
Decreased Glucocorticoid Signaling Potentiates Lipid-Induced Inflammation and Contributes to Insulin Resistance in the Skeletal Muscle of Fructose-Fed Male Rats Exposed to Stress

 ,
,  and
and
Abstract
:1. Introduction
2. Results
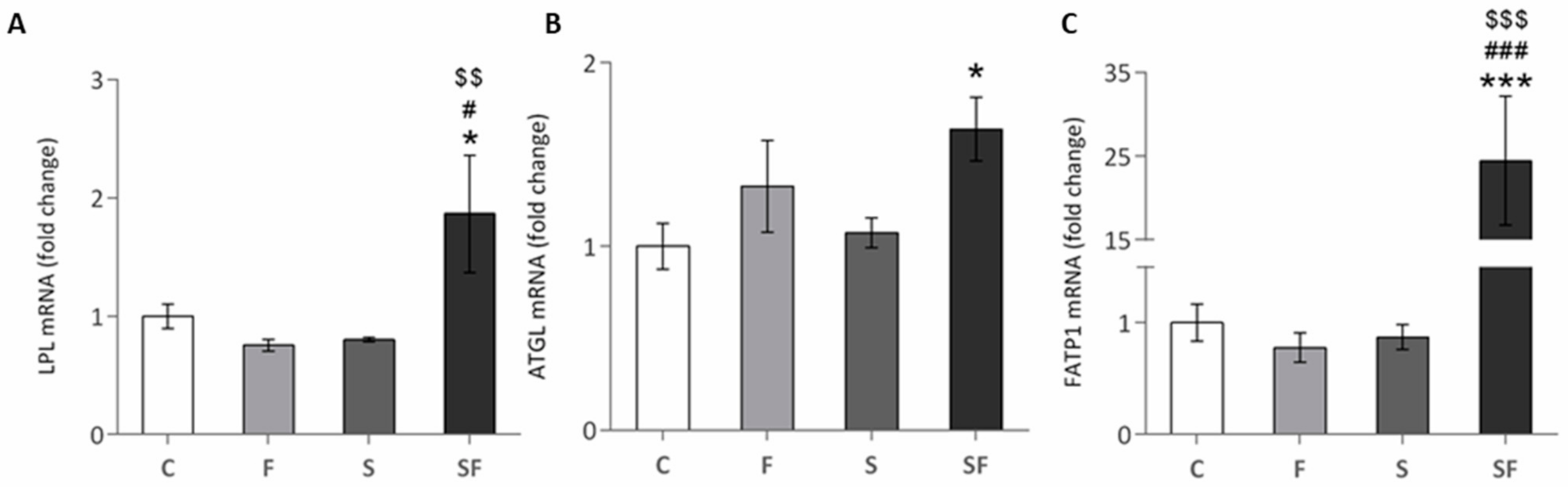
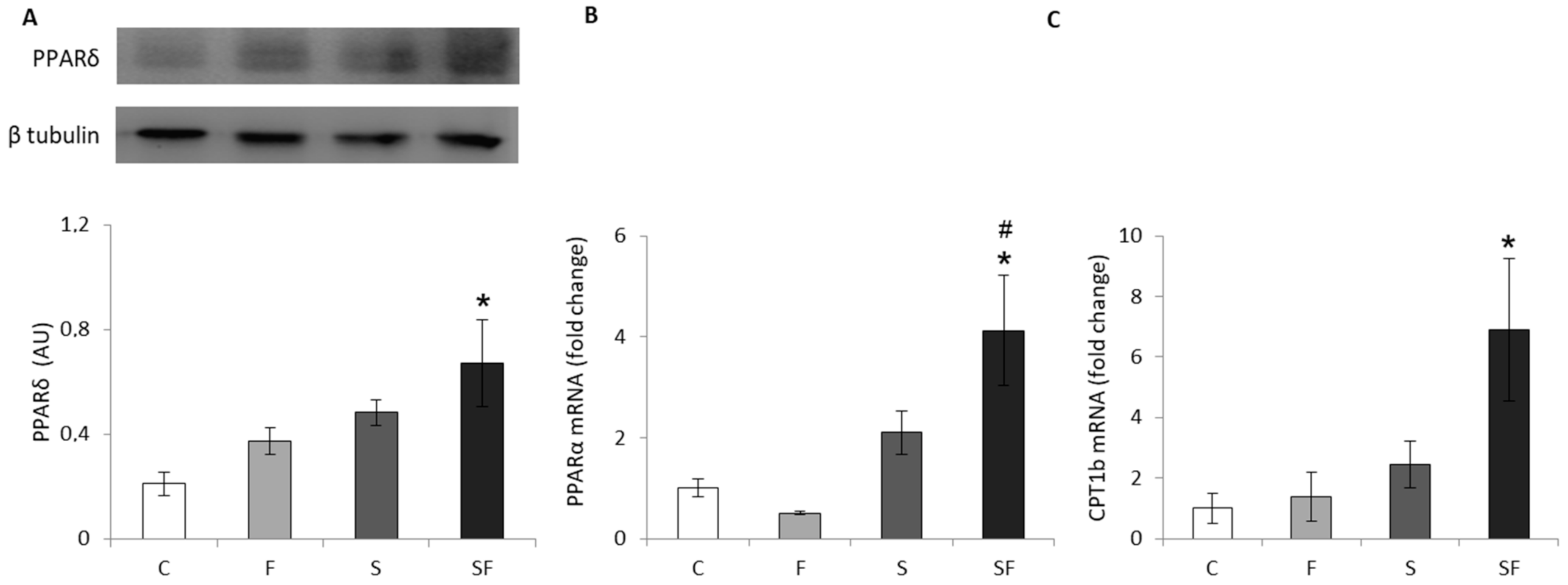
2.1. Lipid Metabolism
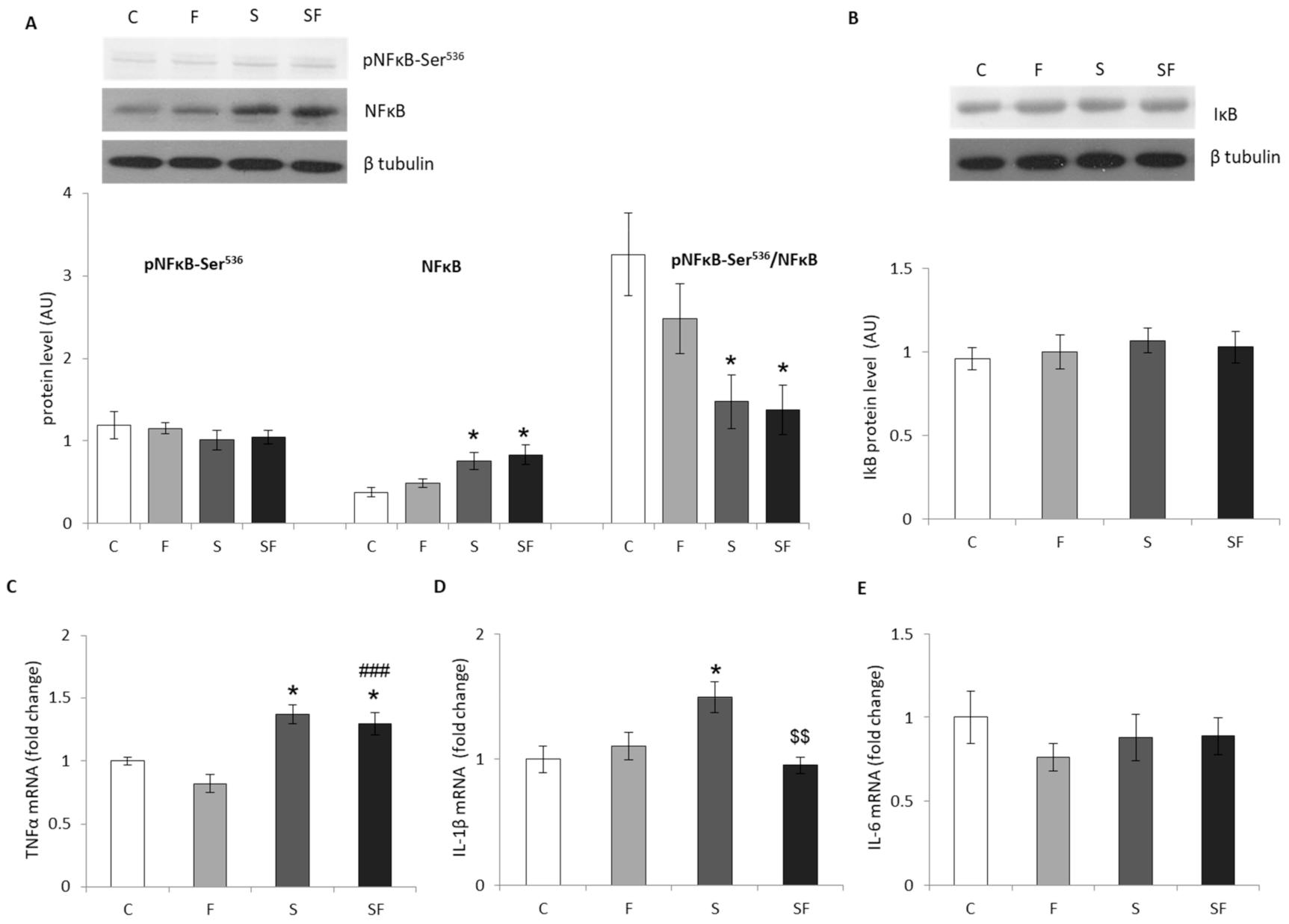
2.2. Inflammation
2.3. Glucocorticoid Signaling Pathway
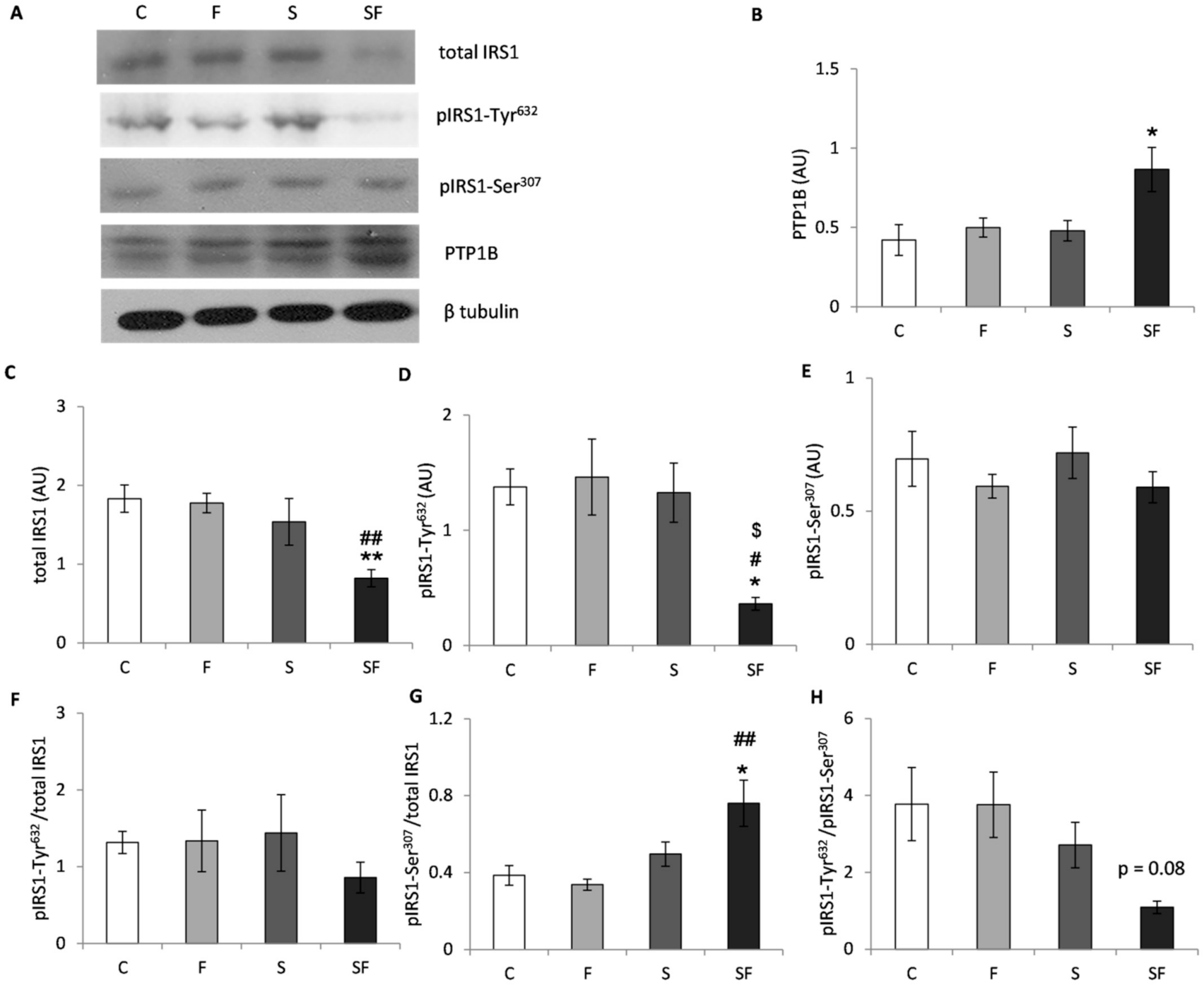
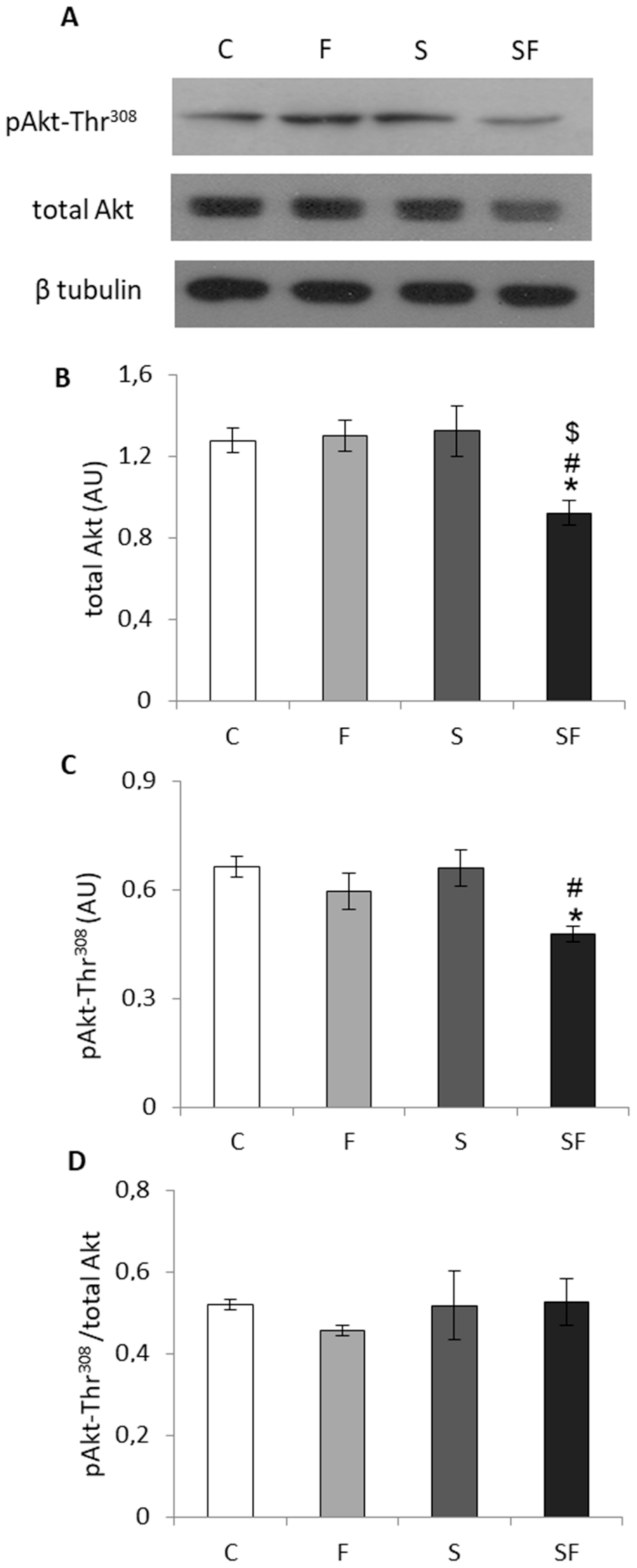
2.4. Insulin Signaling Pathway
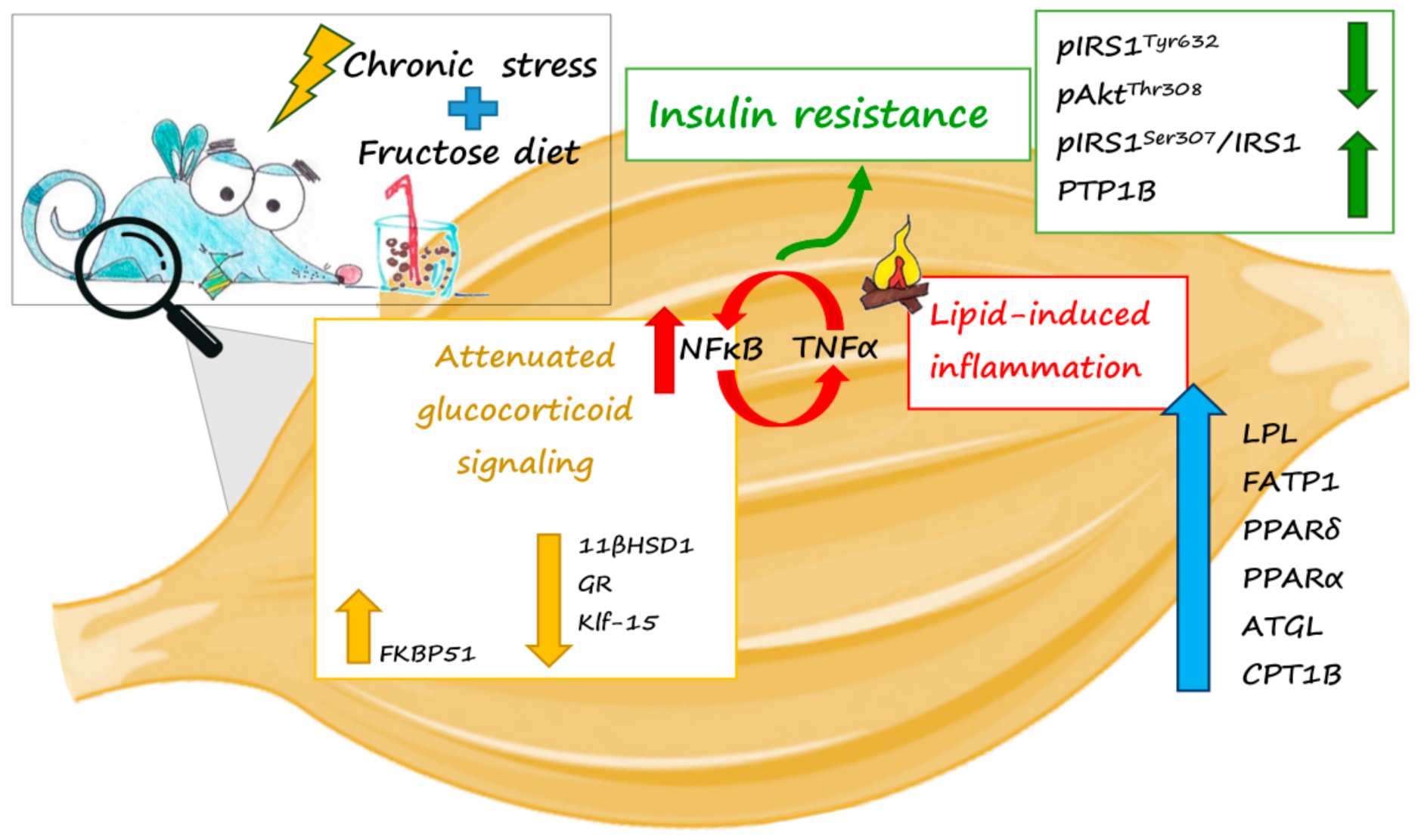
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals and Treatment
4.3. Determination of Plasma Corticosterone Level and Systemic Insulin Sensitivity
4.4. Preparation of Muscle Tissue Extract
4.5. SDS Polyacrylamide Gel Electrophoresis and Western Blot
4.6. RNA Extraction and Reverse Transcription
4.7. Real-Time PCR
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, Y.X.; Xiao, H.B.; Wang, S.S.; Zhao, J.; He, Y.; Wang, W.; Dong, J. Investigation of the relationship between chronic stress and insulin resistance in a Chinese population. J. Epidemiol. 2016, 26, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Fang, C.; Yang, J.; Zhang, H.; Huang, Y.; Xuan, C.; Wang, Y.; Li, S.; Sha, J.; Zha, M.; et al. The effect of noise exposure on insulin sensitivity in mice may be mediated by the JNK/IRS1 pathway. Environ. Health Prev. Med. 2018, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, G.A. Energy and fructose from beverages sweetened with sugar or high-fructose corn syrup pose a health risk for some people. Adv. Nutr. 2013, 4, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Bezerra, R.M.N.; Ueno, M.; Silva, M.S.; Tavares, D.Q.; Carvalho, C.R.O.; Saad, M.J.A. A High Fructose Diet Affects the Early Steps of Insulin Action in Muscle and Liver of Rats. J. Nutr. 2000, 130, 1531–1535. [Google Scholar] [CrossRef] [PubMed]
- Stefanaki, C.; Pervanidou, P.; Boschiero, D.; Chrousos, G.P. Chronic stress and body composition disorders: Implications for health and disease. Hormones 2018, 17, 33–43. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. S2). [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattanavichit, Y.; Chukijrungroat, N.; Saengsirisuwan, V. Sex differences in the metabolic dysfunction and insulin resistance of skeletal muscle glucose transport following high fructose ingestion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R1200–R1212. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of fatty acid uptake and fatty acid β-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 1–22. [Google Scholar] [CrossRef]
