
Muricholic Acids Promote Resistance to Hypercholesterolemia in Cholesterol-Fed Mice

 , ,
, , 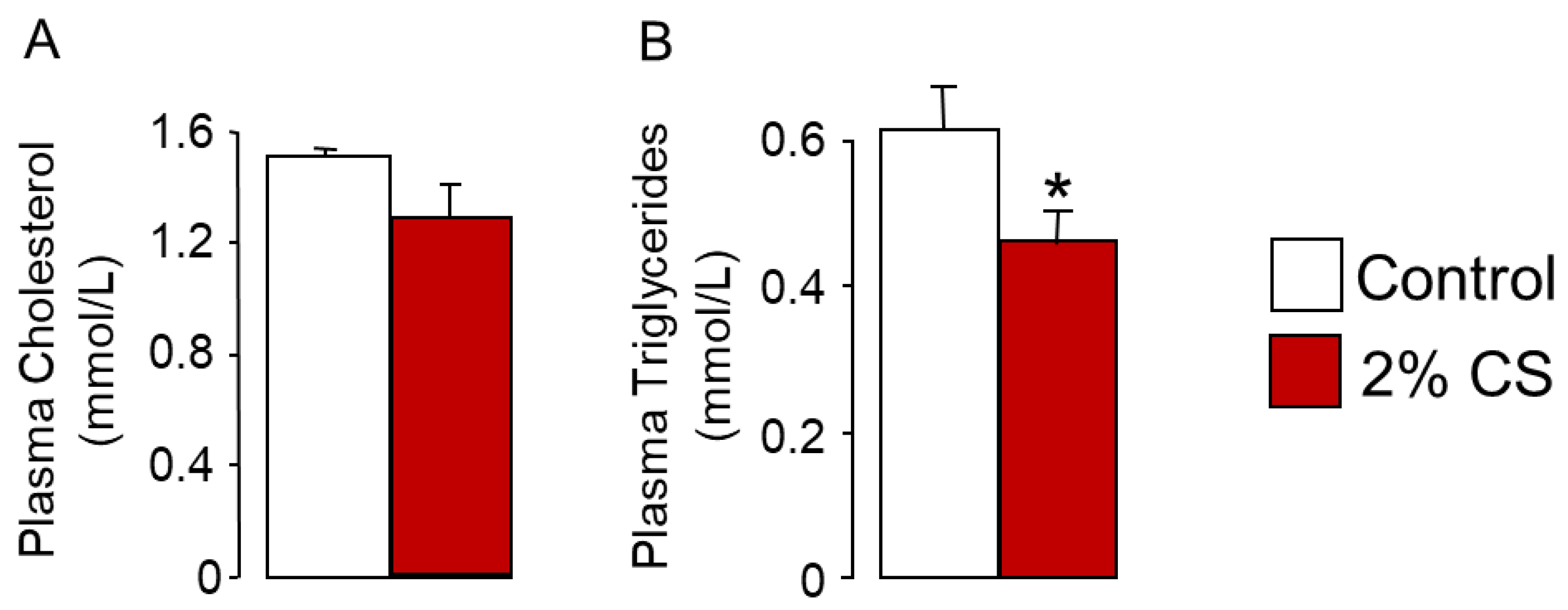{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
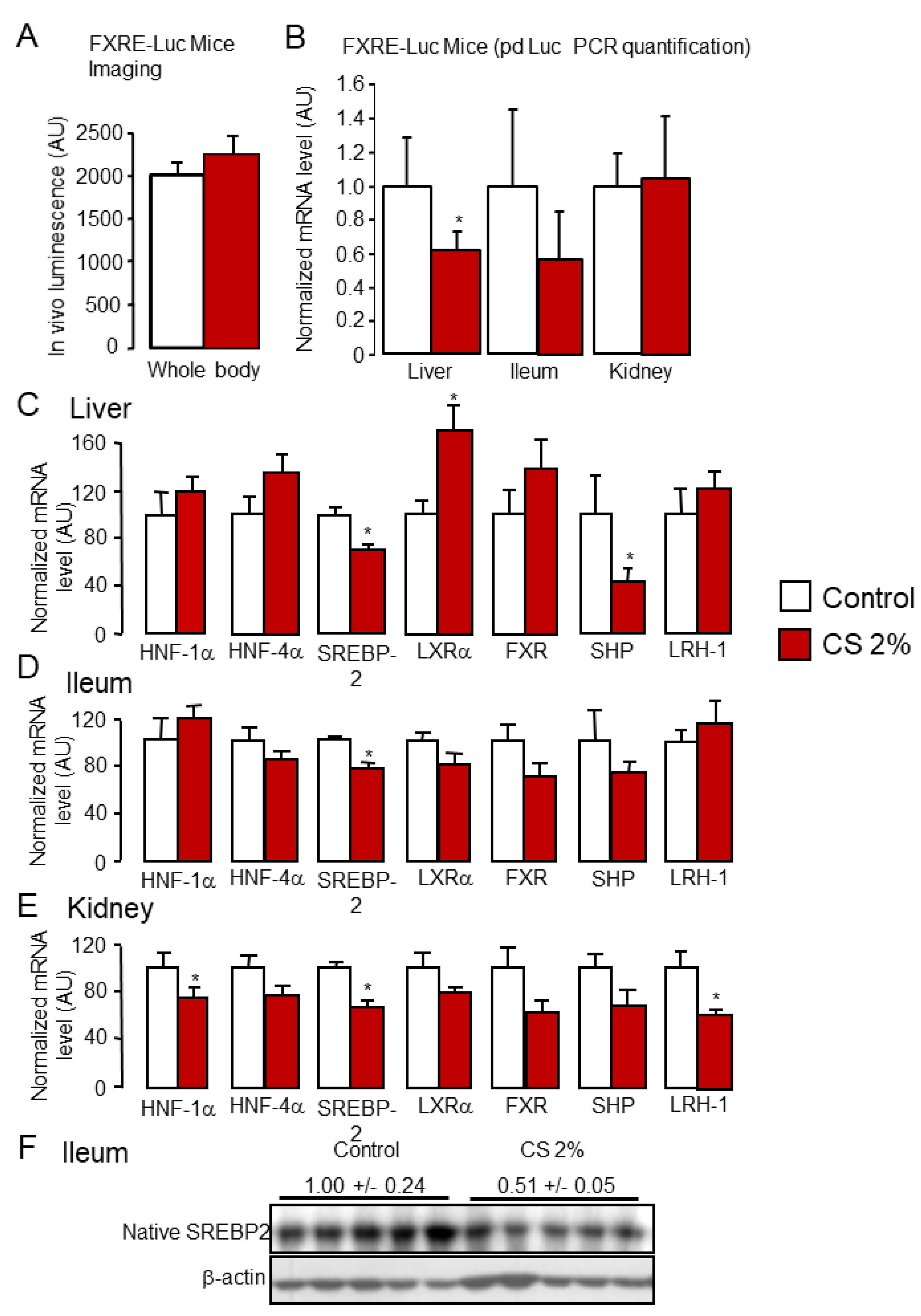
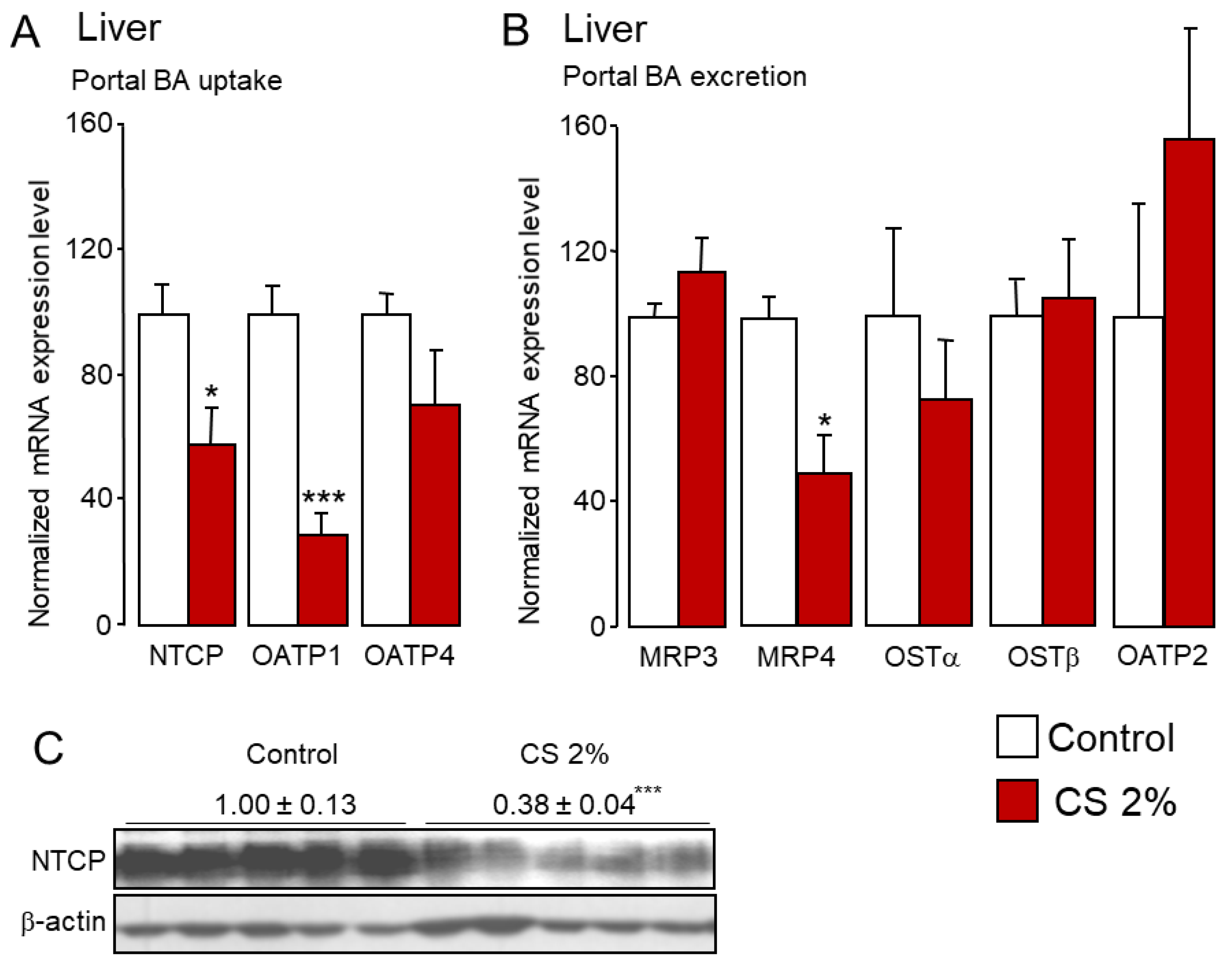
2.1. Cholesterol Feeding Reduces FXR Signaling through Enhanced Hydrophilic Bile Acid Synthesis
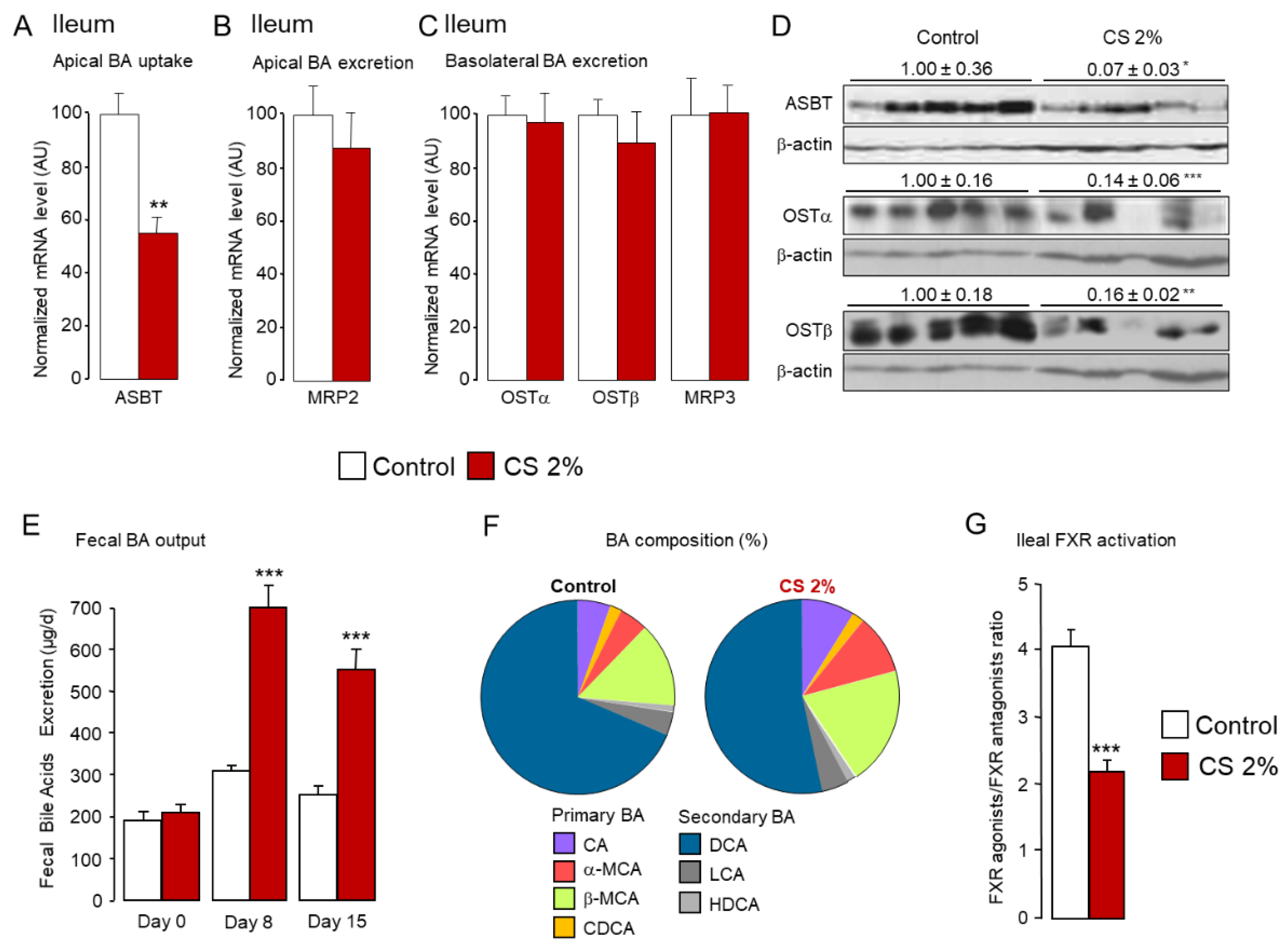
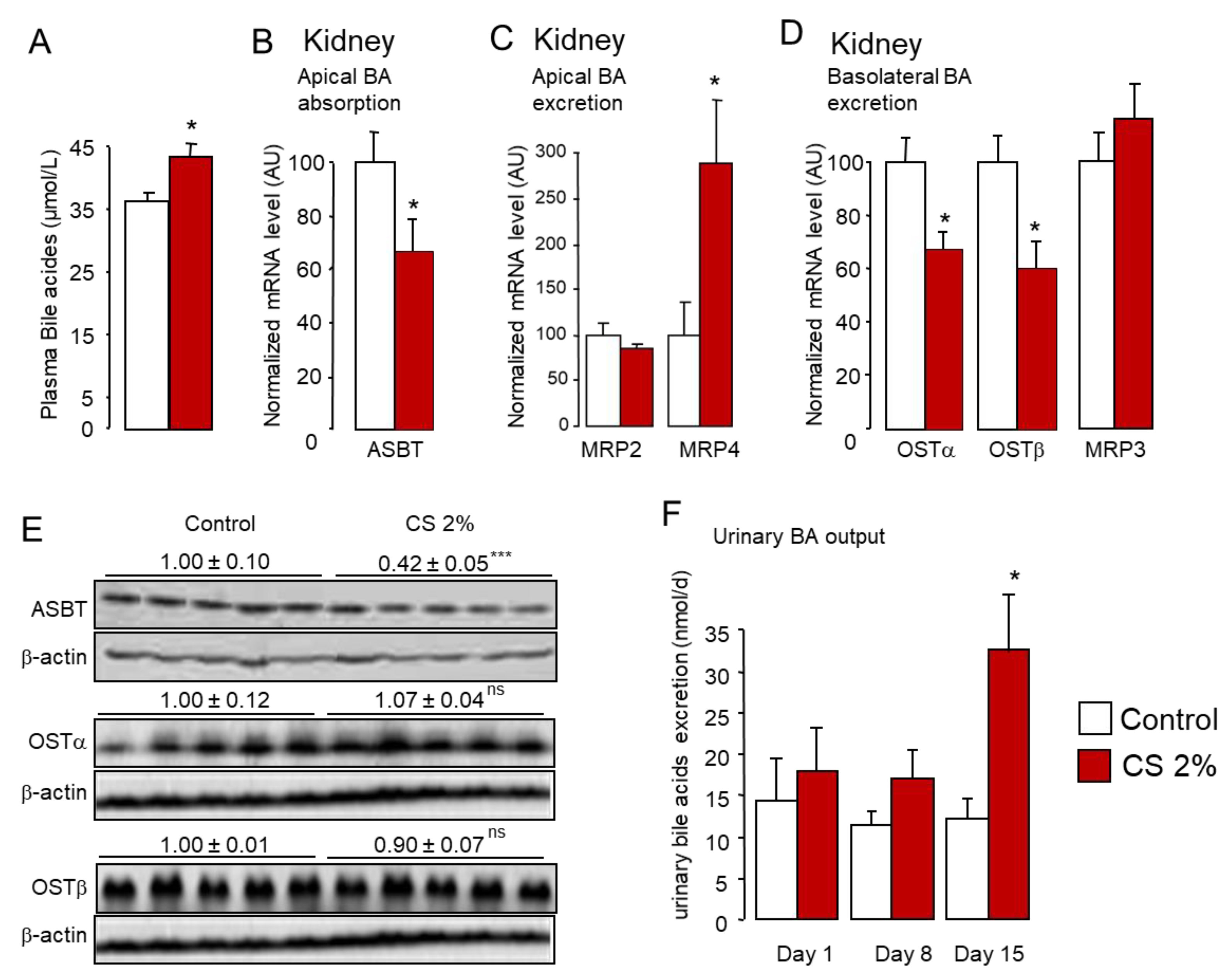
2.2. Cholesterol Feeding Promotes Fecal and Urinary Excretion of De Novo Synthesized Hydrophilic Bile Acids
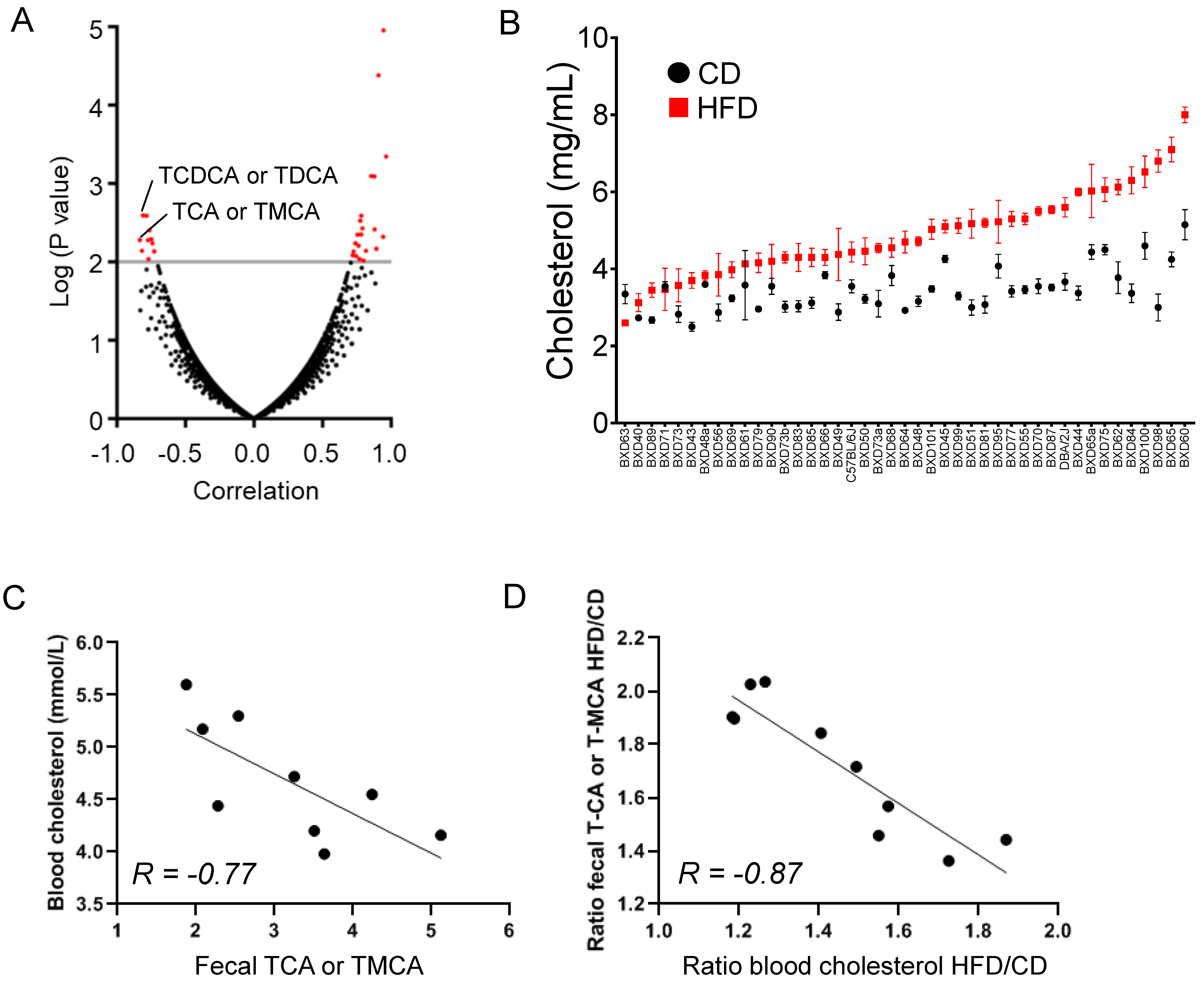
2.3. Fecal Elimination of Hydrophilic BAs Is a Major Route for Body CS Removal upon Dietary CS Overload
2.4. Fecal Cholesterol Elimination Is Increased in CS-Fed Mice


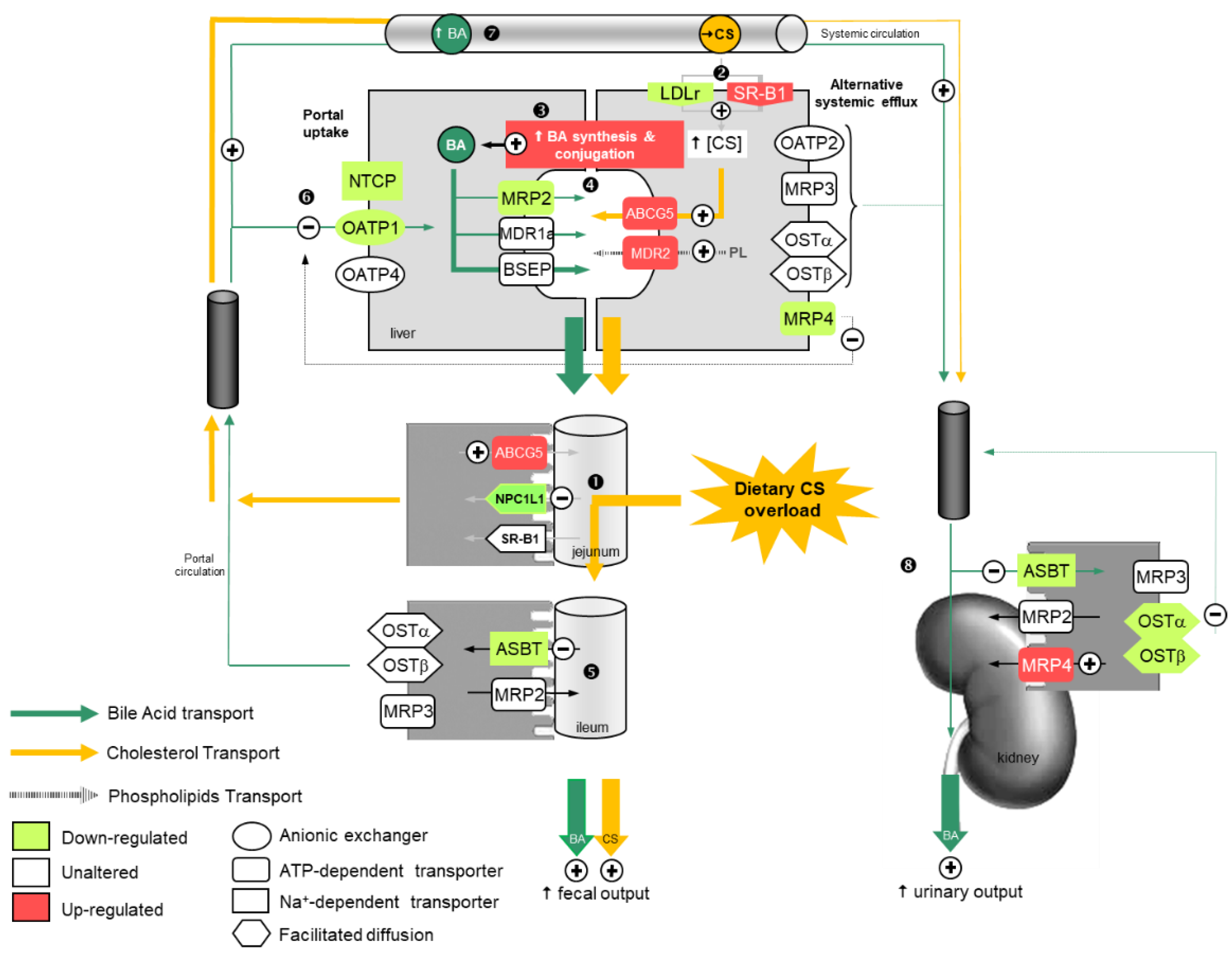
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Quantification of Fecal Bile Acids and Sterols
4.3. Quantification of Liver Lipids and Bile Acids and Liver Enzymatic Activities
4.4. Quantification of Plasma Lipids and Bile Acids
4.5. Western Blotting
4.6. Quantitative Real-Time PCR
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stamler, J.; Stamler, R.; Brown, W.V.; Gotto, A.M.; Greenland, P.; Grundy, S.; Hegsted, D.M.; Luepker, R.V.; Neaton, J.D.; Steinberg, D. Serum cholesterol. Doing the right thing. Circulation 1993, 88, 1954–1960. [Google Scholar] [CrossRef] [Green Version]
- Gould, A.L.; Rossouw, J.E.; Santanello, N.C.; Heyse, J.F.; Furberg, C.D. Cholesterol reduction yields clinical benefit. Circulation 1995, 91, 2274–2282. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.D. A population-based approach to cholesterol control. Am. J. Med. 1997, 102, 23–25. [Google Scholar] [CrossRef]
- Stuart-Shor, E. A public health action plan to prevent heart disease and stroke: The mandate for prevention across the continuum of care and across the lifespan. J. Cardiovasc. Nurs. 2004, 19, 354–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getz, G.S.; Reardon, C.A. Animal models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1104–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, W.; Carballo-Jane, E.; McLaren, D.G.; Mendoza, V.H.; Gagen, K.; Geoghagen, N.S.; McNamara, L.A.; Gorski, J.N.; Eiermann, G.J.; Petrov, A.; et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res. 2012, 53, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Schuster, G.; Parini, P.; Feltkamp, D.; Diczfalusy, U.; Rudling, M.; Angelin, B.; Björkhem, I.; Pettersson, S.; Gustafsson, J.-Å. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRβ-deficient mice. J. Clin. Investig. 2001, 107, 565–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Y.; Levy-Wilson, B.; Goodart, S.; Cooper, A.D. Mice expressing the human CYP7A1 gene in the mouse CYP7A1 knock-out background lack induction of CYP7A1 expression by cholesterol feeding and have increased hypercholesterolemia when fed a high fat diet. J. Biol. Chem. 2002, 277, 42588–42595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiemann, M.; Han, Z.; Soccio, R.; Bollineni, J.; Shefer, S.; Sehayek, E.; Breslow, J.L. Cholesterol feeding of mice expressing cholesterol 7α-hydroxylase increases bile acid pool size despite decreased enzyme activity. Proc. Natl. Acad. Sci. USA 2004, 101, 1846–1851. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gåfvels, M.; Rudling, M.; Murphy, C.; Björkhem, I.; Einarsson, C.; Eggertsen, G. Critical role of cholic acid for development of hypercholesterolemia and gallstones in diabetic mice. Biochem. Biophys. Res. Commun. 2006, 342, 1382–1388. [Google Scholar] [CrossRef]
