
Norepinephrine Protects against Methamphetamine Toxicity through β2-Adrenergic Receptors Promoting LC3 Compartmentalization

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
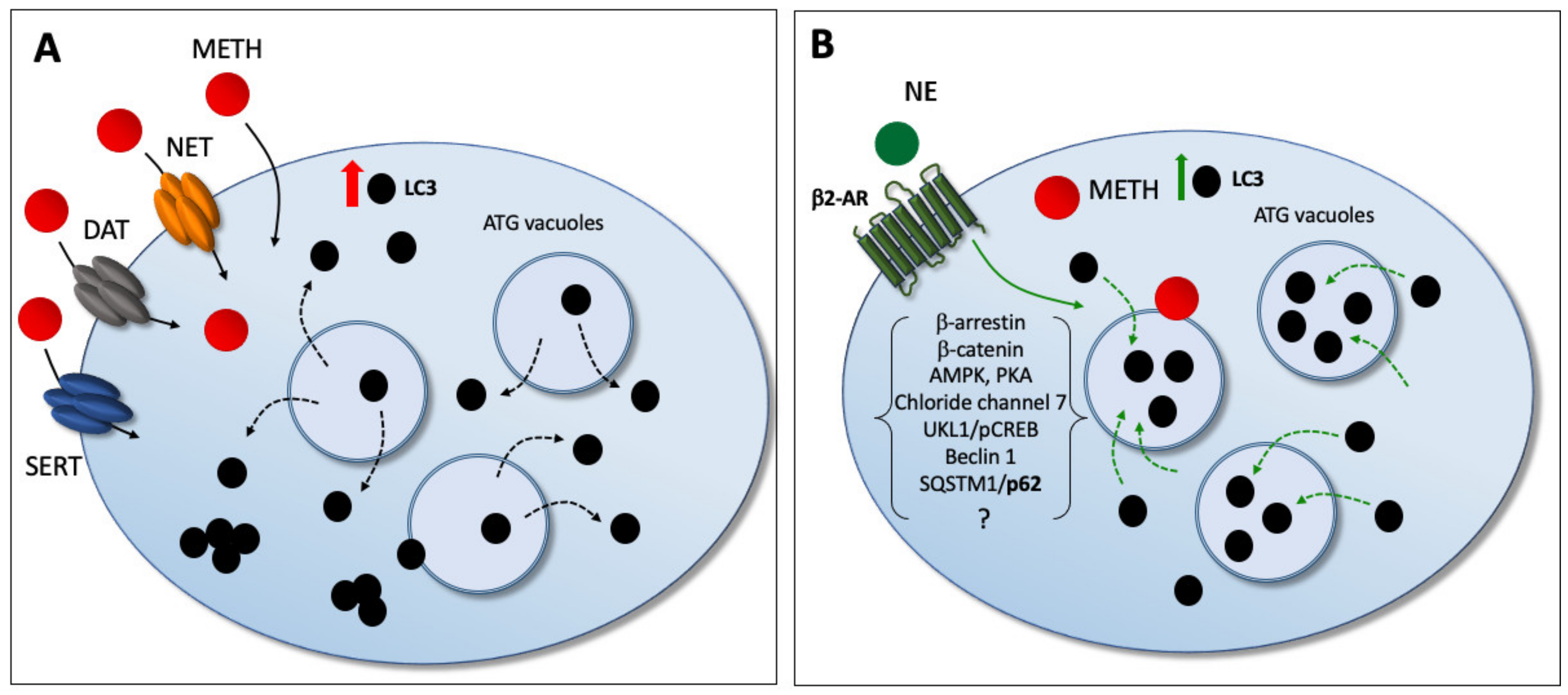{kind=link}
Abstract
:1. Introduction
- (i)
- whether NE directly protects against Meth-induced toxicity;
- (ii)
- whether such an effect is achieved through specific plasma membrane ARs activation, or
- (iii)
- whether NE needs to be taken up in the cell to exert protection;
- (iv)
- whether NE-induced protection counteracts the derangement of specific autophagy compartments and autophagy flux induced by Meth.
2. Results
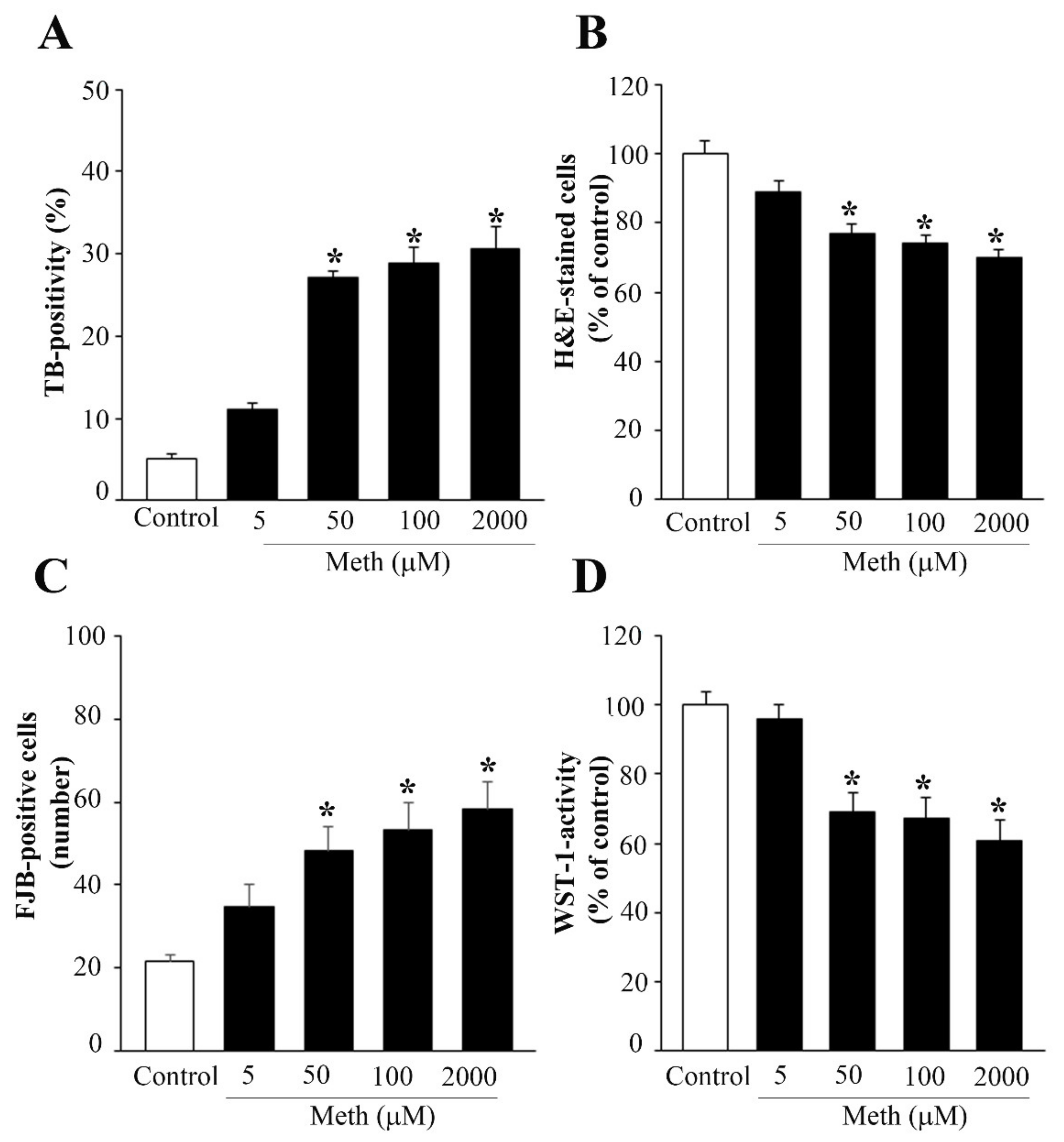
2.1. Pilot Dose–Response Curves on Cell Viability to Select Specific Doses of NE and Meth to Be Administered in the Body of the Study