- Hegarty, B.D.; Furler, S.M.; Ye, J.; Cooney, G.J.; Kraegen, E.W. The role of intramuscular lipid in insulin resistance. Acta Physiol. Scand. 2003, 178, 373–383. [Google Scholar] [CrossRef]
- Samuel, V.T. Fructose induced lipogenesis: From sugar to fat to insulin resistance. Trends Endocrinol. Metab. 2011, 22, 60–65. [Google Scholar] [CrossRef]
- Benetti, E.; Mastrocola, R.; Rogazzo, M.; Chiazza, F.; Aragno, M.; Fantozzi, R.; Collino, M.; Minetto, M.A. High sugar intake and development of skeletal muscle insulin resistance and inflammation in mice: A protective role for PPAR-δ agonism. Mediators Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.; Franke, W.D. Stress and cardiovascular disease risk in female law enforcement officers. Int. Arch. Occup. Environ. Health 2011, 84, 279–286. [Google Scholar] [CrossRef]
- Stoney, C.M.; West, S.G.; Hughes, J.W.; Lentino, L.M.; Finney, M.L.; Falko, J.; Bausserman, L. Acute psychological stress reduces plasma triglyceride clearance. Psychophysiology 2002, 39, 80–85. [Google Scholar] [CrossRef]
- Austin, R.L.; Rune, A.; Bouzakri, K.; Zierath, J.R.; Krook, A. SiRNA-mediated reduction of inhibitor of nuclear factor-κB kinase prevents tumor necrosis factor-α-induced insulin resistance in human skeletal muscle. Diabetes 2008, 57, 2066–2073. [Google Scholar] [CrossRef] [Green Version]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor necrosis factor-α induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 2005, 54, 2939–2945. [Google Scholar] [CrossRef] [Green Version]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Wang, Y.X.; Jiang, C.L. Inflammation: The common pathway of stress-related diseases. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef]
- Rabasa, C.; Dickson, S.L. Impact of stress on metabolism and energy balance. Curr. Opin. Behav. Sci. 2016, 9, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Grad, I.; Picard, D. The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell. Endocrinol. 2007, 275, 2–12. [Google Scholar] [CrossRef]
- Wochnik, G.M.; Rüegg, J.; Abel, G.A.; Schmidt, U.; Holsboer, F.; Rein, T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J. Biol. Chem. 2005, 280, 4609–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denny, W.B.; Valentine, D.L.; Reynolds, P.D.; Smith, D.F.; Scammell, J.G. Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology 2000, 141, 4107–4113. [Google Scholar] [CrossRef]
