.; Suyama, A.; Mabuchi, K. Studies of mortality of atomic bomb survivors. Report 13: Solid cancer and noncancer disease mortality: 1950–1997. Radiat. Res. 2012, 178, 146–172. [Google Scholar] [CrossRef] [Green Version]
- Wakeford, R. The cancer epidemiology of radiation. Oncogene 2004, 23, 6404–6428. [Google Scholar] [CrossRef] [Green Version]
- Boice, J.D., Jr. The linear nonthreshold (LNT) model as used in radiation protection: An NCRP update. Int. J. Radiat. Biol. 2017, 93, 1079–1092. [Google Scholar] [CrossRef]
- Calabrese, E.J. Origin of the linearity no threshold (LNT) dose–response concept. Arch. Toxicol. 2013, 87, 1621–1633. [Google Scholar] [CrossRef]
- Devic, C.; Ferlazzo, M.L.; Berthel, E.; Foray, N. Influence of Individual Radiosensitivity on the Hormesis Phenomenon: Toward a Mechanistic Explanation Based on the Nucleoshuttling of ATM Protein. Dose-Response 2020, 18, 1559325820913784. [Google Scholar] [CrossRef]
- Walter, S.D.; Harbour, J.W. Molecular biology of retinoblastoma. In Recent Advances in Retinoblastoma Treatment; Francis, J.H., Abramson, D.H., Eds.; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Rouas-Freiss, N.; Moreau, P.; LeMaoult, J.; Carosella, E.D. The Dual Role of HLA-G in Cancer. J. Immunol. Res. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, C.; Prasad, A.R.; Nfonsam, V.; Bernstein, H. DNA damage, DNA repair and cancer. In New Research Directions in DNA Repair; Chen, C., Ed.; Intech Publisher: Rijeka, Croatia, 2013; Chapter 16; pp. 413–465. [Google Scholar]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Duesberg, P.H.; Zhou, R.-P.; Goodrich, D. Cancer Genes by Illegitimate Recombination. Ann. N. Y. Acad. Sci. 1989, 567, 259–273. [Google Scholar] [CrossRef]
- Huang, L.; Grim, S.; Smith, L.E.; Kim, P.M.; Nickoloff, J.A.; Goloubeva, O.G.; Morgan, W.F. Ionizing Radiation Induces Delayed Hyperrecombination in Mammalian Cells. Mol. Cell. Biol. 2004, 24, 5060–5068. [Google Scholar] [CrossRef] [Green Version]
- Meyn, M.S. High spontaneous intrachromosomal recombination rates in ataxia-telangiectasia. Science 1993, 260, 1327–1330. [Google Scholar] [CrossRef]
- Albertini, R.J. HPRT mutations in humans: Biomarkers for mechanistic studies. Mutat. Res. Mol. Mech. Mutagen. 2001, 489, 1–16. [Google Scholar] [CrossRef]
- Parshad, R.; Sanford, K.K.; Jones, G.M. Chromatid damage after G2 phase x-irradiation of cells from cancer-prone individuals implicates deficiency in DNA repair. Proc. Natl. Acad. Sci. USA 1983, 80, 5612–5616. [Google Scholar] [CrossRef] [Green Version]
- Riches, A.C.; Bryant, P.E.; Steel, C.M.; Gleig, A.; Robertson, A.J.; Preece, P.E.; Thompson, A.M. Chromosomal radiosensitivity in G2-phase lymphocytes identifies breast cancer patients with distinctive tumour characteristics. Br. J. Cancer 2001, 85, 1157–1161. [Google Scholar] [CrossRef] [Green Version]
- Sanford, K.; Parshad, R.; Gantt, R.; Tarone, R.; Jones, G.; Price, F. Factors Affecting and Significance of G2Chromatin Radiosensitivity in Predisposition to Cancer. Int. J. Radiat. Biol. 1989, 55, 963–981. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Cavanagh, H.; Rogers, K.M. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered. Cancer Clin. Pract. 2015, 13, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, A.; Yoshida, Y.; Tanaka, H.; Arima, M.; Ohno, K. Variable Radiosensitivity in Fibroblasts from Patients with Tuberous Sclerosis. J. Investig. Dermatol. 1985, 84, 77–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, Y.; Hayashi, A.; Arima, M. Rapid rejoining of X-ray-induced DNA single-strand breaks in tuberous sclerosis fibroblasts. Mutat. Res. 1985, 146, 211–218. [Google Scholar]