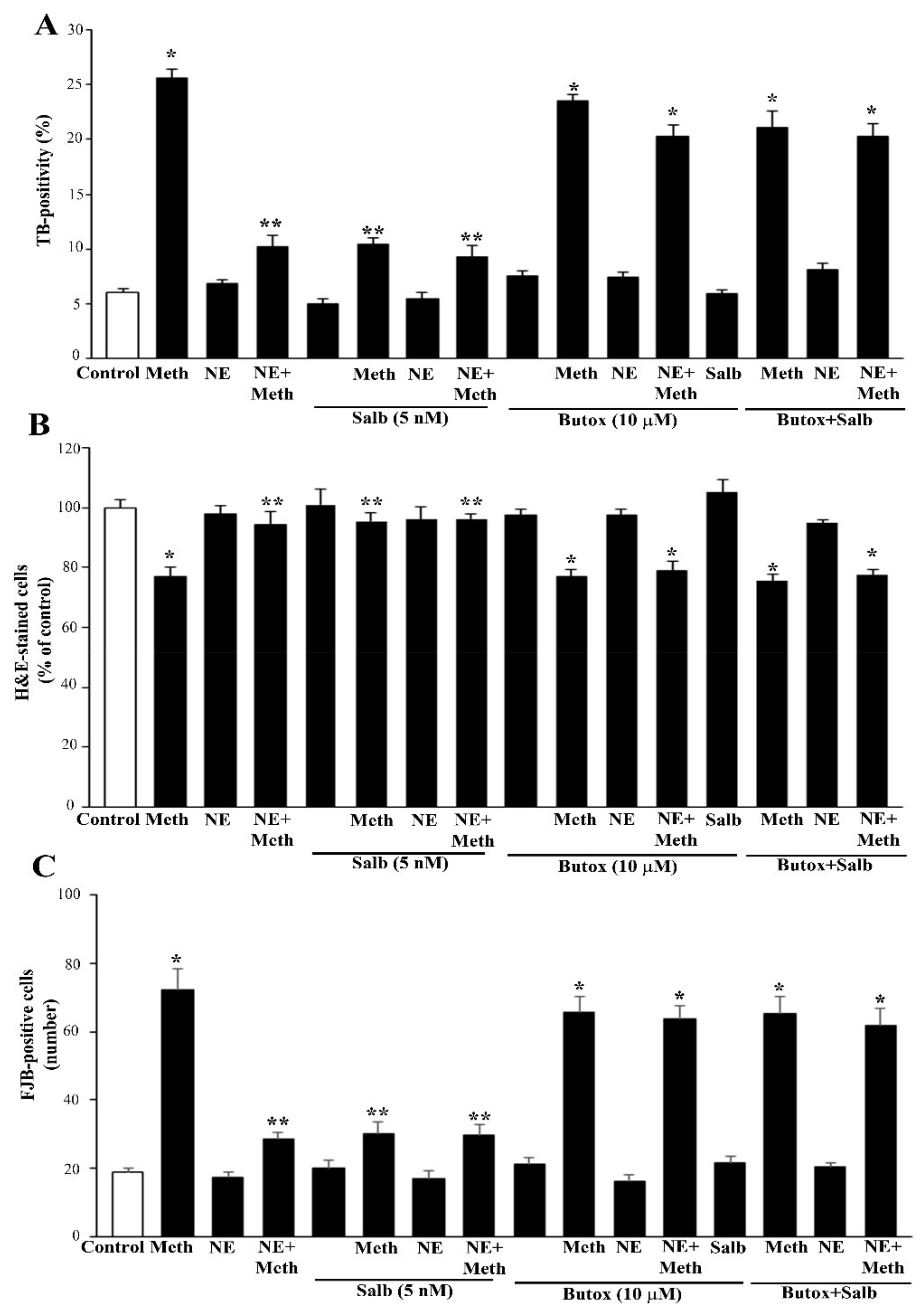
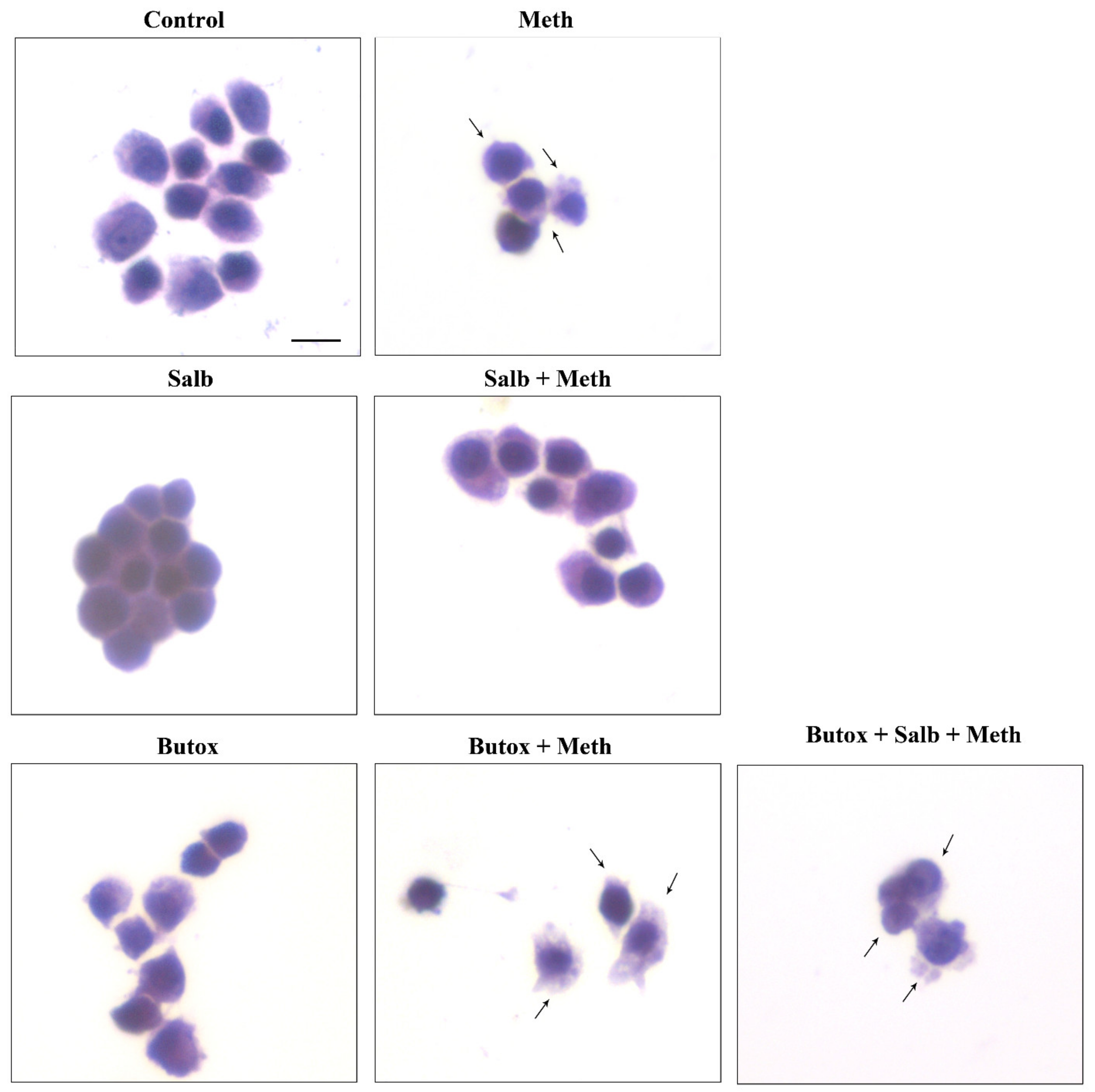
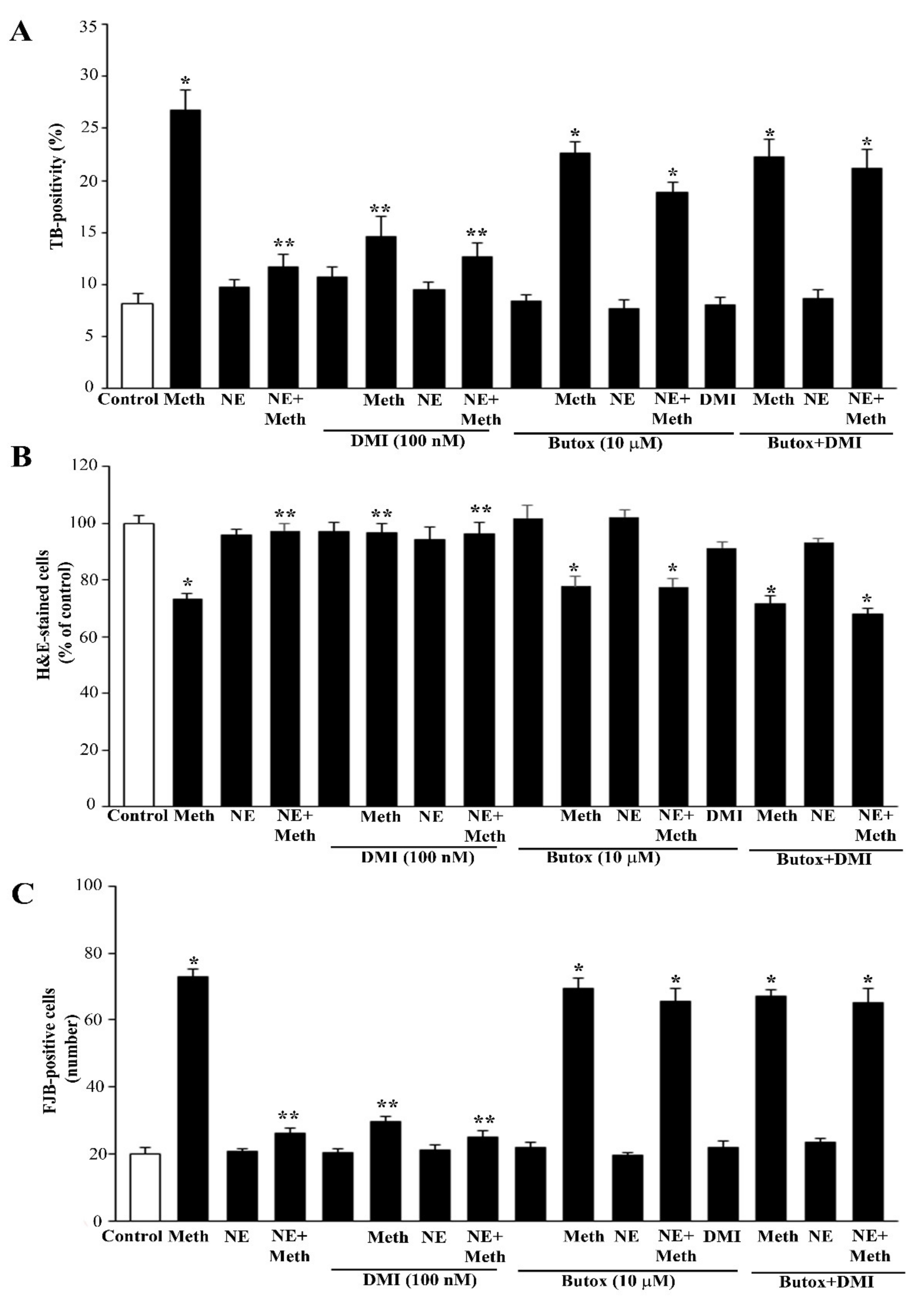