- Scammell, J.G.; Denny, W.B.; Valentine, D.L.; Smiths, D.F. Overexpression of the FK506-binding immunophilin FKBP51 is the common cause of glucocorticoid resistance in three New World primates. Gen. Comp. Endocrinol. 2001, 124, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.C.; Rogoff, D.; McMillan, D.R.; Lavery, G.G. Hexose 6-phosphate dehydrogenase (H6PD) and corticosteroid metabolism. Mol. Cell. Endocrinol. 2007, 265–266, 89–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadwick, J.A.; Swager, S.A.; Lowe, J.; Welc, S.S.; Tidball, J.G.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Rafael-Fortney, J.A. Myeloid cells are capable of synthesizing aldosterone to exacerbate damage in muscular dystrophy. Hum. Mol. Genet. 2016, 25, 5167–5177. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, J.A.; Hauck, J.S.; Lowe, J.; Shaw, J.J.; Guttridge, D.C.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Rafael-Fortney, J.A. Mineralocorticoid receptors are present in skeletal muscle and represent a potential therapeutic target. FASEB J. 2015, 29, 4544–4554. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.; Harris, C.A.; Wang, J.C. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol. Cell. Endocrinol. 2013, 380, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Hermoso, M.A.; Cidlowski, J.A. Putting the Brake on Inflammatory Responses: The Role of Glucocorticoids. IUBMB Life 2003, 55, 497–504. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Ammit, A.J. Role of dual-specificity phosphatase 1 in glucocorticoid-driven antiinflammatory responses. Front. Immunol. 2019, 10, 1446. [Google Scholar] [CrossRef]
- Pradère, J.P.; Hernandez, C.; Koppe, C.; Friedman, R.A.; Luedde, T.; Schwabe, R.F. Negative regulation of NF-κB p65 activity by serine 536 phosphorylation. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Bruce, C.R.; Beale, S.M.; Hoehn, K.L.; So, T.; Rolph, M.S.; Cooney, G.J. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: Evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes 2007, 56, 2085–2092. [Google Scholar] [CrossRef] [Green Version]
- Hancock, C.R.; Han, D.H.; Chen, M.; Terada, S.; Yasuda, T.; Wright, D.C.; Holloszy, J.O. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 7815–7820. [Google Scholar] [CrossRef] [Green Version]
- Carley, A.N.; Severson, D.L. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2005, 1734, 112–126. [Google Scholar] [CrossRef]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Ui, J.Y.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef] [Green Version]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The subtle balance between lipolysis and lipogenesis: A critical point in metabolic homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [Green Version]