- Wang, J.; Einarsson, C.; Murphy, C.; Parini, P.; Björkhem, I.; Gåfvels, M.; Eggertsen, G. Studies on LXR- and FXR-mediated effects on cholesterol homeostasis in normal and cholic acid-depleted mice. J. Lipid Res. 2006, 47, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, M.; Matsuda, Y.; Nomoto, M.; Takamatsu, Y.; Sato, N.; Hamatsu, M.; Dawson, P.A.; Gonzalez, F.J.; Yamazoe, Y. Cholesterol feeding prevents hepatic accumulation of bile acids in cholic acid-fed farnesoid X receptor (FXR)-null mice: FXR-independent suppression of intestinal bile acid absorption. Drug Metab. Dispos. 2009, 37, 338–344. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Pan, L.; Li, H.; Shang, Q.; Honda, A.; Shefer, S.; Bollineni, J.; Matsuzaki, Y.; Tint, G.S.; Salen, G. Dietary cholesterol stimulates CYP7A1 in rats because farnesoid X receptor is not activated. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G730–G735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, H.; Jiao, R.; Peng, C.; Wong, Y.M.; Yeung, V.S.Y.; Huang, Y.; Chen, Z.-Y. Choosing hamsters but not rats as a model for studying plasma cholesterol-lowering activity of functional foods. Mol. Nutr. Food Res. 2009, 53, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Turley, S.D.; Spady, D.K.; Dietschy, J.M. Regulation of fecal bile acid excretion in male golden Syrian hamsters fed a cereal-based diet with and without added cholesterol. Hepatology 1997, 25, 797–803. [Google Scholar] [CrossRef]
- Fernandez, M.L.; Wilson, T.A.; Conde, K.; Vergara-Jimenez, M.; Nicolosi, R.J. Hamsters and guinea pigs differ in their plasma lipoprotein cholesterol distribution when fed diets varying in animal protein, soluble fiber, or cholesterol content. J. Nutr. 1999, 129, 1323–1332. [Google Scholar] [CrossRef] [Green Version]
- Kolodgie, F.D.; Katocs, A.S.; Largis, E.E.; Wrenn, S.M.; Cornhill, J.F.; Herderick, E.E.; Lee, S.J.; Virmani, R. Hypercholesterolemia in the rabbit induced by feeding graded amounts of low-level cholesterol: Methodological considerations regarding individual variability in response to dietary cholesterol and development of lesion type. ATVB 1996, 16, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Salen, G.; Shefer, S.; Tint, G.S.; Kren, B.T.; Nguyen, L.B.; Steer, C.J.; Chen, T.S.; Salen, L.; Greenblatt, D. Increased bile acid pool inhibits cholesterol 7 alpha-hydroxylase in cholesterol-fed rabbits. Gastroenterology 1997, 113, 1958–1965. [Google Scholar] [CrossRef]
- Xu, G.; Salen, G.; Shefer, S.; Tint, G.S.; Nguyen, L.B.; Parker, T.T.; Chen, T.S.; Roberts, J.; Kong, X.; Greenblatt, D. Regulation of classic and alternative bile acid synthesis in hypercholesterolemic rabbits: Effects of cholesterol feeding and bile acid depletion. J. Lipid Res. 1998, 39, 1608–1615. [Google Scholar] [CrossRef]
- Xu, G.; Shneider, B.L.; Shefer, S.; Nguyen, L.B.; Batta, A.K.; Tint, G.S.; Arrese, M.; Thevananther, S.; Ma, L.; Stengelin, S.; et al. Ileal bile acid transport regulates bile acid pool, synthesis, and plasma cholesterol levels differently in cholesterol-fed rats and rabbits. J. Lipid Res. 2000, 41, 298–304. [Google Scholar] [CrossRef]