2.2. NE Does Protect against Meth-Induced PC12 Cell Death
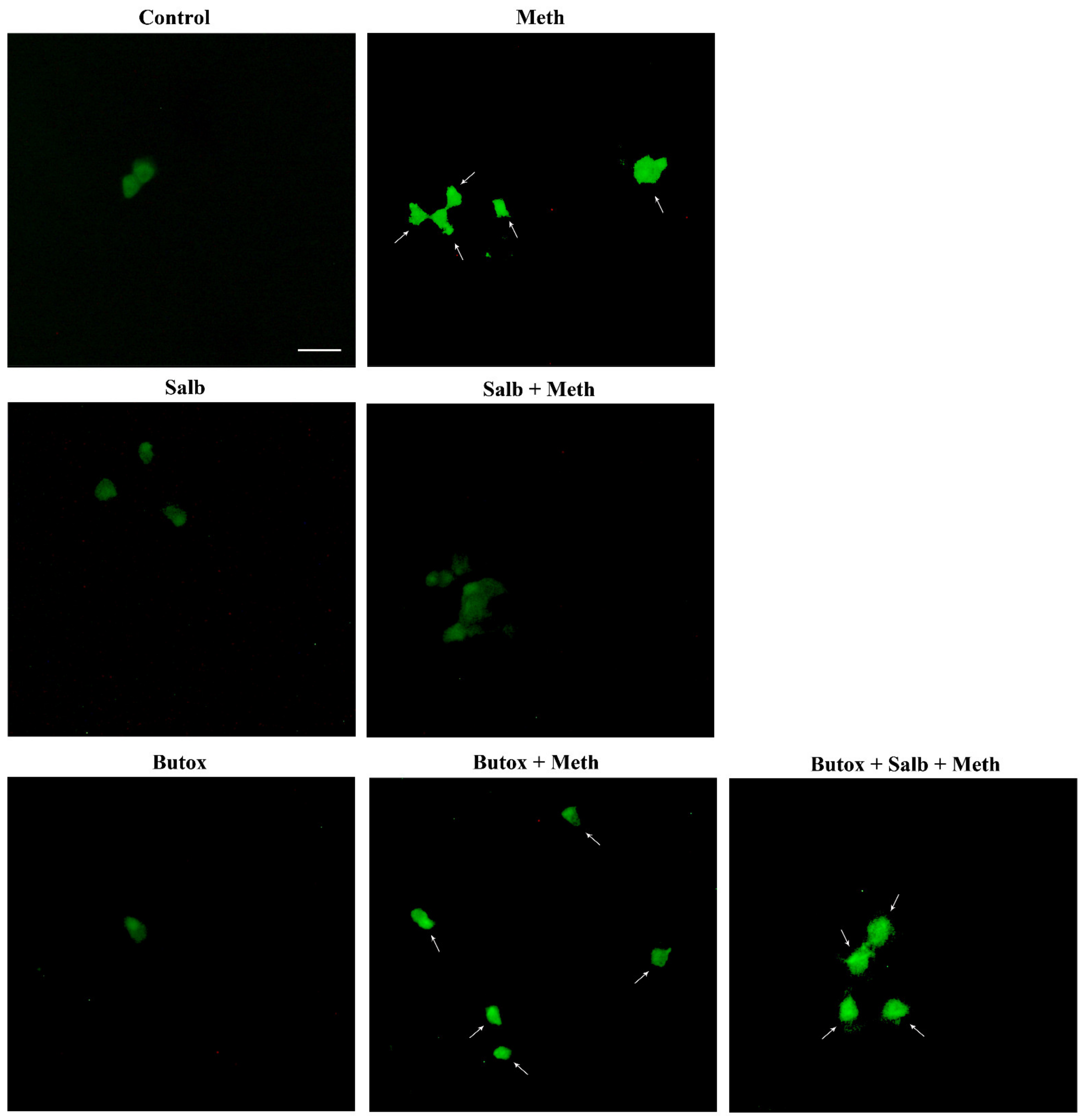
2.3. Effects of Various Agonists and Antagonists Acting on AR