- Meex, R.C.R.; Hoy, A.J.; Mason, R.M.; Martin, S.D.; McGee, S.L.; Bruce, C.R.; Watt, M.J. ATGL-mediated triglyceride turnover and the regulation of mitochondrial capacity in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E960–E970. [Google Scholar] [CrossRef] [Green Version]
- Bursać, B.; Djordjevic, A.; Veličković, N.; Milutinović, D.V.; Petrović, S.; Teofilović, A.; Gligorovska, L.; Preitner, F.; Tappy, L.; Matić, G. Involvement of glucocorticoid prereceptor metabolism and signaling in rat visceral adipose tissue lipid metabolism after chronic stress combined with high-fructose diet. Mol. Cell. Endocrinol. 2018, 476, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M.; Way, J.M.; Tanner, C.J.; Winegar, D.A.; Kliewer, S.A.; Houmard, J.A.; Kraus, W.E.; Lynis Dohm, G. Peroxisome proliferator-activated receptor-α regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002, 51, 901–909. [Google Scholar] [CrossRef] [Green Version]
- De Lange, P.; Lombardi, A.; Silvestri, E.; Goglia, F.; Lanni, A.; Moreno, M. Peroxisome proliferator-activated receptor delta: A conserved director of lipid homeostasis through regulation of the oxidative capacity of muscle. PPAR Res. 2008, 2008, 172676. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A potential link between muscle peroxisome proliferator- activated receptor-α signaling and obesity-related diabetes. Cell Metab. 2005, 1, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Cresser, J.; Bonen, A.; Chabowski, A.; Stefanyk, L.E.; Gulli, R.; Ritchie, I.; Dyck, D.J. Oral administration of a PPAR-δ agonist to rodents worsens, not improves, maximal insulin-stimulated glucose transport in skeletal muscle of different fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Perseghin, G.; Caumo, A.; Caloni, M.; Testolin, G.; Luzi, L. Incorporation of the fasting plasma FFA concentration into QUICKI improves its association with insulin sensitivity in nonobese individuals. J. Clin. Endocrinol. Metab. 2001, 86, 4776–4781. [Google Scholar] [CrossRef] [PubMed]
- Koves, T.R.; Li, P.; An, J.; Akimoto, T.; Slentz, D.; Ilkayeva, O.; Dohm, G.L.; Yan, Z.; Newgard, C.B.; Muoio, D.M. Peroxisome proliferator-activated receptor-γ co-activator 1α-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J. Biol. Chem. 2005, 280, 33588–33598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.B.