- Henske, E.P.; Jozwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nature reviews. Dis. Primers 2016, 2, 16035. [Google Scholar] [CrossRef] [PubMed]
- Ferlazzo, M.L.; Bach-Tobdji, M.K.E.; Djerad, A.; Sonzogni, L.; Devic, C.; Granzotto, A.; Bodgi, L.; Bachelet, J.-T.; Djefal-Kerrar, A.; Hennequin, C.; et al. Radiobiological Characterization of Tuberous Sclerosis: A Delay in the Nucleo-Shuttling of ATM May Be Responsible for Radiosensitivity. Mol. Neurobiol. 2017, 55, 4973–4983. [Google Scholar] [CrossRef] [PubMed]
- Woods, W.G.; McKenzie, B.; Letourneau, M.A.; Byrne, T.D. Sensitivity of Cultured Skin Fibroblasts from Patients with Neurofibromatosis to DNA-Damaging Agents. Ann. N. Y. Acad. Sci. 1986, 486, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Krone, W.; Nothdurft, W.; Reisacher, A.; Gall, H. Cell-culture studies on neurofibromatosis (von Recklinghausen’s disease). III. Experiments on X-ray sensitivity. Arch. Dermatol. Res. 1985, 277, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Hafez, M.; Sharaf, L.; Abd el-Nabi, S.M.; el-Wehedy, G. Evidence of chromosomal instability in neurofibromatosis. Cancer 1985, 55, 2424–2436. [Google Scholar] [CrossRef]
- Hannan, M.A.; Sackey, K.; Sigut, D. Cellular radiosensitivity of patients with different types of neurofibromatosis. Cancer Genet. Cytogenet. 1993, 66, 120–125. [Google Scholar] [CrossRef]
- Ferner, R.E. Neurofibromatosis 1 and neurofibromatosis 2: A twenty first century perspective. Lancet Neurol. 2007, 6, 340–351. [Google Scholar] [CrossRef]
- Vulin, A.; Sedkaoui, M.; Moratille, S.; Sevenet, N.; Soularue, P.; Rigaud, O.; Guibbal, L.; Dulong, J.; Jeggo, P.; Deleuze, J.-F.; et al. Severe PATCHED1 Deficiency in Cancer-Prone Gorlin Patient Cells Results in Intrinsic Radiosensitivity. Int. J. Radiat. Oncol. 2018, 102, 417–425. [Google Scholar] [CrossRef]
- Thalakoti, S.; Geller, T. Basal cell nevus syndrome or Gorlin syndrome. Handb. Clin. Neurol. 2015, 132, 119–128. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Chen, Z.; Li, Y.; Luo, X.; Li, Y. Effect of high energy electron beam on proteolysis and antioxidant activity of rice proteins. Food Funct. 2020, 11, 871–882. [Google Scholar] [CrossRef]
- Lakhdar, I.M.; Ferlazzo, M.L.; Al Choboq, J.; Berthel, E.; Sonzogni, L.; Devic, C.; Granzotto, A.; Thariat, J.; Foray, N. Fibroblasts from Retinoblastoma Patients Show Radiosensitivity Linked to Abnormal Localization of the ATM Protein. Curr. Eye Res. 2020, 1–12. [Google Scholar] [CrossRef]
- Foray, N.; Randrianarison, V.; Marot, D.; Perricaudet, M.; Lenoir, G.; Feunteun, J. Gamma-rays-induced death of human cells carrying mutations of BRCA1 or BRCA2. Oncogene 1999, 18, 7334–7342. [Google Scholar] [CrossRef] [Green Version]
- Ferlazzo, M.; Berthel, E.; Granzotto, A.; Devic, C.; Sonzogni, L.; Bachelet, J.-T.; Pereira, S.; Bourguignon, M.; Sarasin, A.; Mezzina, M.; et al. Some mutations in the xeroderma pigmentosum D gene may lead to moderate but significant radiosensitivity associated with a delayed radiation-induced ATM nuclear localization. Int. J. Radiat. Biol. 2019, 96, 394–410. [Google Scholar] [CrossRef]
- Kim, S.-T.; Lim, D.-S.; Canman, C.E.; Kastan, M.B. Substrate Specificities and Identification of Putative Substrates of ATM Kinase Family Members. J. Biol. Chem. 1999, 274, 37538–37543. [Google Scholar] [CrossRef] [Green Version]
- Foray, N.; Marot, D.; Gabriel, A.; Randrianarison, V.; Carr, A.M.; Perricaudet, M.; Ashworth, A.; Jeggo, P. A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J. 2003, 22, 2860–2871. [Google Scholar] [CrossRef] [Green Version]