- Nishina, P.M.; Verstuyft, J.; Paigen, B. Synthetic low and high fat diets for the study of atherosclerosis in the mouse. J. Lipid Res. 1990, 31, 859–869. [Google Scholar] [CrossRef]
- Ishida, B.Y.; Blanche, P.J.; Nichols, A.V.; Yashar, M.; Paigen, B. Effects of atherogenic diet consumption on lipoproteins in mouse strains C57BL/6 and C3H. J. Lipid Res. 1991, 32, 559–568. [Google Scholar] [CrossRef]
- Li-Hawkins, J.; Gåfvels, M.; Olin, M.; Lund, E.G.; Andersson, U.; Schuster, G.; Björkhem, I.; Russell, D.W.; Eggertsen, G. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Investig. 2002, 110, 1191–1200. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Hagi, A.; Inoue, Y. The relationship between plasma high density lipoprotein cholesterol levels and cholesteryl ester transfer protein activity in six species of healthy experimental animals. Biol. Pharm. Bull. 2001, 24, 579–581. [Google Scholar] [CrossRef] [Green Version]
- Dietschy, J.M.; Turley, S.D. Control of cholesterol turnover in the mouse. J. Biol. Chem. 2002, 277, 3801–3804. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.C.; Agellon, L.B.; Walsh, A.; Breslow, J.L.; Tall, A. Dietary cholesterol increases transcription of the human cholesteryl ester transfer protein gene in transgenic mice. Dependence on natural flanking sequences. J. Clin. Investig. 1992, 90, 1290–1295. [Google Scholar] [CrossRef] [Green Version]
- Trauner, M.; Boyer, J.L. Bile salt transporters: Molecular characterization, function, and regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullak-Ublick, G.A.; Stieger, B.; Meier, P.J. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 2004, 126, 322–342. [Google Scholar] [CrossRef]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol. Rev. 2020, 101, 683–731. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Landrier, J.-F.; Gaillard, D.; Grober, J.; Monnot, M.-C.; Athias, A.; Besnard, P. Cholesterol dependent downregulation of mouse and human apical sodium dependent bile acid transporter (ASBT) gene expression: Molecular mechanism and physiological consequences. Gut 2006, 55, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Baghdasaryan, A.; Chiba, P.; Trauner, M. Clinical application of transcriptional activators of bile salt transporters. Mol. Asp. Med. 2014, 37, 57–76. [Google Scholar] [CrossRef]
- Ishibashi, S.; Schwarz, M.; Frykman, P.K.; Herz, J.; Russell, D.W. Disruption of cholesterol 7α-hydroxylase gene in mice, I. Postnatal lethality reversed by bile acid and vitamin supplementation. J. Biol. Chem. 1996, 271, 18017–18023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, M.; Lund, E.G.; Setchell, K.D.R.; Kayden, H.J.; Zerwekh, J.E.; Björkhem, I.; Herz, J.; Russell, D.W. Disruption of cholesterol 7α-hydroxylase gene in mice. J. Biol. Chem. 1996, 271, 18024–18031. [Google Scholar] [CrossRef] [Green Version]
- Duane, W.C.; Ginsberg, R.L.; Bennion, L.J. Effects of fasting on bile acid metabolism and biliary lipid composition in man. J. Lipid Res. 1976, 17, 211–219. [Google Scholar] [CrossRef]
- Muraca, M.; Vilei, M.T.; Miconi, L.; Petrin, P.; Antoniutti, M.; Pedrazzoli, S. A simple method for the determination of lipid composition of human bile. J. Lipid Res. 1991, 32, 371–374. [Google Scholar] [CrossRef]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [Green Version]