on Meth-Induced PC12 Cell Death
2.4. Is There a Protective Role for Intracellular NE?
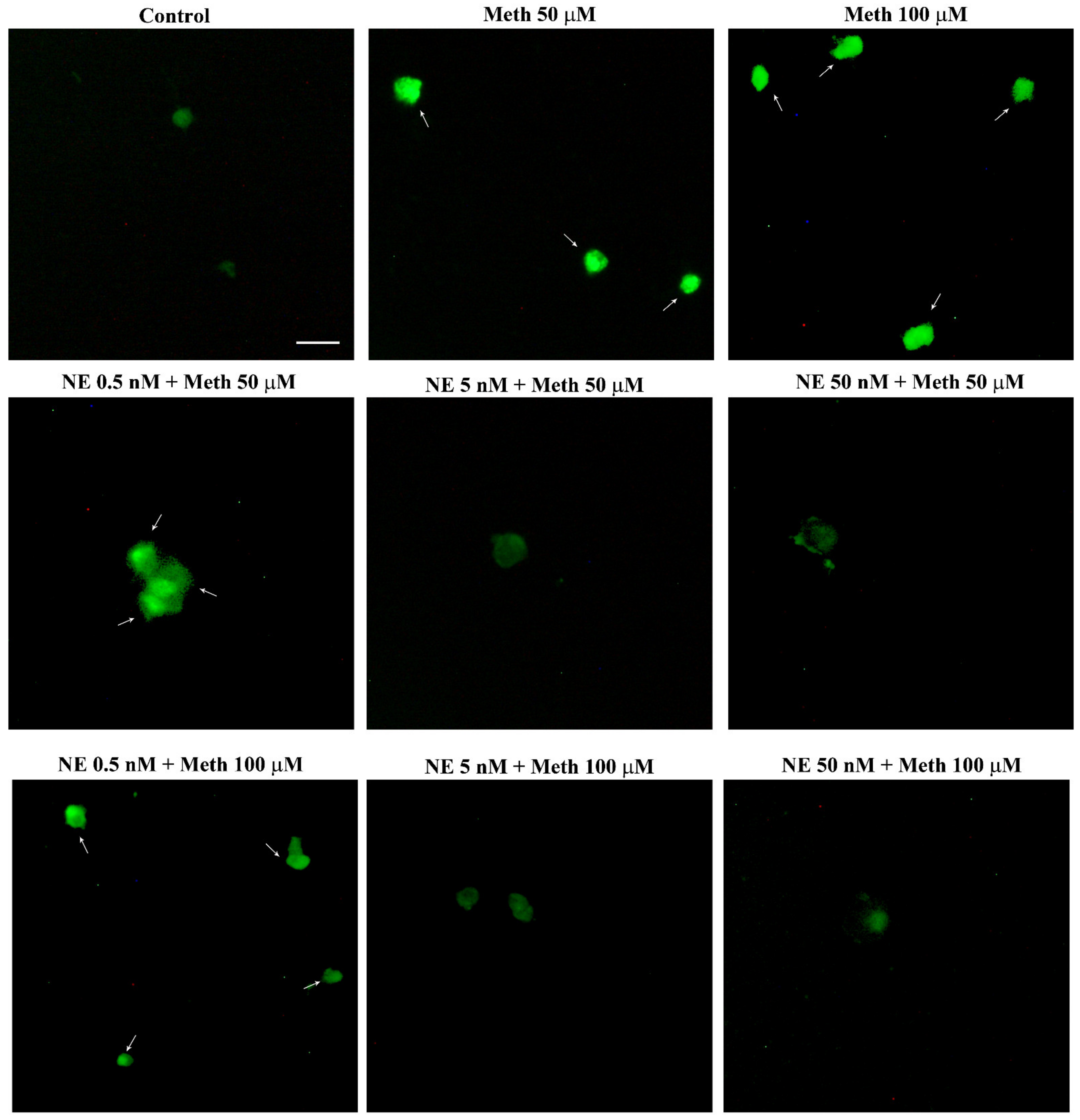