- Belkacemi, Y.; Colson-Durand, L.; Granzotto, A.; Husheng, S.; To, N.H.; Majdoul, S.; Guet, S.; Hervé, M.-L.; Fonteneau, G.; Diana, C.; et al. The Henri Mondor Procedure of Morbidity and Mortality Review Meetings: Prospective Registration of Clinical, Dosimetric, and Individual Radiosensitivity Data of Patients With Severe Radiation Toxicity. Int. J. Radiat. Oncol. 2016, 96, 629–636. [Google Scholar] [CrossRef]
- Pereira, S.; Bodgi, L.; Duclos, M.; Canet, A.; Ferlazzo, M.L.; Devic, C.; Granzotto, A.; Deneuve, S.; Vogin, G.; Foray, N. Fast and binary assay for predicting radiosensitivity based on the nucleoshuttling of ATM protein: Development, validation and performances. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 353–360. [Google Scholar] [CrossRef]
- Deneuve, S.; Mirjolet, C.; Bastogne, T.; Duclos, M.; Retif, P.; Zrounba, P.; Roux, P.-E.; Poupart, M.; Vogin, G.; Foray, N.; et al. Proof of Concept of a Binary Blood Assay for Predicting Radiosensitivity. Cancers 2021, 13, 2477. [Google Scholar] [CrossRef]
- Bodgi, L.; Foray, N. The nucleo-shuttling of the ATM protein as a basis for a novel theory of radiation response: Resolution of the linear-quadratic model*. Int. J. Radiat. Biol. 2016, 92, 117–131. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Syndromes | Mutated Genes | Major Defective Mechanism | Prevalence per 100,000 | SF2 (%) | Cancer Predisposition | Aging Neurodegeneration | Immuno- Deficiency | Subcellular Localization of the Protein |
|---|---|---|---|---|---|---|---|---|
| Ataxia telangiectasia | Homoz ATM mutations | DSB signaling and repair | ~1 | 1–5 | Leukemia, Lymphoma | No | Yes | Nucleus Cytoplasm |
| Ligase IV syndrome | Homoz LIG4 mutations | NHEJ | Few cases | 2–6 | Leukemia, Lymphoma | No | Yes | Nucleus |
| Nijmegen’s syndrome | Homoz NBS1 mutations | DSB signaling and repair | ~1 | 5–9 | Leukemia, Lymphoma | No | Yes | Nucleus |
| Hutchinson-Gilford Progeria syndrome | Heteroz* LMNA mutations | Nuclear membrane | 0.12–0.25 | 8–19 | No | Yes | No? | Inner nuclear membrane |
| Agamma-globulinemia Bruton’s disease | X-linked homoz BTK mutations | V(D)J recombination | 1.4–2.8 | 10 | No Some cases of colorectal cancer due to infections | No | Yes | Nucleus Cytoplasm |
| Hypogamma-globulinemia Lig I deficiency | compound heteroz LIGI mutations | NER | one case | 11 | No | No | Yes | Nucleus Golgi apparatus Vesicles |
| ICF syndrome | Homoz, compound heteroz, DNMT3B mutations | DNA methylation | ~50 cases | 14 | No? | Yes? | Yes | Nucleus but also cytoplasm in mutated cells |
| Glutathione synthetase deficiency | most compound heteroz GSS mutations | Glutathione cycle | ~70 cases ~0.1 | 14 | No | Cerebellar degeneration in some severe cases | No? | Nucleus |
| NBSLD Syndrome | Homoz, compound heteroz RAD50 mutations | Few cases | 15 | No? | Yes? | No | Nucleus | |
| ATLD Syndrome | Homoz or compound heteroz MRE11 mutations | Few cases | 15–30 | No | Yes? | No | Nucleus Cytoplasm | |
| Cockayne’s syndrome | Homoz or compound heteroz CS mutations | NER/TCR | 0.4 | 15–30 | No | Yes | No | Nucleus |
| Xeroderma pigmentosum | Homoz or compound heteroz XP mutations | NER/TCR | 0.4 to 1 | 15–30 | Skin cancer | Yes | No | Nucleus only, except for XPD (both nucleus and cytoplasm) |
| Usher’s syndrome | Homoz USH mutations | 3–5 | 16 | No | Yes? | No | Cytoplasm | |
| Huntington’s disease | Heteroz (gain-of-function) HTT mutations | DNA methylation | 4–7 | 19 | No | Yes | No | Nucleus Cytoplasm |
| Duchesne’s dystrophy | X-linked DMD mutations | 1–9 | 16–28 | No | Yes | No | Cytoplasm | |
| Fanconi Anemia | Homoz or heteroz X-linked FANC (A to D) mutations | 1 | 15–40 | Leukemia, squamous cell carcinoma Breast cancer | No | Yes | Nucleus only, except for FANCD both nucleus and cytoplasm | |