- de Boer, J.F.; Verkade, E.; Mulder, N.L.; de Vries, H.D.; Huijkman, N.; Koehorst, M.; Boer, T.; Wolters, J.C.; Bloks, V.W.; van de Sluis, B.; et al. A human-like bile acid pool induced by deletion of hepatic Cyp2c70 modulates effects of FXR activation in mice. J. Lipid Res. 2020, 61, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef]
- Heuman, D.M. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J. Lipid Res. 1989, 30, 719–730. [Google Scholar] [CrossRef]
- Thakare, R.; Alamoudi, J.A.; Gautam, N.; Rodrigues, A.D.; Alnouti, Y. Species differences in bile acids, I. Plasma and urine bile acid composition. J. Appl. Toxicol. 2018, 38, 1323–1335. [Google Scholar] [CrossRef]
- Chiang, J.Y.; Kimmel, R.; Stroup, D. Regulation of cholesterol 7alpha-hydroxylase gene (CYP7A1) Transcription by the liver orphan receptor (LXRalpha). Gene 2001, 262, 257–265. [Google Scholar] [CrossRef]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Brendel, C.; Schoonjans, K.; Botrugno, O.A.; Treuter, E.; Auwerx, J. The small heterodimer partner interacts with the liver X receptor alpha and represses its transcriptional activity. Mol. Endocrinol. 2002, 16, 2065–2076. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Gupta, S.; Pandak, W.M.; Hylemon, P.B. LXR alpha is the dominant regulator of CYP7A1 transcription. Biochem. Biophys. Res. Commun. 2002, 293, 338–343. [Google Scholar] [CrossRef]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Pan, L.; Erickson, S.K.; Forman, B.M.; Shneider, B.L.; Ananthanarayanan, M.; Li, X.; Shefer, S.; Balasubramanian, N.; Ma, L.; et al. Removal of the bile acid pool upregulates cholesterol 7alpha-hydroxylase by deactivating FXR in rabbits. J. Lipid Res. 2002, 43, 45–50. [Google Scholar] [CrossRef]
- Xu, G.; Li, H.; Pan, L.-X.; Shang, Q.; Honda, A.; Ananthanarayanan, M.; Erickson, S.K.; Shneider, B.L.; Shefer, S.; Bollineni, J.; et al. FXR-Mediated down-regulation of CYP7A1 dominates LXRalpha in long-term cholesterol-fed NZW rabbits. J. Lipid Res. 2003, 44, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cooper, A.D.; Levy-Wilson, B. Hepatocyte nuclear factor 1 binds to and transactivates the human but not the rat CYP7A1 promoter. Biochem. Biophys. Res. Commun. 1999, 260, 829–834. [Google Scholar] [CrossRef]
- Agellon, L.B.; Drover, V.A.B.; Cheema, S.K.; Gbaguidi, G.F.; Walsh, A. Dietary cholesterol fails to stimulate the human cholesterol 7alpha-hydroxylase gene (CYP7A1) in transgenic mice. J. Biol. Chem. 2002, 277, 20131–20134. [Google Scholar] [CrossRef] [Green Version]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlström, A.; Kovatcheva-Datchary, P.; Ståhlman, M.; Khan, M.-T.; Bäckhed, F.; Marschall, H.-U. Induction of farnesoid X receptor signaling in germ-free mice colonized with a human microbiota. J. Lipid Res. 2017, 58, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Houten, S.M.; Volle, D.H.; Cummins, C.L.; Mangelsdorf, D.J.; Auwerx, J. In vivo imaging of farnesoid X receptor activity reveals the ileum as the primary bile acid signaling tissue. Mol. Endocrinol. 2007, 21, 1312–1323. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Salem, M.; Yousef, I.M.; Tuchweber, B.; Lam, P.; Childs, S.J.; Helgason, C.D.; Ackerley, C.; Phillips, M.J.; Ling, V. Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc. Natl. Acad. Sci. USA 2001, 98, 2011–2016. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kobayashi, Y.; Gabazza, E.C.; Higuchi, K.; Kamisako, T.; Kuroda, M.; Takeuchi, K.; Iwasa, M.; Kaito, M.; Adachi, Y. Increased renal expression of bilirubin glucuronide transporters in a rat model of obstructive jaundice. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G656–G662. [Google Scholar] [CrossRef] [Green Version]