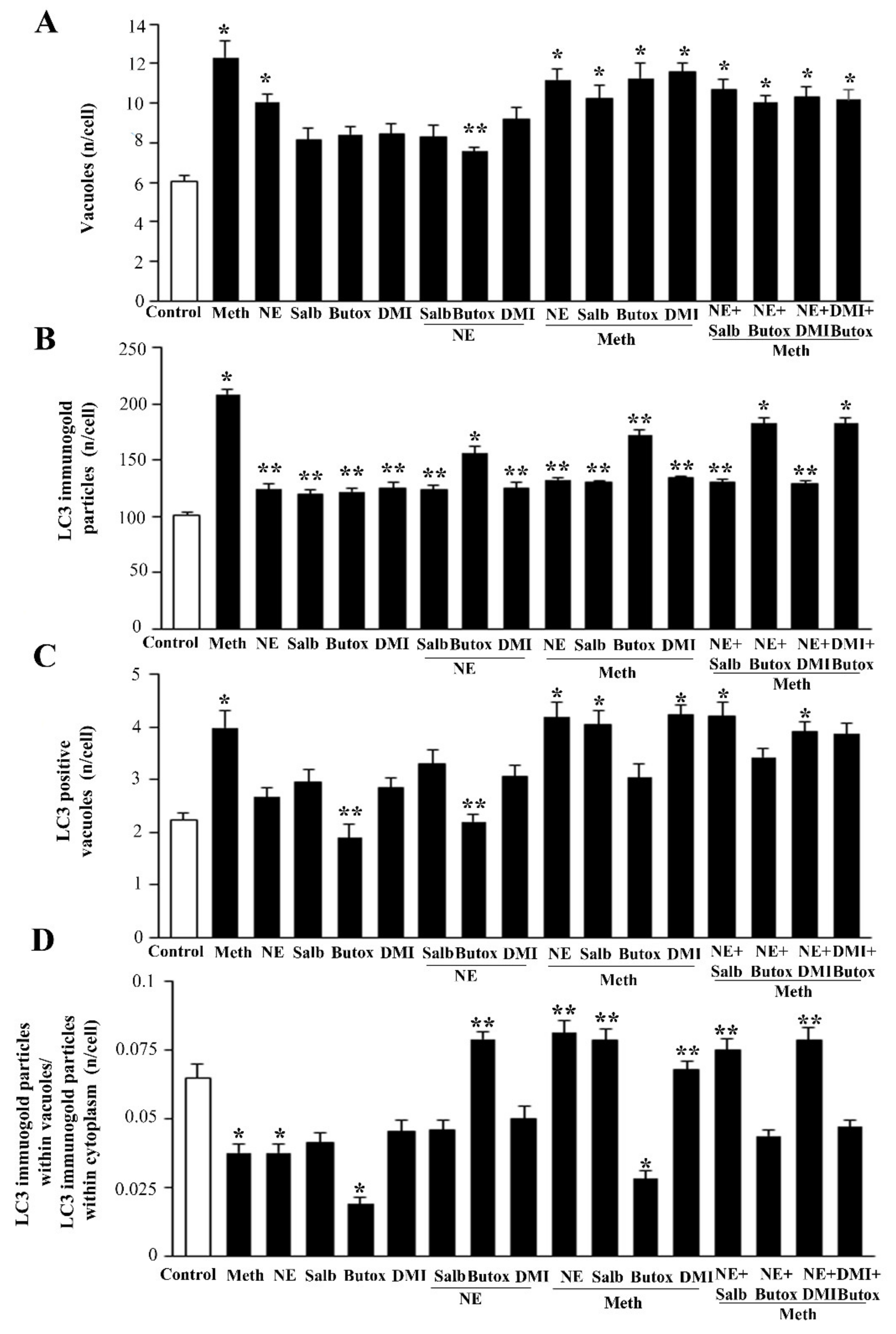
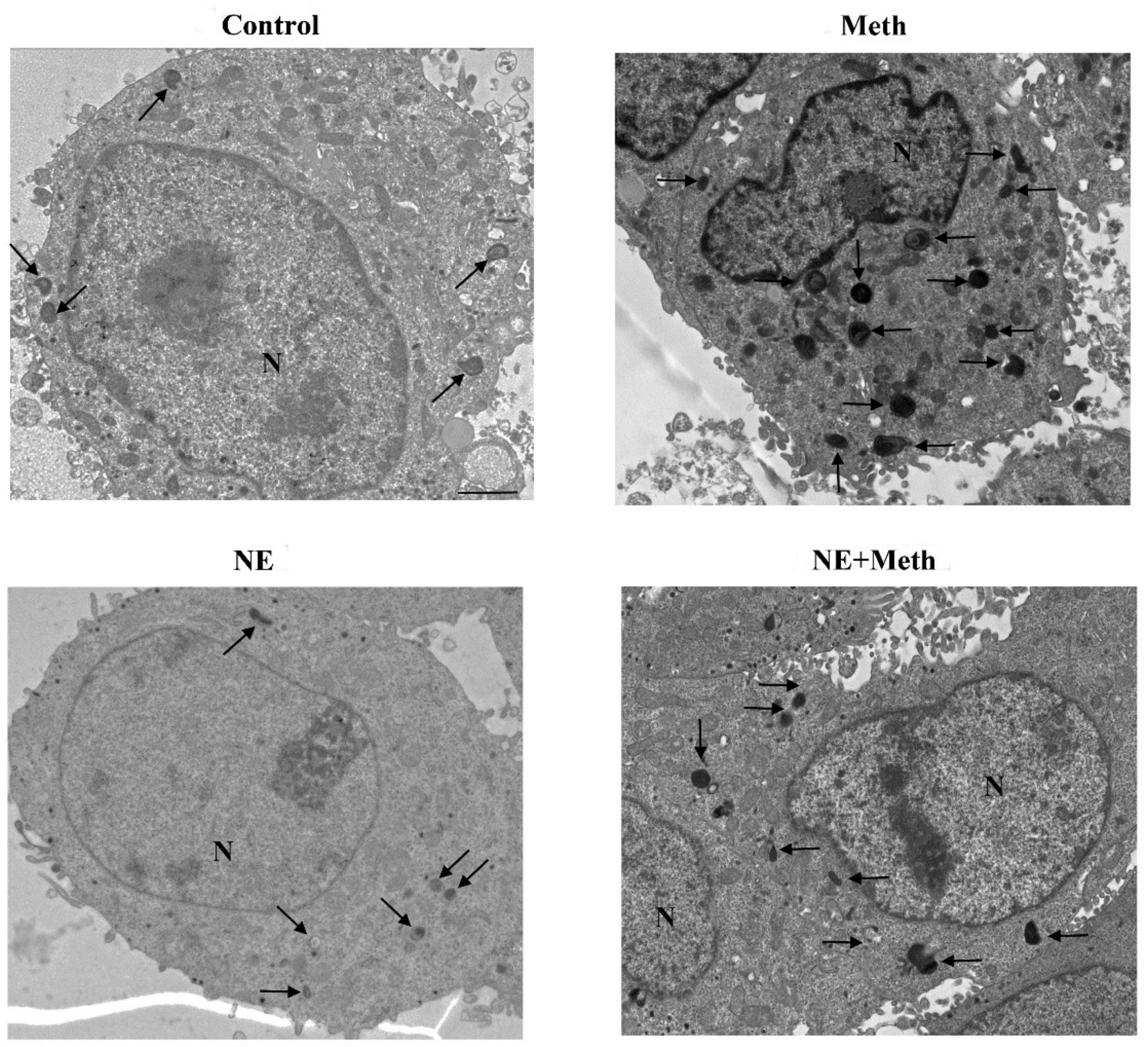
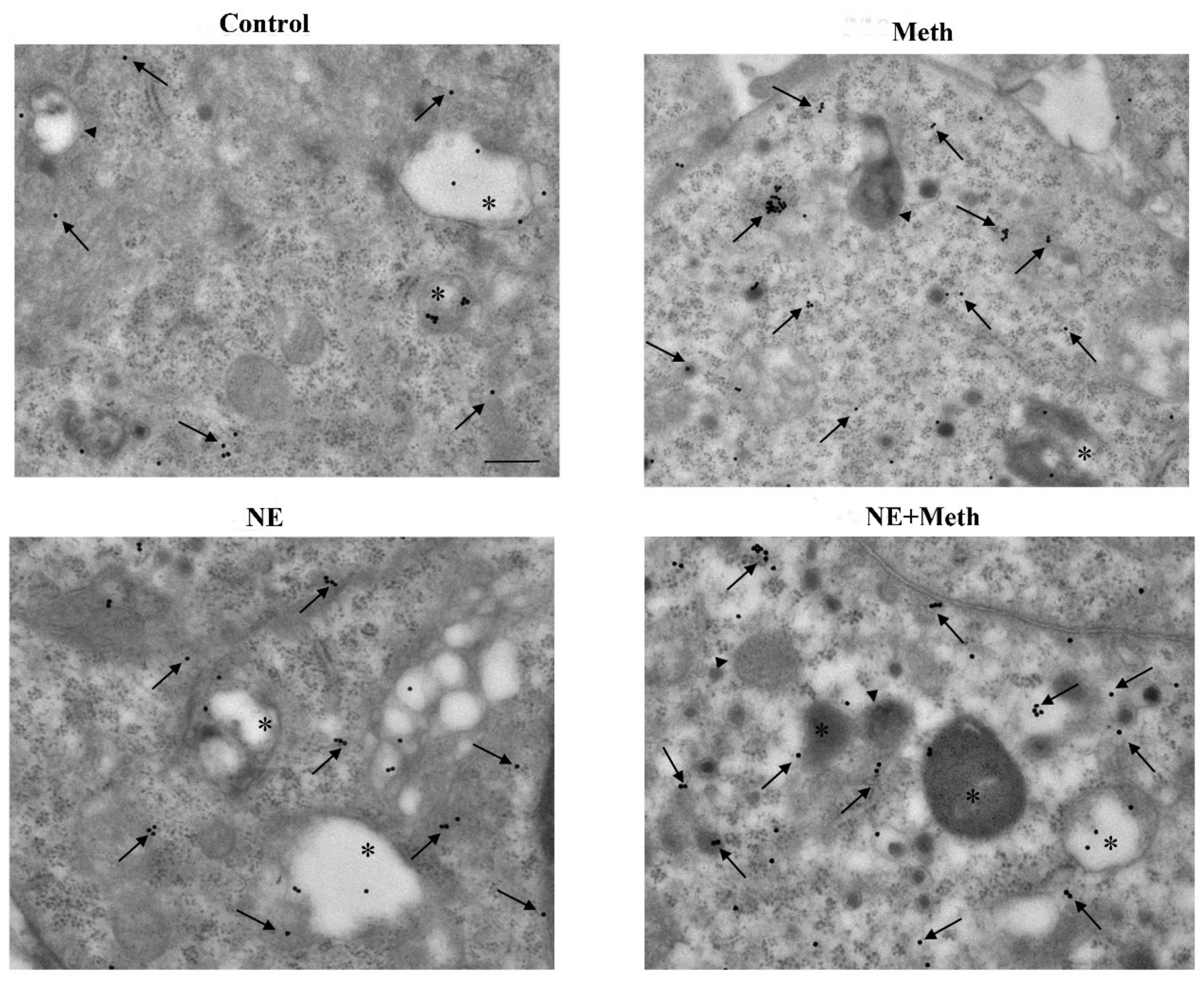
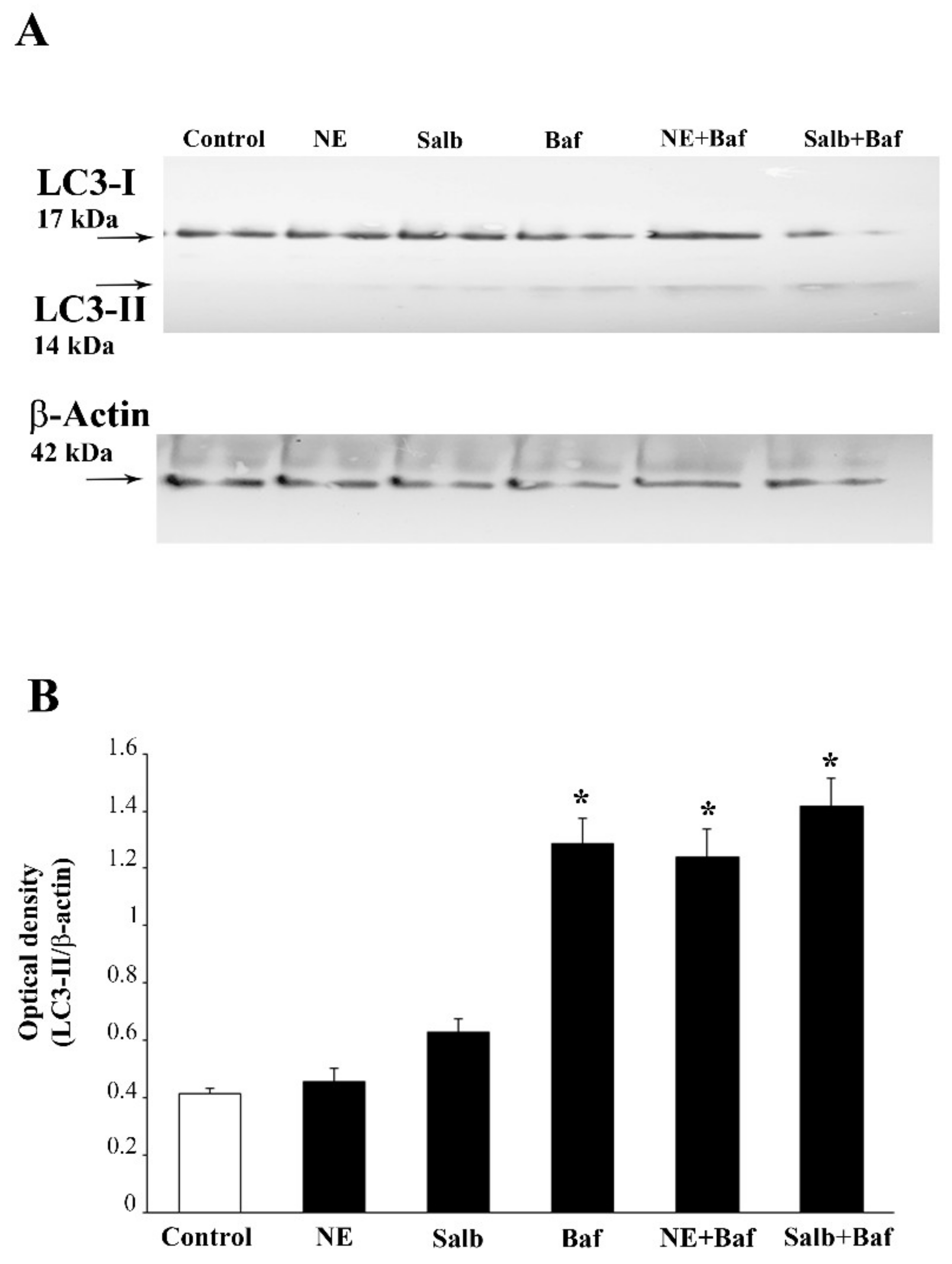