| Bloom’s Syndrome | Homoz or compound heteroz BLM mutations | HR/TLS | 0.5–2 | 15–40 | leukemia, lymphoma | No | Yes | Nucleus Cytoplasm |
| Gorlin’s (NF2) syndrome | Heteroz or de novo PTCH1 mutations | 1–9 | 12–30 | Non-melanoma skin cancer | No | No | Golgi apparatus Cytoplasm domains | |
| Tuberous sclerosis Complex syndrome | Heteroz TSC mutations | DSB signaling and repair | 4–10 | 24 | CMS and PMS tumors | No | No | Cytoplasm |
| Von Recklinghausen (NF1) syndrome | Heteroz or de novo NF1 mutations | DSB signaling and repair | 200–300 | 15–35 | CMS and PMS tumors | No | No | Nucleus Cytoplasm |
| Li-Fraumeni syndrome | Heteroz p53 mutations | Cell cycle regulation | 4–10 | 20-50 | breast, brain, leukemia, sarcoma | No | No | Nucleus Cytoplasm |
| Gardner’s syndrome | Heteroz APC mutations | Cell adhesion | 2.2–3.2 | 18–30 | Mainly colorectal cancer | No | No | Nucleus Golgi apparatus |
| Turcot’s syndrome | Homoz, compound heteroz, heteroz MLH mutations | MMR | ~150 cases | 21–30 | Mainly colorectal cancer | No | No | Nucleus |
| Hereditary retinoblastoma | Heteroz RB1 mutations | Cell cycle regulation | 5–7 | 25–35 | Retinoblastoma, sarcoma, melanoma, lung and breast cancer | No | No | Nucleus but also cytoplasm in mutated cells |
| Hereditary breast/ovary cancer | Heteroz BRCA2 mutations | HR | ~125 | 20–40 | Breast/ovary cancer | No | No | Nucleus Cytoplasm |
| Hereditary breast/ovary cancer | Heteroz BRCA1 mutations | HR | ~333 | 30–50 | Breast/ovary cancer | No | No | Nucleus Cytoplasm |
| AT heterozygotes | Heteroz ATM mutations | DSB signaling and repair | 1000 | 20–55 | High risk of breast cancer | No | No | Nucleus Cytoplasm |
| Werner syndrome | Homoz or compound heteroz WRN mutations | HR/TLS | 2.5–5 | 20–55 | some rare cancers | Yes | No | Nuclear Cytoplasm for some mutations |
| Rothmund-Thomson syndrome | Homoz or compound heteroz RecQL4mutations | HR/TLS | ~300 cases | 30–50 | osteosarcoma | Yes | No | Nucleus Cytoplasm |
| Severe combined immunodeficiency | Homoz or compound heteroz Cernnunos or Artemis mutations | V(D)J recombination NHEJ | ~33 | 30–50 | Some rare lymphoma | No | Yes | Nucleus |
| Down’s syndrome | Chromosome 21 trisomy | 100–150 | 25 | High risk of ALL and AML | Yes | Yes | - | |
| Lynch’s syndrome | Heteroz MLH1, MSH2/6, hPMS2 mutations | MMR | 100–125 | 30–50 | Mainly Colorectal cancer | No | No | Nucleus |
| Alzheimer’s disease | 2000–4000 | No? | Yes | No | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Nachef, L.; Al-Choboq, J.; Restier-Verlet, J.; Granzotto, A.; Berthel, E.; Sonzogni, L.; Ferlazzo, M.L.; Bouchet, A.; Leblond, P.; Combemale, P.; et al. Human Radiosensitivity and Radiosusceptibility: What Are the Differences? Int. J. Mol. Sci. 2021, 22, 7158. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137158
El-Nachef L, Al-Choboq J, Restier-Verlet J, Granzotto A, Berthel E, Sonzogni L, Ferlazzo ML, Bouchet A, Leblond P, Combemale P, et al. Human Radiosensitivity and Radiosusceptibility: What Are the Differences? International Journal of Molecular Sciences. 2021; 22(13):7158. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137158
Chicago/Turabian StyleEl-Nachef, Laura, Joelle Al-Choboq, Juliette Restier-Verlet, Adeline Granzotto, Elise Berthel, Laurène Sonzogni, Mélanie L. Ferlazzo, Audrey Bouchet, Pierre Leblond, Patrick Combemale, and et al. 2021. "Human Radiosensitivity and Radiosusceptibility: What Are the Differences?" International Journal of Molecular Sciences 22, no. 13: 7158. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137158