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.H.; Hoppel, C.L.; Lok, K.H.; Zhao, L.; Wong, S.W.; Minkler, P.E.; Hwang, D.H.; Newman, J.W.; Garvey, W.T. Plasma Acylcarnitine Profiles Suggest Incomplete Long-Chain Fatty Acid β-Oxidation and Altered Tricarboxylic Acid Cycle Activity in Type 2 Diabetic African-American Women. J. Nutr. 2009, 139, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Warfel, J.D.; Bermudez, E.M.; Mendoza, T.M.; Ghosh, S.; Zhang, J.; Elks, C.M.; Mynatt, R.; Vandanmagsar, B. Mitochondrial fat oxidation is essential for lipid-induced inflammation in skeletal muscle in mice. Sci. Rep. 2016, 6, 37941. [Google Scholar] [CrossRef] [Green Version]
- Wicks, S.E.; Vandanmagsar, B.; Haynie, K.R.; Fuller, S.E.; Warfel, J.D.; Stephens, J.M.; Wang, M.; Han, X.; Zhang, J.; Noland, R.C.; et al. Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, E3300–E3309. [Google Scholar] [CrossRef] [Green Version]
- Pivonello, R.; De Leo, M.; Vitale, P.; Cozzolino, A.; Simeoli, C.; De Martino, M.C.; Lombardi, G.; Colao, A. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology 2010, 92, 77–81. [Google Scholar] [CrossRef]
- Hellhammer, D.H.; Wade, S. Endocrine correlates of stress vulnerability. Psychother. Psychosom. 1993, 60, 8–17. [Google Scholar] [CrossRef]
- Fries, E.; Hesse, J.; Hellhammer, J.; Hellhammer, D.H. A new view on hypocortisolism. Psychoneuroendocrinology 2005, 30, 1010–1016. [Google Scholar] [CrossRef]
- Sarapultsev, A.; Sarapultsev, P.; Dremencov, E.; Komelkova, M.; Tseilikman, O.; Tseilikman, V. Low glucocorticoids in stress-related disorders: The role of inflammation. Stress 2020, 23, 651–661. [Google Scholar] [CrossRef]
- MacEdo, I.C.; Medeiros, L.F.; Oliveira, C.; Oliveira, C.M.; Rozisky, J.R.; Scarabelot, V.L.; Souza, A.; Silva, F.R.; Santos, V.S.; Cioato, S.G.; et al. Cafeteria diet-induced obesity plus chronic stress alter serum leptin levels. Peptides 2012, 38, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Adam, T.C.; Epel, E.S. Stress, eating and the reward system. Physiol. Behav. 2007, 91, 449–458. [Google Scholar] [CrossRef]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. USA 2014, 111, E2482–E2491. [Google Scholar] [CrossRef] [Green Version]
- Semjonous, N.M.; Sherlock, M.; Jeyasuria, P.; Parker, K.L.; Walker, E.A.; Stewart, P.M.; Lavery, G.G. Hexose-6-phosphate dehydrogenase contributes to skeletal muscle homeostasis independent of 11β-hydroxysteroid dehydrogenase type 1. Endocrinology 2011, 152, 93–102. [Google Scholar] [CrossRef]
