EVOO Polyphenols Relieve Synergistically Autophagy Dysregulation in a Cellular Model of Alzheimer’s Disease



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
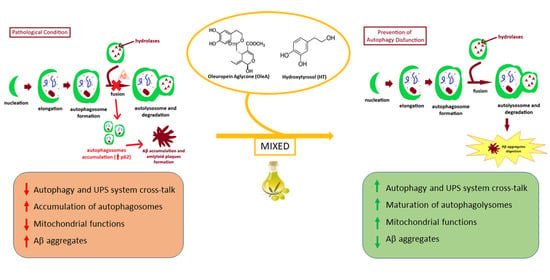
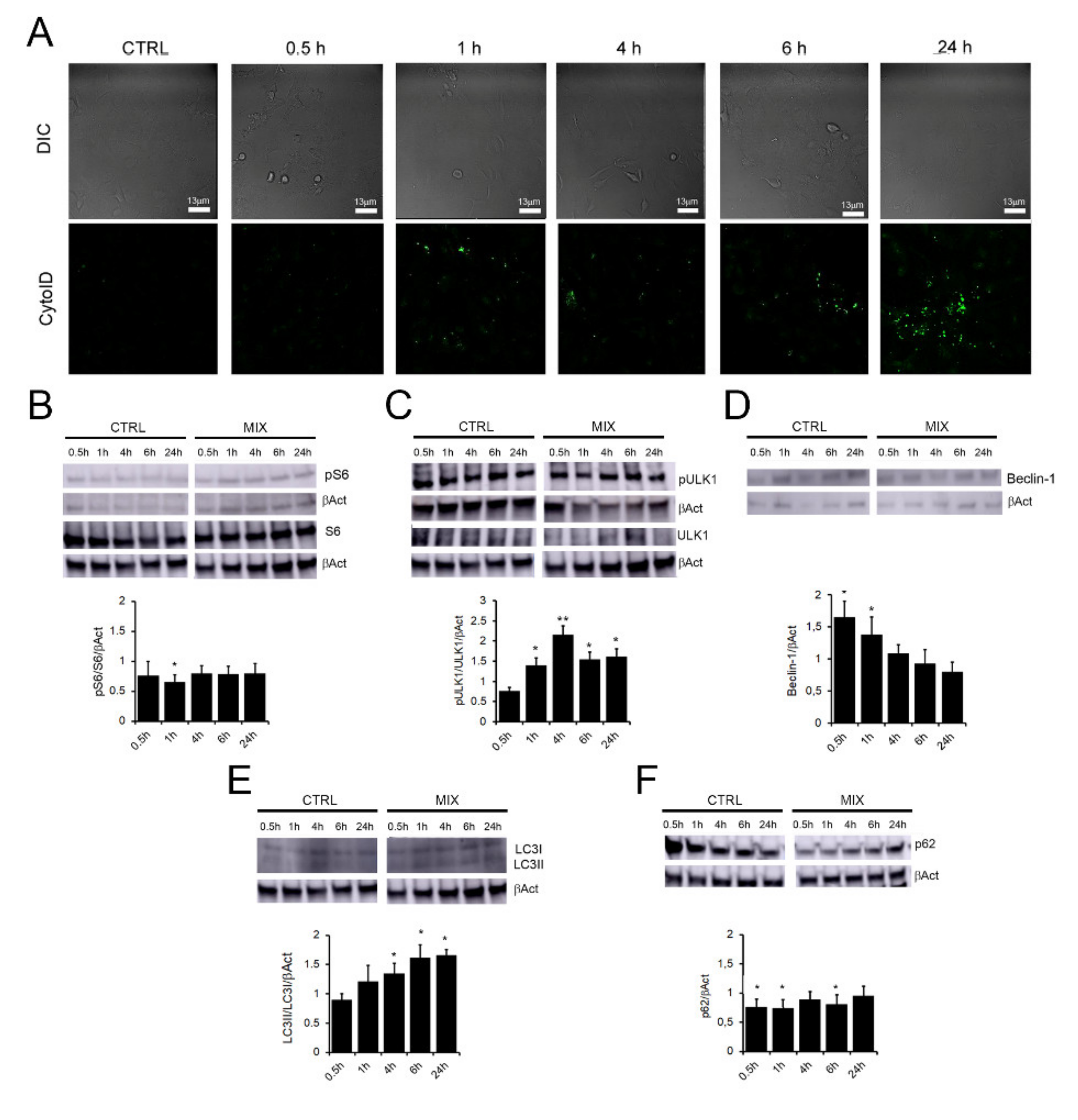
2.1. OleA/HT Mixtures Trigger Autophagy
2.2. Cells Pre-Treatment with the MIX Prevents Aβ Oligomers Cytotoxicity
2.3. The MIX Favours Aβ1−42 Oligomers Digestion by Autophagy
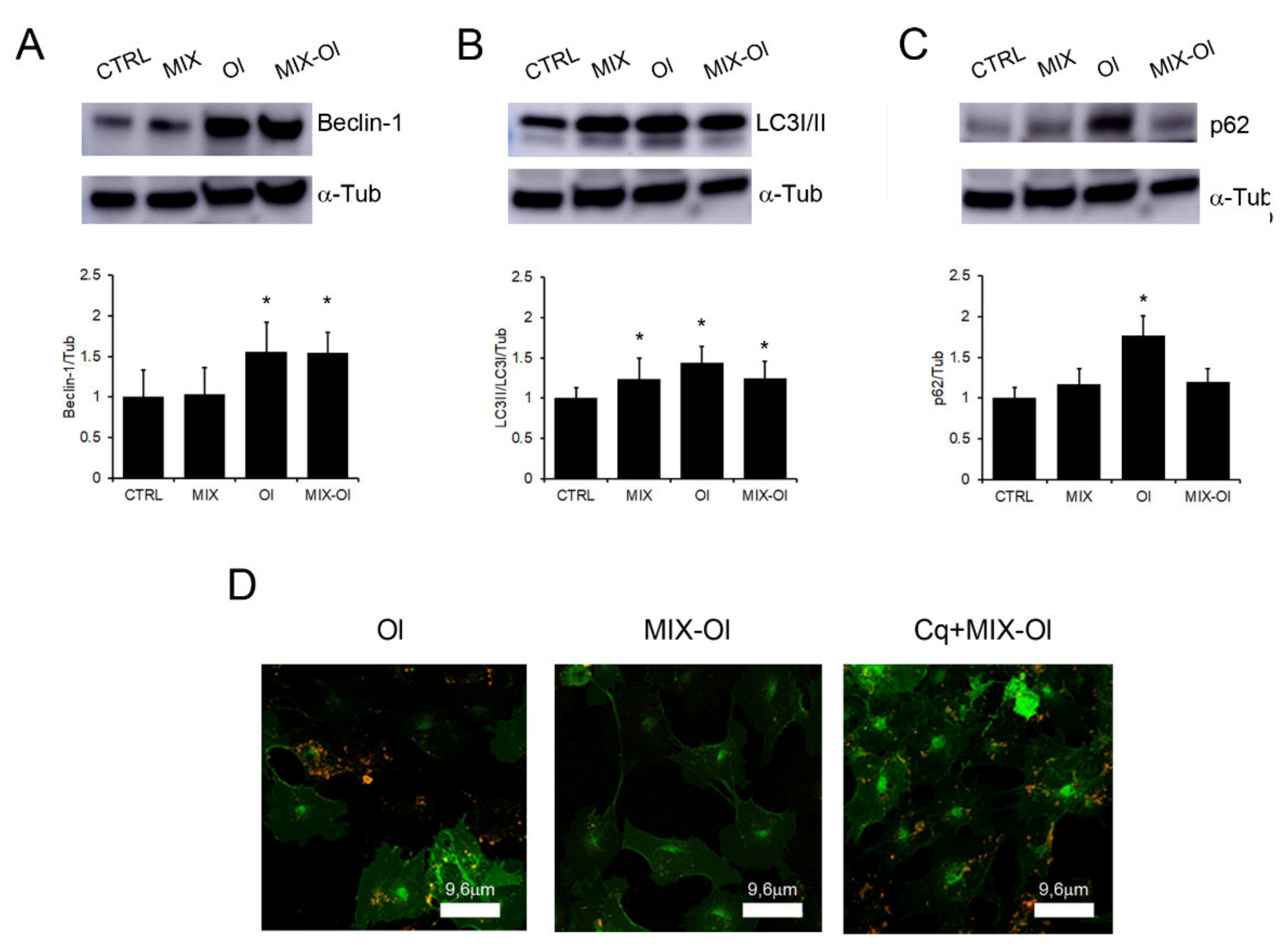
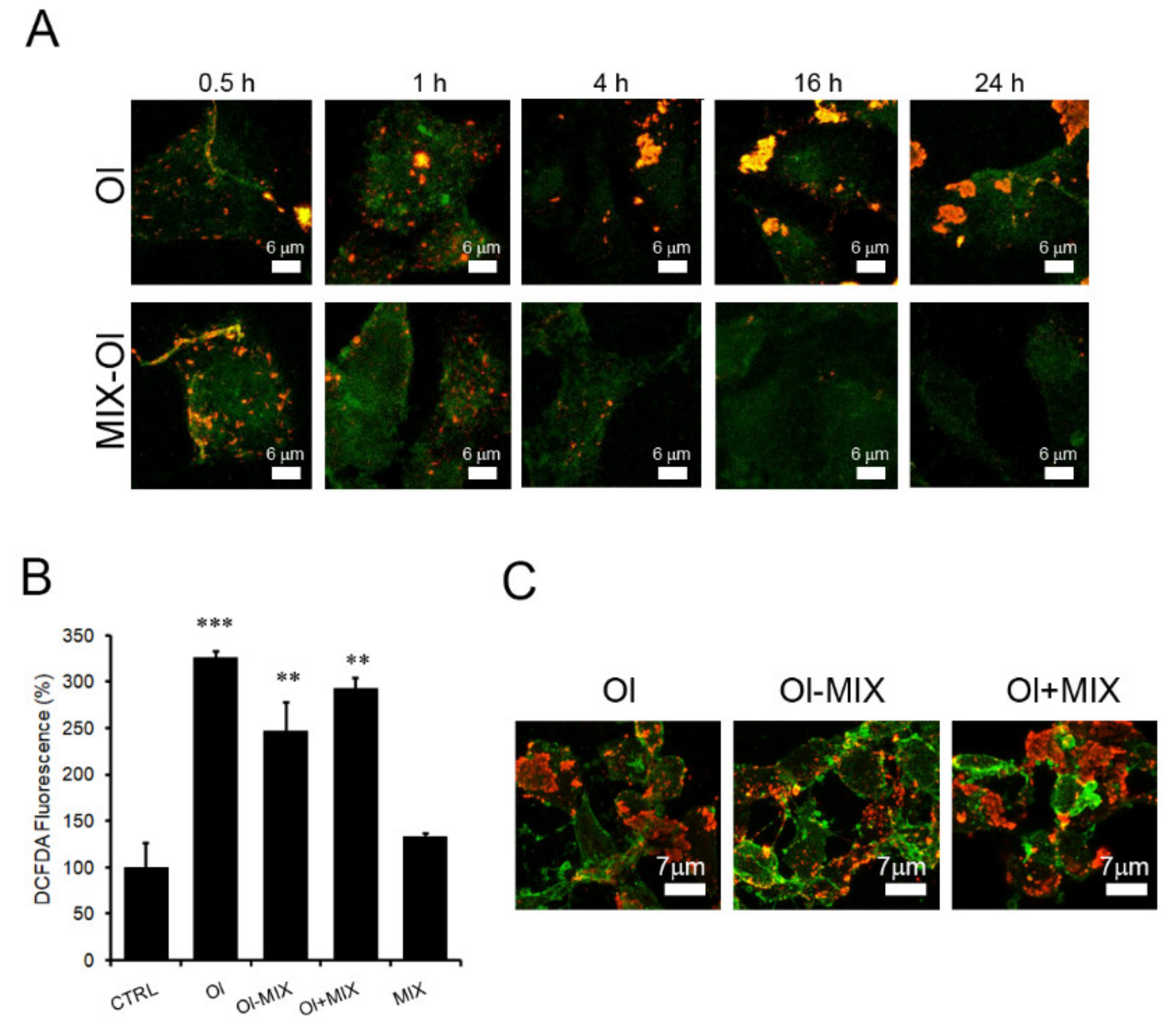
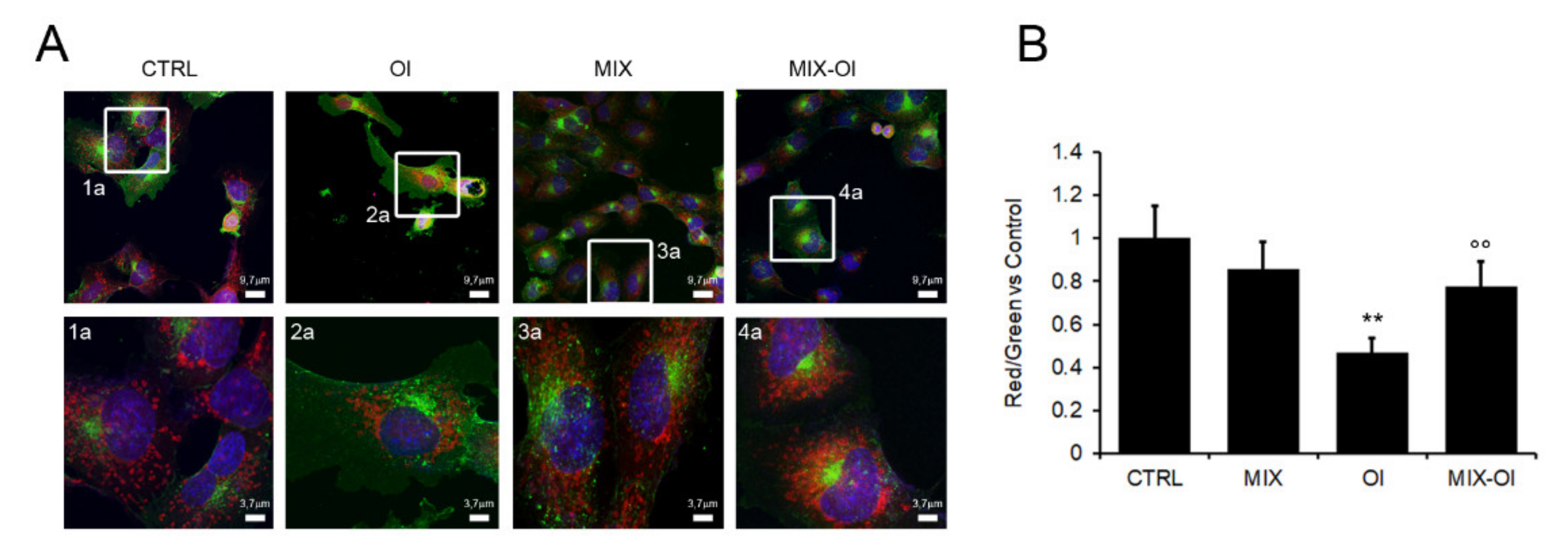
2.4. The MIX Preserves the Autophagic Flux
2.5. The MIX Affects Mitochondria Functions
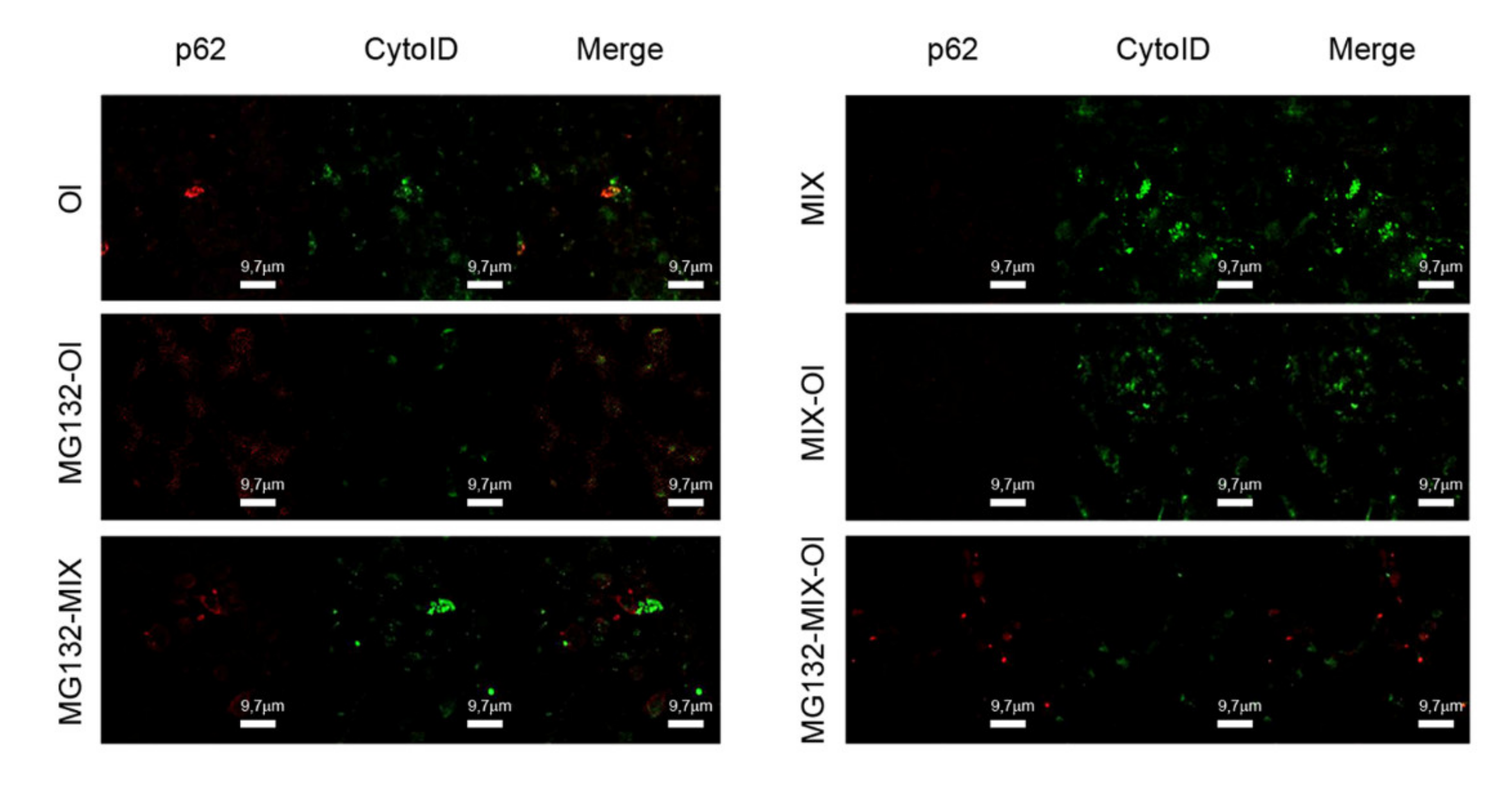
2.6. Effects of the MIX on the Cross-Talk between Autophagy and the Ubiquitin Proteasome System
3. Discussion
4. Materials and Methods
4.1. Aβ1–42 Aggregation
4.2. Preparation of Oleuropein Aglycone and Hydroxytyrosol Samples
4.3. Cell Culture
4.4. MTT Assay
4.5. ROS Determination
4.6. Western-Blotting
4.7. Autophagosome Detection
4.8. Mitochondrial Membrane Potential
4.9. Quantification of Active Mitochondria
4.10. Immunofluorescence
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Alzheimer Association. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar]