2.5. NE Counteracts Meth-Induced Dissipation of LC3 from Autophagy Vacuoles
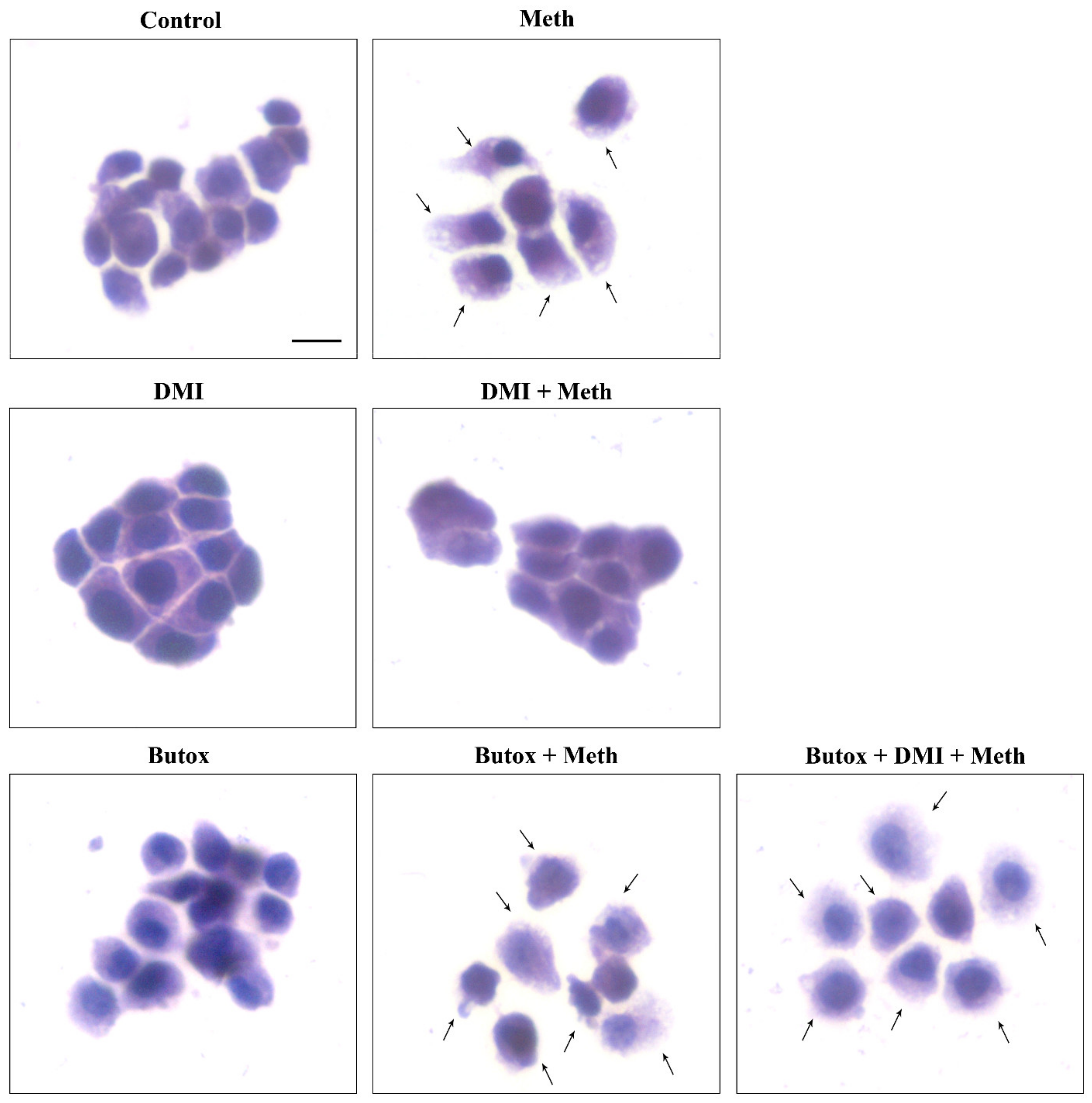
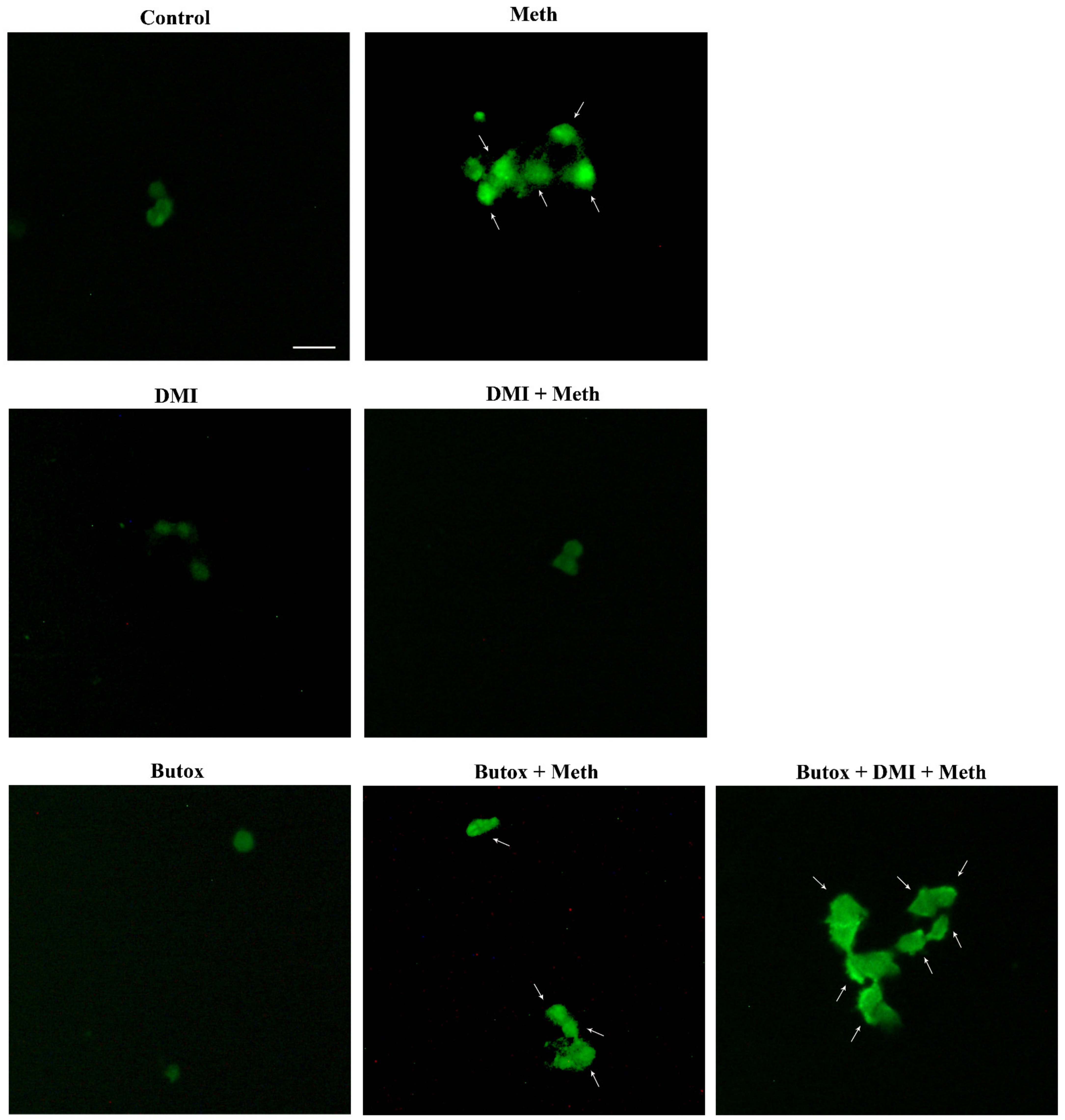