- Zielinska, A.E.; Fletcher, R.S.; Sherlock, M.; Doig, C.L.; Lavery, G.G. Cellular and genetic models of H6PDH and 11β-HSD1 function in skeletal muscle. Cell Biochem. Funct. 2017, 35, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Loerz, C.; Maser, E. The cortisol-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 in skeletal muscle in the pathogenesis of the metabolic syndrome. J. Steroid Biochem. Mol. Biol. 2017, 174, 65–71. [Google Scholar] [CrossRef]
- Teofilović, A.; Brkljačić, J.; Djordjevic, A.; VojnovićMilutinović, D.; Tappy, L.; Matić, G.; Veličković, N. Impact of insulin and glucocorticoid signalling on hepatic glucose homeostasis in the rat exposed to high-fructose diet and chronic stress. Int. J. Food Sci. Nutr. 2020, 71, 815–825. [Google Scholar] [CrossRef]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.I.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasiljević, A.; Veličković, N.; Bursać, B.; Djordjevic, A.; Milutinović, D.V.; Nestorović, N.; Matić, G. Enhanced prereceptor glucocorticoid metabolism and lipogenesis impair insulin signaling in the liver of fructose-fed rats. J. Nutr. Biochem. 2013, 24, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.S.; Doig, C.L.; Hussain, Z.; O’Leary, M.; Morgan, S.A.; Pearson, M.J.; Naylor, A.; Jones, S.W.; Filer, A.; Stewart, P.M.; et al. 11β-Hydroxysteroid dehydrogenase type 1 within muscle protects against the adverse effects of local inflammation. J. Pathol. 2016, 240, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, A.E.; Gray, M.; Brownstein, D.G.; Salter, D.M.; Sawatzky, D.A.; Clay, S.; Gilmour, J.S.; Seckl, J.R.; Savill, J.S.; Chapman, K.E. 11β-hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology 2012, 153, 234–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Vazquez, I.; Fernández-Veledo, S.; De Alvaro, C.; Rondinone, C.M.; Valverde, A.M.; Lorenzo, M. Protein-tyrosine phosphatase 1B-deficient myocytes show increased insulin sensitivity and protection against tumor necrosis factor-α-induced insulin resistance. Diabetes 2007, 56, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Häusl, A.S.; Balsevich, G.; Gassen, N.C.; Schmidt, M.V. Focus on FKBP51: A molecular link between stress and metabolic disorders. Mol. Metab. 2019, 29, 170–181. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Joëls, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef]
- Schweiger, M.; Schreiber, R.; Haemmerle, G.; Lass, A.; Fledelius, C.; Jacobsen, P.; Tornqvist, H.; Zechner, R.; Zimmermann, R. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J. Biol. Chem. 2006, 281, 40236–40241. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Zhou, Q.L.; Coleman, K.A.; Chouinard, M.; Boese, Q.; Czech, M.P. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc. Natl. Acad. Sci. USA 2003, 100, 7569–7574. [Google Scholar] [CrossRef] [Green Version]