- Shneider, B.L.; Dawson, P.A.; Christie, D.M.; Hardikar, W.; Wong, M.H.; Suchy, F.J. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J. Clin. Investig. 1995, 95, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Craddock, A.L.; Love, M.W.; Daniel, R.W.; Kirby, L.C.; Walters, H.C.; Wong, M.H.; Dawson, P.A. Expression and transport properties of the human ileal and renal sodium-dependent bile acid transporter. Am. J. Physiol. 1998, 274, G157–G169. [Google Scholar] [CrossRef]
- Dawson, P.A.; Haywood, J.; Craddock, A.L.; Wilson, M.; Tietjen, M.; Kluckman, K.; Maeda, N.; Parks, J.S. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J. Biol. Chem. 2003, 278, 33920–33927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballatori, N.; Christian, W.V.; Lee, J.Y.; Dawson, P.A.; Soroka, C.J.; Boyer, J.L.; Madejczyk, M.S.; Li, N. OSTalpha-OSTbeta: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005, 42, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Hubbert, M.; Haywood, J.; Craddock, A.L.; Zerangue, N.; Christian, W.V.; Ballatori, N. The heteromeric organic solute transporter alpha-beta, ostalpha-ostbeta, is an ileal basolateral bile acid transporter. J. Biol. Chem. 2005, 280, 6960–6968. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi, A.; Hinoshita, E.; Iwamoto, Y.; Kohno, K.; Kuwano, M.; Uchiumi, T. Enhanced expression of the human multidrug resistance protein 3 by bile salt in human enterocytes. A transcriptional control of a plausible bile acid transporter. J. Biol. Chem. 2001, 276, 46822–46829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rost, D.; Mahner, S.; Sugiyama, Y.; Stremmel, W. Expression and localization of the multidrug resistance-associated protein 3 in rat small and large intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G720–G726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankenberg, T.; Rao, A.; Chen, F.; Haywood, J.; Shneider, B.L.; Dawson, P.A. Regulation of the mouse organic solute transporter alpha-beta, ostalpha-ostbeta, by bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G912–G922. [Google Scholar] [CrossRef] [Green Version]
- Landrier, J.-F.; Eloranta, J.J.; Vavricka, S.R.; Kullak-Ublick, G.A. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and -beta genes. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G476–G485. [Google Scholar] [CrossRef] [Green Version]
- Meier, P.J.; Stieger, B. Bile salt transporters. Annu. Rev. Physiol. 2002, 64, 635–661. [Google Scholar] [CrossRef]
- Denson, L.A.; Sturm, E.; Echevarria, W.; Zimmerman, T.L.; Makishima, M.; Mangelsdorf, D.J.; Karpen, S.J. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology 2001, 121, 140–147. [Google Scholar] [CrossRef]
- Chen, F.; Ma, L.; Dawson, P.A.; Sinal, C.J.; Sehayek, E.; Gonzalez, F.J.; Breslow, J.; Ananthanarayanan, M.; Shneider, B.L. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J. Biol. Chem. 2003, 278, 19909–19916. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, T.; Li, F.; Gonzalez, F.J. FXR signaling in the enterohepatic system. Mol. Cell. Endocrinol. 2013, 368, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.; Hagenbuch, B.; Fried, M.; Meier, P.J.; Kullak-Ublick, G.A. Role of liver-enriched transcription factors and nuclear receptors in regulating the human, mouse, and rat NTCP gene. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G752–G761. [Google Scholar] [CrossRef] [Green Version]