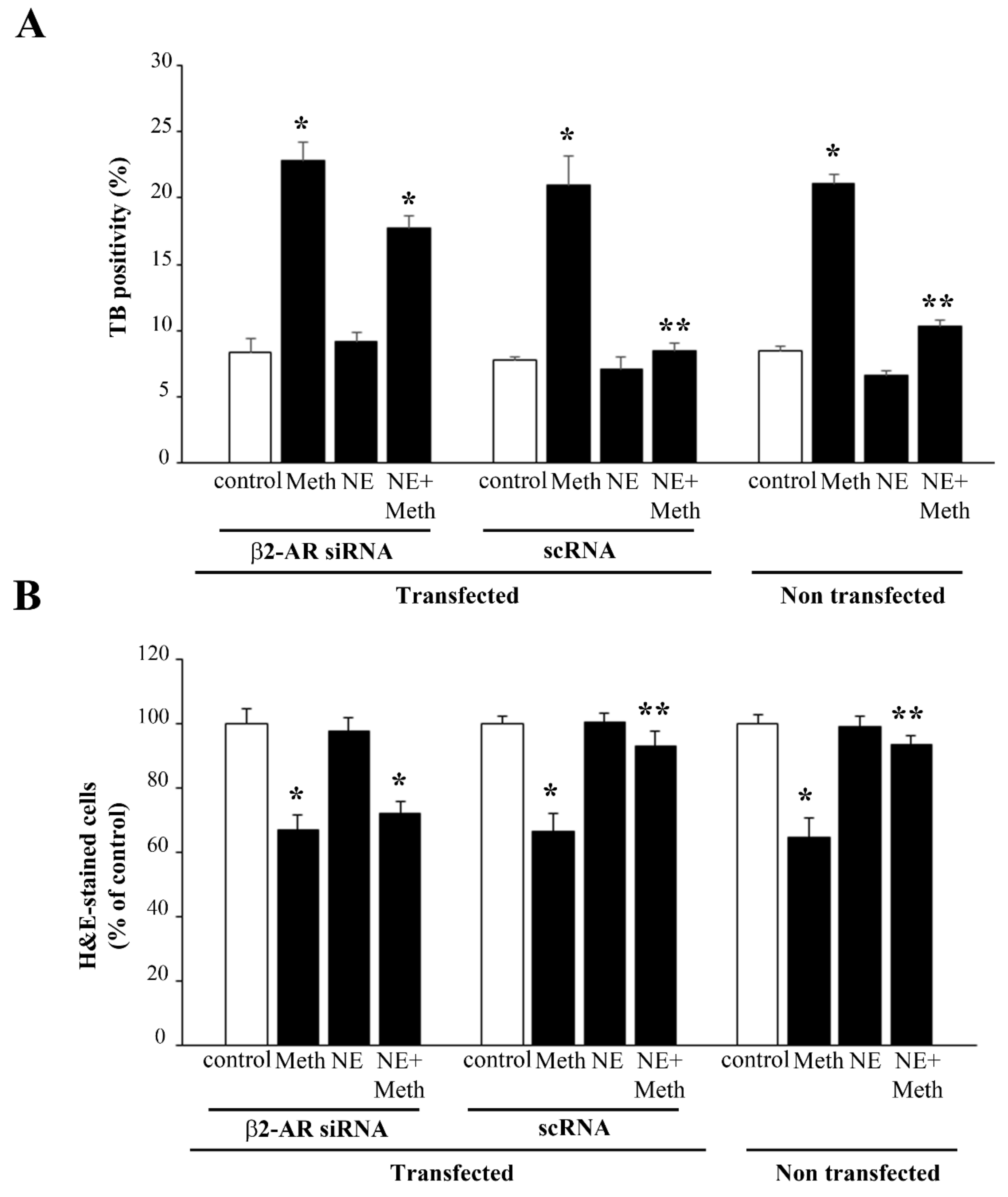
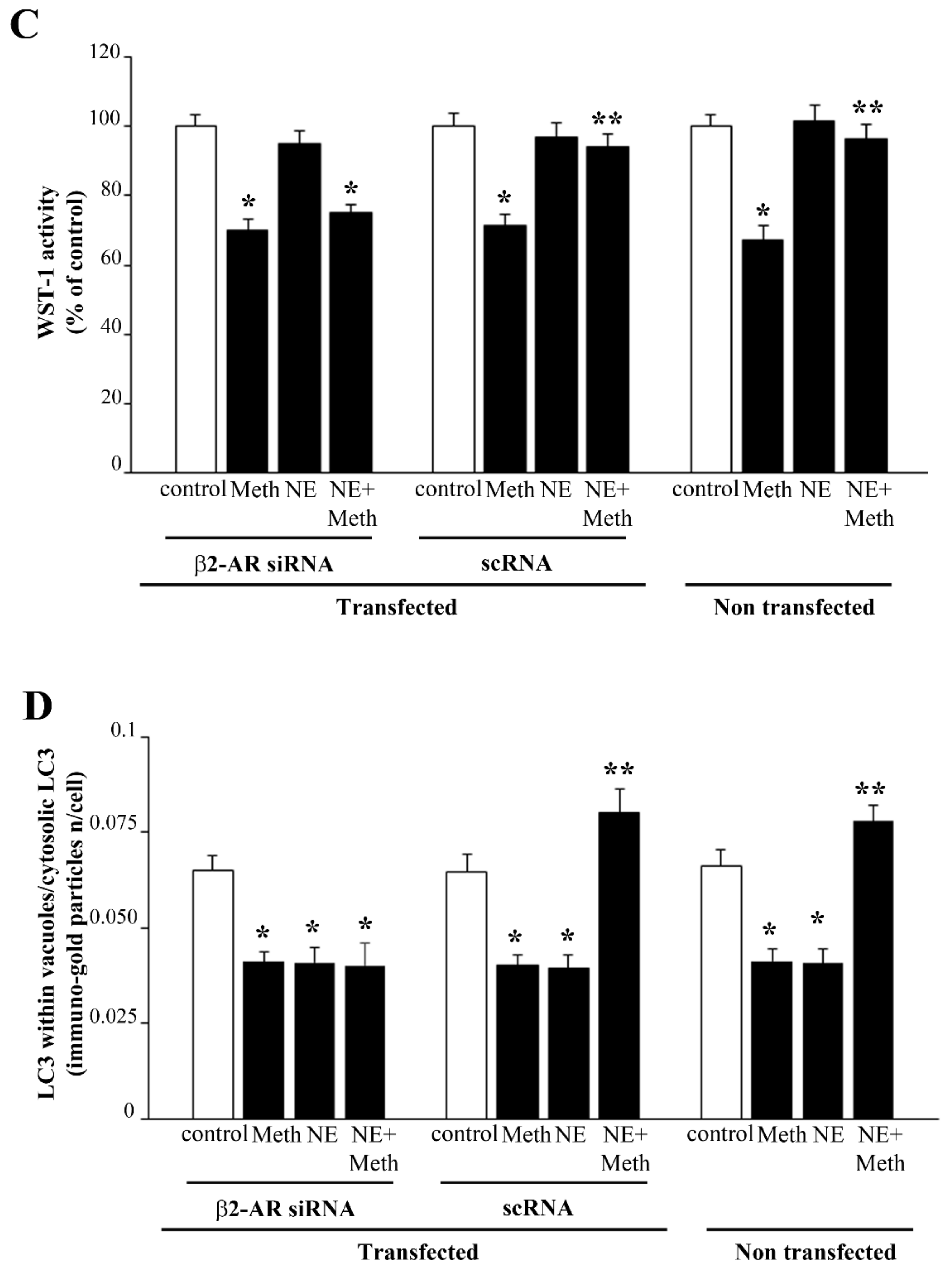
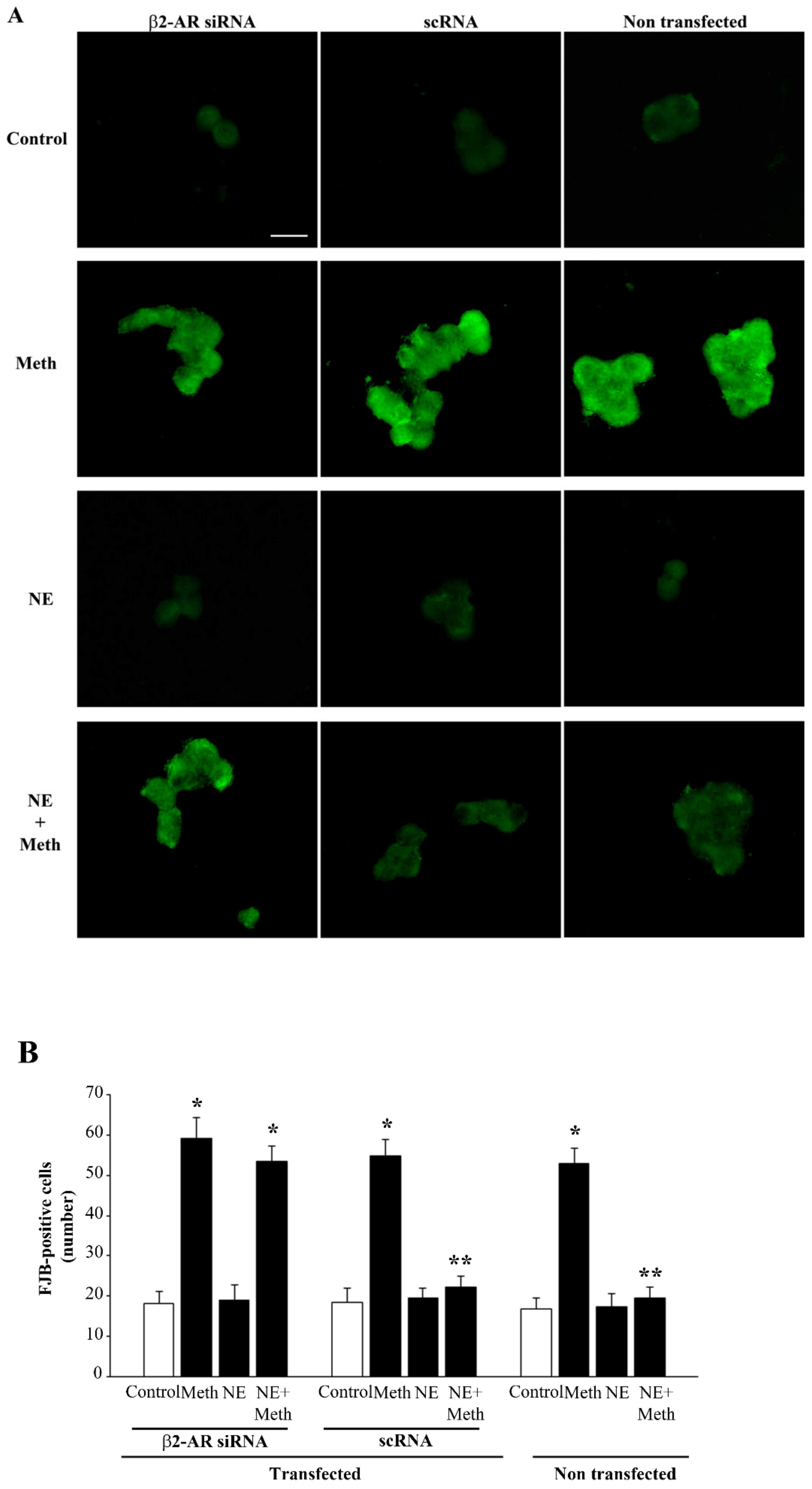