- Prince, M.; Comas-Herrera, A.; Knapp, M.; Guerchet, M.; Karagiannidou, M. World Alzheimer report 2016 improving healthcare for people living with dementia. Coverage, quality and costs now and in the future. Alzheimer’s Dis. Int. 2016, 1–140. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Knopman, D.S. Lowering of amyloid-β by β-secretase inhibitors—Some informative failures. N. Engl. J. Med. 2019, 380, 1476–1478. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Vizcarra, J.A.; Marsili, L.; Lang, A.E.; Simon, D.K.; Merola, A.; Josephs, K.A.; Fasano, A.; Morgante, F.; Savica, R.; et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 2019, 92, 329–337. [Google Scholar] [CrossRef]
- Donohue, M.C.; Sperling, R.A.; Petersen, R.; Sun, C.K.; Weiner, M.W.; Aisen, P.S. Alzheimer’s Disease Neuroimaging Initiative. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA 2017, 317, 2305–2316. [Google Scholar] [CrossRef]
- Gardener, H.; Caunca, M.R. Mediterranean Diet in Preventing Neurodegenerative Diseases. Curr. Nutr. Rep. 2018, 7, 10–20. [Google Scholar] [CrossRef]
- Leri, M.; Scuto, M.; Ontario, M.L.; Calabrese, V.; Calabrese, E.J.; Bucciantini, M.; Stefani, M. Healthy Effects of Plant Polyphenols: Molecular Mechanisms. Int. J. Mol. Sci. 2020, 21, 1250. [Google Scholar] [CrossRef] [Green Version]