- Bouzakri, K.; Roques, M.; Gual, P.; Espinosa, S.; Guebre-Egziabher, F.; Riou, J.P.; Laville, M.; Le Marchand-Brustel, Y.; Tanti, J.F.; Vidal, H. Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes 2003, 52, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Sanghez, V.; Cubuk, C.; Sebastián-Leon, P.; Carobbio, S.; Dopazo, J.; Vidal-Puig, A.; Bartolomucci, A. Chronic subordination stress selectively downregulates the insulin signaling pathway in liver and skeletal muscle but not in adipose tissue of male mice. Stress 2016, 19, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Veličković, N.; Teofilović, A.; Ilić, D.; Djordjevic, A.; Vojnović Milutinović, D.; Petrović, S.; Preitner, F.; Tappy, L.; Matić, G. Modulation of hepatic inflammation and energy-sensing pathways in the rat liver by high-fructose diet and chronic stress. Eur. J. Nutr. 2019, 58, 1829–1845. [Google Scholar] [CrossRef] [Green Version]
- Kovačević, S.; Nestorov, J.; Matić, G.; Elaković, I. Chronic stress combined with a fructose diet reduces hypothalamic insulin signaling and antioxidative defense in female rats. Neuroendocrinology 2019, 108. [Google Scholar] [CrossRef]
- Romic, S.; Djordjevic, A.; Tepavcevic, S.; Culafic, T.; Stojiljkovic, M.; Bursac, B.; Stanisic, J.; Kostic, M.; Gligorovska, L.; Koricanac, G. Effects of a fructose-rich diet and chronic stress on insulin signaling and regulation of glycogen synthase kinase-3 beta and the sodium–potassium pump in the hearts of male rats. Food Funct. 2020, 11, 1455–1466. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Haj, F.G.; Kim, Y.B.; Kim, H.J.; Shulman, G.I.; Kim, J.K.; Neel, B.G.; Kahn, B.B. Transgenic overexpression of protein-tyrosine phosphatase 1B in muscle causes insulin resistance, but overexpression with leukocyte antigen-related phosphatase does not additively impair insulin action. J. Biol. Chem. 2004, 279, 24844–24851. [Google Scholar] [CrossRef] [Green Version]
- Dadke, S.S.; Li, H.C.; Kusari, A.B.; Begum, N.; Kusari, J. Elevated expression and activity of protein-tyrosine phosphatase 1B in skeletal muscle of insulin-resistant type II diabetic Goto-Kakizaki rats. Biochem. Biophys. Res. Commun. 2000, 274, 583–589. [Google Scholar] [CrossRef]