- Slitt, A.L.; Allen, K.; Morrone, J.; Aleksunes, L.M.; Chen, C.; Maher, J.M.; Manautou, J.E.; Cherrington, N.J.; Klaassen, C.D. Regulation of transporter expression in mouse liver, kidney, and intestine during extrahepatic cholestasis. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 637–647. [Google Scholar] [CrossRef] [Green Version]
- Ballatori, N.; Fang, F.; Christian, W.V.; Li, N.; Hammond, C.L. Ostalpha-Ostbeta is required for bile acid and conjugated steroid disposition in the intestine, kidney, and liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G179–G186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halilbasic, E.; Claudel, T.; Trauner, M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J. Hepatol. 2013, 58, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Williams, E.G.; Dubuis, S.; Mottis, A.; Jovaisaite, V.; Houten, S.M.; Argmann, C.A.; Faridi, P.; Wolski, W.; Kutalik, Z.; et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 2014, 158, 1415–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Mitsche, M.A.; Lütjohann, D.; Cohen, J.C.; Xie, X.-S.; Hobbs, H.H. Relative roles of ABCG5/ABCG8 in liver and intestine. J. Lipid Res. 2015, 56, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Langheim, S.; Yu, L.; von Bergmann, K.; Lütjohann, D.; Xu, F.; Hobbs, H.H.; Cohen, J.C. ABCG5 and ABCG8 require MDR2 for secretion of cholesterol into bile. J. Lipid Res. 2005, 46, 1732–1738. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Berge, K.E.; Pomajzl, C.; Richardson, J.A.; Hobbs, H.; Mangelsdorf, D.J. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J. Biol. Chem. 2002, 277, 18793–18800. [Google Scholar] [CrossRef] [Green Version]
- Altmann, S.W.; Davis, H.R.; Zhu, L.-J.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.N.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef] [Green Version]
- Altmann, S.W.; Davis, H.R.; Yao, X.; Laverty, M.; Compton, D.S.; Zhu, L.; Crona, J.H.; Caplen, M.A.; Hoos, L.M.; Tetzloff, G.; et al. The identification of intestinal scavenger receptor class B, type I (SR-BI) by expression cloning and its role in cholesterol absorption. Biochim. Biophys. Acta 2002, 1580, 77–93. [Google Scholar] [CrossRef]
- Wang, D.Q.; Lammert, F.; Cohen, D.E.; Paigen, B.; Carey, M.C. Cholic acid aids absorption, biliary secretion, and phase transitions of cholesterol in murine cholelithogenesis. Am. J. Physiol. 1999, 276, G751–G760. [Google Scholar] [CrossRef]
- Wang, D.Q.-H.; Tazuma, S.; Cohen, D.E.; Carey, M.C. Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: Studies in the gallstone-susceptible mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G494–G502. [Google Scholar] [CrossRef] [Green Version]
- Reynier, M.O.; Montet, J.C.; Gerolami, A.; Marteau, C.; Crotte, C.; Montet, A.M.; Mathieu, S. Comparative effects of cholic, chenodeoxycholic, and ursodeoxycholic acids on micellar solubilization and intestinal absorption of cholesterol. J. Lipid Res. 1981, 22, 467–473. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Carey, M.C. The hydrophobic-hydrophilic balance of bile salts. Inverse correlation between reverse-phase high performance liquid chromatographic mobilities and micellar cholesterol-solubilizing capacities. J. Lipid Res. 1982, 23, 70–80. [Google Scholar] [CrossRef]