- Féart, C.; Samieri, C.; Allès, B.; Barberger-Gateau, P. Potential benefits of adherence to the Mediterranean diet on cognitive health. Proc. Nutr. Soc. 2013, 72, 140–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berr, C.; Portet, F.; Carriere, I.; Akbaraly, T.N.; Feart, C.; Gourlet, V.; Combe, N.; Barberger-Gateau, P.; Ritchie, K. Olive oil and cognition: Results from the three-city study. Dement. Geriatr. Cogn. Disord. 2009, 28, 357–364. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Luccarini, I.; Grossi, C.; Rigacci, S.; Coppi, E.; Pugliese, A.M.; Pantano, D.; la Marca, G.; Ed Dami, T.; Berti, A.; Stefani, M.; et al. Oleuropein aglycone protects against pyroglutamylated-3 amyloid-β toxicity: Biochemical, epigenetic and functional correlates. Neurobiol. Aging 2015, 36, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Kostomoiri, M.; Fragkouli, A.; Sagnou, M.; Skaltsounis, L.A.; Pelecanou, M.; Tsilibary, E.C.; Tzinia, A.K. Oleuropein, an anti-oxidant polyphenol constituent of olive promotes α-secretase cleavage of the amyloid precursor protein (AαPP). Cell. Mol. Neurobiol. 2013, 33, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Grossi, C.; Rigacci, S.; Ambrosini, S.; Ed Dami, T.; Luccarini, I.; Traini, C.; Failli, P.; Berti, A.; Casamenti, F.; Stefani, M. The polyphenol oleuropein aglycone protects TgCRND8 mice against Aβ plaque pathology. PLoS ONE 2013, 8, e71702. [Google Scholar] [CrossRef]