- Stull, A.J.; Wang, Z.Q.; Zhang, X.H.; Yu, Y.; Johnson, W.D.; Cefalu, W.T. Skeletal muscle protein tyrosine phosphatase 1B regulates insulin sensitivity in African Americans. Diabetes 2012, 61, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Zabolotny, J.M.; Kim, Y.B.; Welsh, L.A.; Kershaw, E.E.; Neel, B.G.; Kahn, B.B. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J. Biol. Chem. 2008, 283, 14230–14241. [Google Scholar] [CrossRef] [Green Version]
- Joëls, M.; Karst, H.; Alfarez, D.; Heine, V.M.; Qin, Y.; van Riel, E.; Verkuyl, M.; Lucassen, P.J.; Krugers, H.J. Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress 2004, 7, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Ventura, E.E.; Davis, J.N.; Goran, M.I. Sugar content of popular sweetened beverages based on objective laboratory analysis: Focus on fructose content. Obesity 2011, 19, 868–874. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Fructose | Stress | Stress + Fructose | Two-Way ANOVA | |||
|---|---|---|---|---|---|---|---|
| Fructose | Stress | Interaction | |||||
| Corticosterone (ng/mL) | 114.00 ± 31.81 | 107.70 ± 16.82 | 742.20 ± 120.90 *** | 219.10 ± 32.29 $$$ | p < 0.001 | p < 0.0001 | p < 0.001 |
| Corticosterone (ng/mg) | 0.113 ± 0.024 | 0.291 ± 0.065 | 0.356 ± 0.075 * | 0.424 ± 0.064 ** | NS | p < 0.01 | NS |
| R-QUICKI | 0.172 ± 0.003 | 0.169 ± 0.003 | 0.177 ± 0.004 | 0.164 ± 0.003 $ | p < 0.05 | NS | NS |
| QUICKI | 0.168 ± 0.003 | 0.167 ± 0.003 | 0.174 ± 0.003 | 0.165 ± 0.002 | NS | NS | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirif, A.Z.; Kovačević, S.; Brkljačić, J.; Teofilović, A.; Elaković, I.; Djordjevic, A.; Matić, G. Decreased Glucocorticoid Signaling Potentiates Lipid-Induced Inflammation and Contributes to Insulin Resistance in the Skeletal Muscle of Fructose-Fed Male Rats Exposed to Stress. Int. J. Mol. Sci. 2021, 22, 7206. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137206
Shirif AZ, Kovačević S, Brkljačić J, Teofilović A, Elaković I, Djordjevic A, Matić G. Decreased Glucocorticoid Signaling Potentiates Lipid-Induced Inflammation and Contributes to Insulin Resistance in the Skeletal Muscle of Fructose-Fed Male Rats Exposed to Stress. International Journal of Molecular Sciences. 2021; 22(13):7206. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137206
Chicago/Turabian StyleShirif, Abdulbaset Zidane, Sanja Kovačević, Jelena Brkljačić, Ana Teofilović, Ivana Elaković, Ana Djordjevic, and Gordana Matić. 2021. "Decreased Glucocorticoid Signaling Potentiates Lipid-Induced Inflammation and Contributes to Insulin Resistance in the Skeletal Muscle of Fructose-Fed Male Rats Exposed to Stress" International Journal of Molecular Sciences 22, no. 13: 7206. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137206