- Wang, D.Q.-H.; Tazuma, S. Effect of beta-muricholic acid on the prevention and dissolution of cholesterol gallstones in C57L/J mice. J. Lipid Res. 2002, 43, 1960–1968. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, M.; Davis, D.L.; Vick, B.R.; Russell, D.W. Genetic analysis of cholesterol accumulation in inbred mice. J. Lipid. Res. 2001, 42, 1812–1819. [Google Scholar] [CrossRef]
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; Malloy, M.J.; et al. Human cholesterol 7α-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Investig. 2002, 110, 109–117. [Google Scholar] [CrossRef]
- Erickson, S.K.; Lear, S.R.; Deane, S.; Dubrac, S.; Huling, S.L.; Nguyen, L.; Bollineni, J.S.; Shefer, S.; Hyogo, H.; Cohen, D.E.; et al. Hypercholesterolemia and changes in lipid and bile acid metabolism in male and female cyp7A1-deficient mice. J. Lipid Res. 2003, 44, 1001–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Fujimori, T.; Furuya, A.; Satoh, J.; Nabeshima, Y.; Nabeshima, Y. Impaired negative feedback suppression of bile acid synthesis in mice lacking ΒKlotho. J. Clin. Investig. 2005, 115, 2202–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, M.; Russell, D.W.; Dietschy, J.M.; Turley, S.D. Marked reduction in bile acid synthesis in cholesterol 7alpha-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. J. Lipid Res. 1998, 39, 1833–1843. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Vales, C.; Lee, F.Y.; Lee, H.; Lusis, A.J.; Edwards, P.A. FXR deficiency causes reduced atherosclerosis in Ldlr−/− Mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2316–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, G.L.; Santamarina-Fojo, S.; Akiyama, T.E.; Amar, M.J.A.; Paigen, B.J.; Brewer, B.; Gonzalez, F.J. Effects of FXR in foam-cell formation and atherosclerosis development. Biochim. Biophys. Acta 2006, 1761, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Hanniman, E.A.; Lambert, G.; McCarthy, T.C.; Sinal, C.J. Loss of functional farnesoid X receptor increases atherosclerotic lesions in apolipoprotein E-deficient mice. J. Lipid Res. 2005, 46, 2595–2604. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Sun, L.; Hu, X.; Wang, X.; Xu, F.; Chen, B.; Liang, X.; Xia, J.; Wang, P.; Aibara, D.; et al. Suppressing the intestinal farnesoid X receptor/sphingomyelin phosphodiesterase 3 axis decreases atherosclerosis. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Mahaney, P.E.; Borges-Marcucci, L.; Lai, K.; Wang, S.; Krueger, J.A.; Gardell, S.J.; Huard, C.; Martinez, R.; Vlasuk, G.P.; et al. A synthetic farnesoid X receptor (FXR) agonist promotes cholesterol lowering in models of dyslipidemia. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G543–G552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urizar, N.L.; Liverman, A.B.; Dodds, D.T.; Silva, F.V.; Ordentlich, P.; Yan, Y.; Gonzalez, F.J.; Heyman, R.A.; Mangelsdorf, D.J.; Moore, D.D. A natural product that lowers cholesterol as an antagonist ligand for FXR. Science 2002, 296, 1703–1706. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Van Natta, M.L.; Connelly, M.A.; Vuppalanchi, R.; Neuschwander-Tetri, B.A.; Tonascia, J.; Guy, C.; Loomba, R.; Dasarathy, S.; Wattacheril, J.; et al. Impact of obeticholic acid on the lipoprotein profile in patients with non-alcoholic steatohepatitis. J. Hepatol. 