- Nardiello, P.; Pantano, D.; Lapucci, A.; Stefani, M.; Casamenti, F. Diet supplementation with hydroxytyrosol ameliorates brain pathology and respores cognitive functions in a mouse model of amylod β deposition. J. Alzheimer’s Dis. 2018, 61, 1161–1172. [Google Scholar] [CrossRef]
- Silva, R.F.M.; Pogačnik, L. Polyphenols from Food and Natural Products: Neuroprotection and Safety. Antioxidants 2020, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Rigacci, S.; Miceli, C.; Nediani, C.; Berti, A.; Cascella, R.; Pantano, D.; Nardiello, P.; Luccarini, I.; Casamenti, F.; Stefani, M. Oleuropein aglycone induces autophagy via the AMPK/mTOR signalling pathway: A mechanistic insight. Oncotarget 2015, 6, 35344–35357. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M.; Leri, M.; Stefani, M.; Melki, R.; Zecchi-Orlandini, S.; Nosi, D. The Amphipathic GM1 Molecule Stabilizes Amyloid Aggregates, Preventing their Cytotoxicity. Biophys. J. 2020, 119, 326–336. [Google Scholar] [CrossRef]
- Bucciantini, M.; Nosi, D.; Forzan, M.; Russo, E.; Calamai, M.; Pieri, L.; Formigli, L.; Quercioli, F.; Soria, S.; Pavone, F.; et al. Toxic effects of amyloid fibrils on cell membranes: The importance of ganglioside GM1. FASEB J. 2012, 26, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Leri, M.; Natalello, A.; Bruzzone, E.; Stefani, M.; Bucciantini, M. Oleuropein aglycone and hydroxytyrosol interfere differently with toxic Aβ 1-42 aggregation. Food Chem. Toxicol. 2019, 129, 1–12. [Google Scholar] [CrossRef]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 04. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S401–S412. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Cecarini, V.; Bonfili, L.; Cuccioloni, M.; Mozzicafreddo, M.; Rossi, G.; Buizza, L.; Uberti, D.; Angeletti, M.; Eleuteri, A.M. Crosstalk between the ubiquitin-proteasome system and autophagy in a human cellular model of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 1741–1751. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Ciechanover, A.; Livneh, I. p62 at the crossroad of the ubiquitin-proteasome system and autophagy. Oncotarget 2016, 7, 83833–83834. [Google Scholar] [CrossRef]
- Tanji, K.; Miki, Y.; Ozaki, T.; Maruyama, A.; Yoshida, H.; Mimura, J.; Matsumiya, T.; Mori, F.; Imaizumi, T.; Itoh, K.; et al. Phosphorylation of serine 349 of p62 in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2014, 2, 50. [Google Scholar] [CrossRef] [Green Version]
- Höhn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, H.A.; González, A.E.; Cerda-Troncoso, C.; Shaughnessy, R.; Otth, C.; Soza, A.; Burgos, P.V. Interplay Between the Autophagy-Lysosomal Pathway and the Ubiquitin-Proteasome System: A Target for Therapeutic Development in Alzheimer’s Disease. Front. Cell Neurosci. 2018, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Lünemann, J.D.; Schmidt, J.; Dalakas, M.C.; Münz, C. Macroautophagy as a pathomechanism in sporadic inclusion body myositis. Autophagy 2007, 3, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Attarwala, I.Y.; Xie, X.Q. SQSTM1/p62: A Potential Target for Neurodegenerative Disease. ACS Chem. Neurosci. 2019, 10, 2094–2114. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, S.A.; Romero-Ramos, M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front. Cell Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M. Structural features and cytotoxicity of amyloid oligomers: Implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog. Neurobiol. 2012, 99, 226–245. [Google Scholar] [CrossRef] [PubMed]
- Rigacci, S.; Guidotti, V.; Bucciantini, M.; Parri, M.; Nediani, C.; Cerbai, E.; Stefani, M.; Berti, A. Oleuropein aglycon prevents cytotoxic amyloid aggregation of human amylin. J. Nutr. Biochem. 