2020, 72, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskar, M.G.; Eriksson, M.; Rudling, M.; Angelin, B. Treatment with the natural FXR agonist chenodeoxycholic acid reduces clearance of plasma LDL whilst decreasing circulating PCSK9, lipoprotein(a) and apolipoprotein C-III. J. Intern. Med. 2017, 281, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Briand, F.; Brousseau, E.; Quinsat, M.; Burcelin, R.; Sulpice, T. Obeticholic acid raises LDL-cholesterol and reduces HDL-cholesterol in the diet-induced NASH (DIN) hamster model. Eur. J. Pharmacol. 2018, 818, 449–456. [Google Scholar] [CrossRef]
- Poupon, R.E.; Ouguerram, K.; Chrétien, Y.; Verneau, C.; Eschwège, E.; Magot, T.; Poupon, R. Cholesterol-lowering effect of ursodeoxycholic acid in patients with primary biliary cirrhosis. Hepatology 1993, 17, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R.; Krasowski, M.D. Bile salts of vertebrates: Structural variation and possible evolutionary significance. J. Lipid Res. 2010, 51, 226–246. [Google Scholar] [CrossRef] [Green Version]
- Batta, A.K.; Salen, G.; Rapole, K.R.; Batta, M.; Batta, P.; Alberts, D.; Earnest, D. Highly simplified method for gas-liquid chromatographic quantitation of bile acids and sterols in human stool. J. Lipid Res. 1999, 40, 1148–1154. [Google Scholar] [CrossRef]
- Keller, S.; Jahreis, G. Determination of underivatised sterols and bile acid trimethyl silyl ether methyl esters by gas chromatography-mass spectrometry-single ion monitoring in faeces. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 813, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Gambert, P.; Lallemant, C.; Archambault, A.; Maume, B.F.; Padieu, P. Assessment of serum cholesterol by two methods: Gas-liquid chromatography on a capillary column and chemical ionization-mass fragmentography with isotopic dilution of [3,4-13C] cholesterol as internal standard. J. Chromatogr. 1979, 162, 1–6. [Google Scholar] [CrossRef]
- Becart, I.; Chevalier, C.; Biesse, J.P. Quantitative analysis of phospholipids by HPLC with a light scattering evaporating detector—Application to raw materials for cosmetic use. J. High Resolut. Chromatogr. 1990, 13, 126–129. [Google Scholar] [CrossRef]
- Souidi, M.; Parquet, M.; Férézou, J.; Lutton, C. Modulation of cholesterol 7α-hydroxylase and sterol 27-hydroxylase activities by steroids and physiological conditions in hamster. Life Sci. 1999, 64, 1585–1593. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaillard, D.; Masson, D.; Garo, E.; Souidi, M.; Pais de Barros, J.-P.; Schoonjans, K.; Grober, J.; Besnard, P.; Thomas, C. Muricholic Acids Promote Resistance to Hypercholesterolemia in Cholesterol-Fed Mice. Int. J. Mol. Sci. 2021, 22, 7163. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137163
Gaillard D, Masson D, Garo E, Souidi M, Pais de Barros J-P, Schoonjans K, Grober J, Besnard P, Thomas C. Muricholic Acids Promote Resistance to Hypercholesterolemia in Cholesterol-Fed Mice. International Journal of Molecular Sciences. 2021; 22(13):7163. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137163
Chicago/Turabian StyleGaillard, Dany, David Masson, Erwan Garo, Maamar Souidi, Jean-Paul Pais de Barros, Kristina Schoonjans, Jacques Grober, Philippe Besnard, and Charles Thomas. 2021. "Muricholic Acids Promote Resistance to Hypercholesterolemia in Cholesterol-Fed Mice" International Journal of Molecular Sciences 22, no. 13: 7163. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137163