2010, 21, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Zafra-Gómez, A.; Luzón-Toro, B.; Capel-Cuevas, S.; Morales, J.C. Stability of Hydroxytyrosol in Aqueous Solutions at Different Concentration, Temperature and with Different Ionic Content: A Study Using UPLC-MS. Food Nutr. Sci. 2011, 2, 1114–1120. [Google Scholar] [CrossRef] [Green Version]
- Nosi, D.; Mercatelli, R.; Chellini, F.; Soria, S.; Pini, A.; Formigli, L.; Quercioli, F. A molecular imaging analysis of Cx43 association with Cdo during skeletal myoblast differentiation. J. Biophotonics 2012, 6, 612–621. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leri, M.; Bertolini, A.; Stefani, M.; Bucciantini, M. EVOO Polyphenols Relieve Synergistically Autophagy Dysregulation in a Cellular Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7225. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137225
Leri M, Bertolini A, Stefani M, Bucciantini M. EVOO Polyphenols Relieve Synergistically Autophagy Dysregulation in a Cellular Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(13):7225. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137225
Chicago/Turabian StyleLeri, Manuela, Andrea Bertolini, Massimo Stefani, and Monica Bucciantini. 2021. "EVOO Polyphenols Relieve Synergistically Autophagy Dysregulation in a Cellular Model of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 13: 7225. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137225