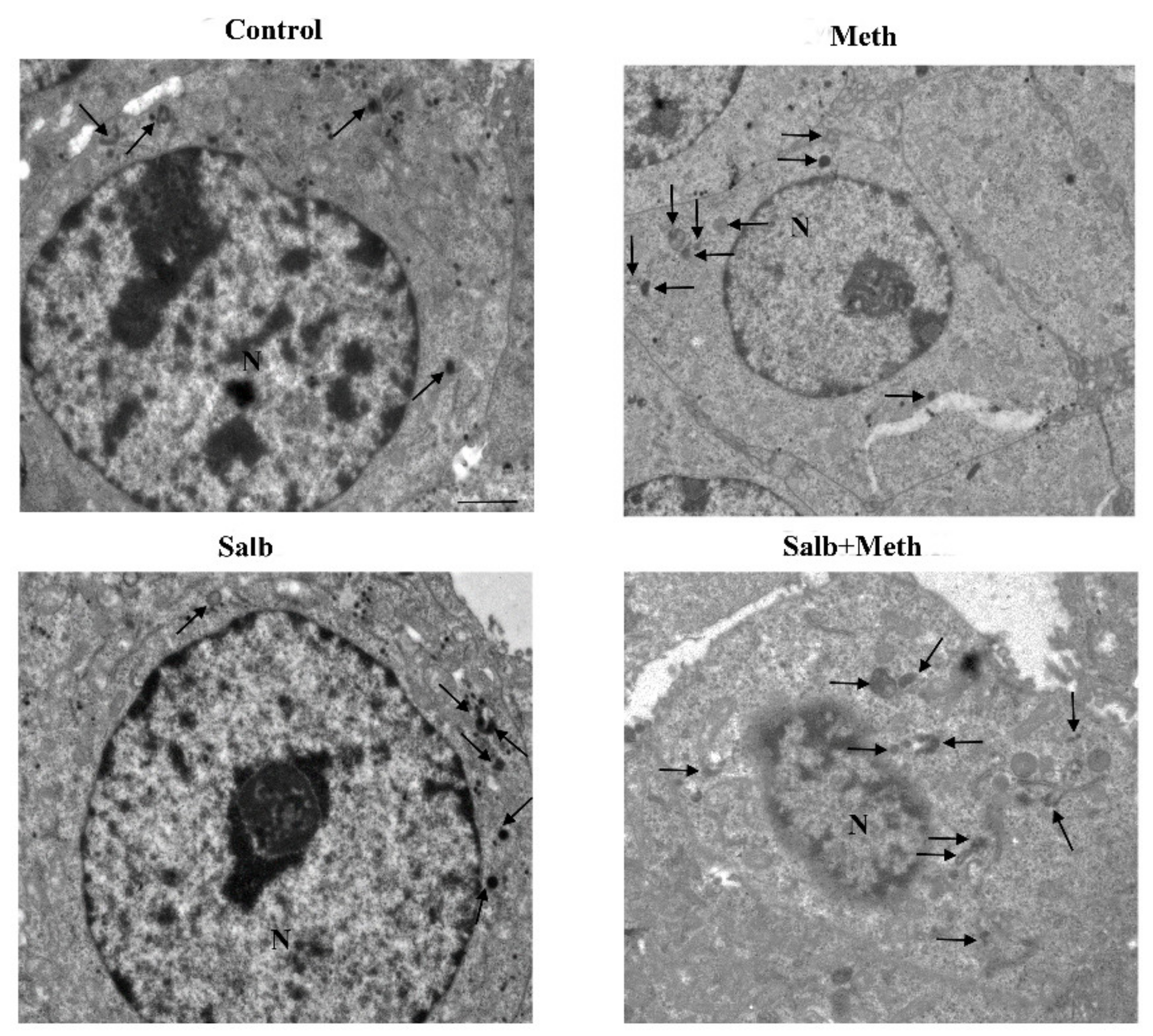
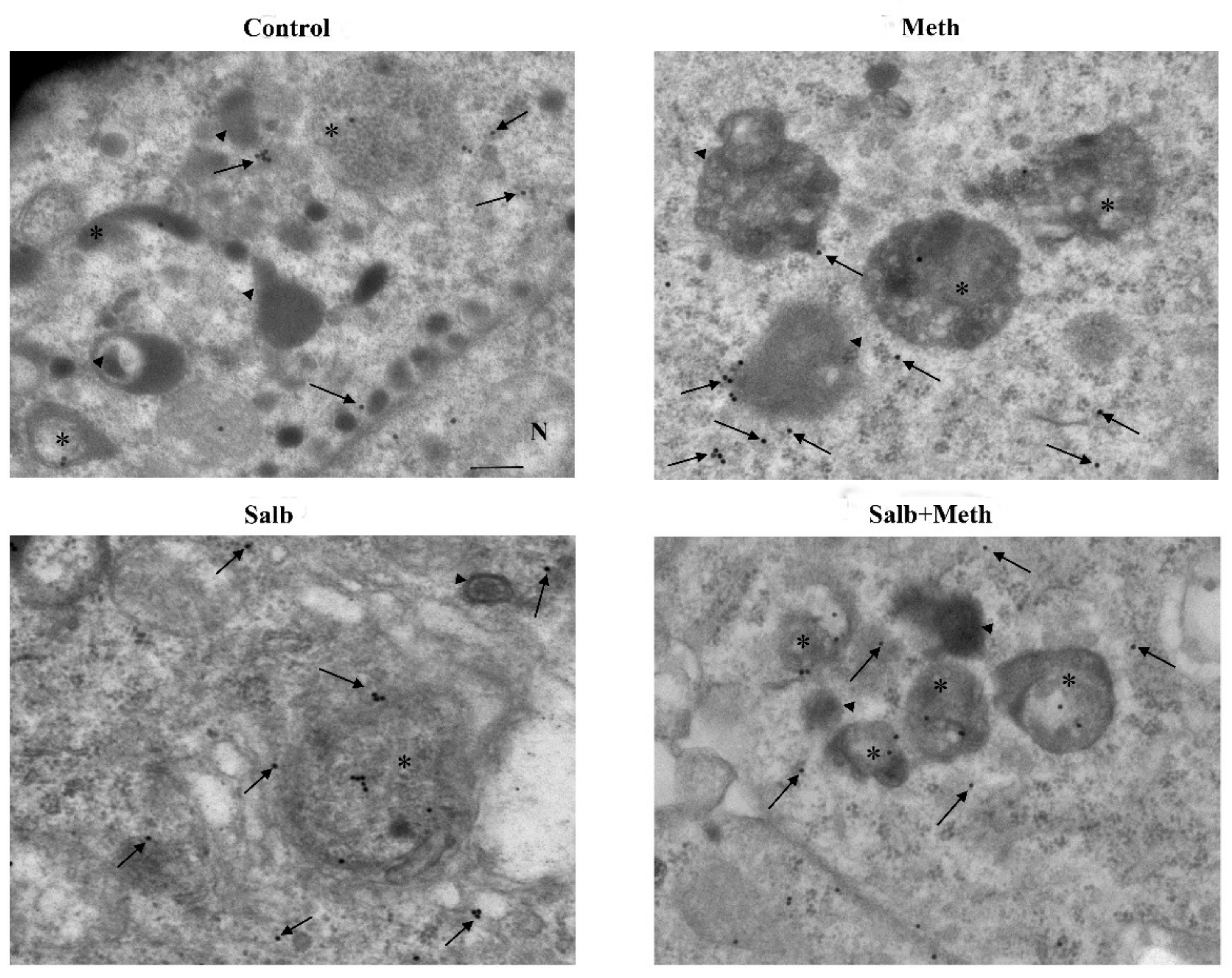
2.6. β2-AR Activity Counteracts Meth-Induced LC3 Dissipation
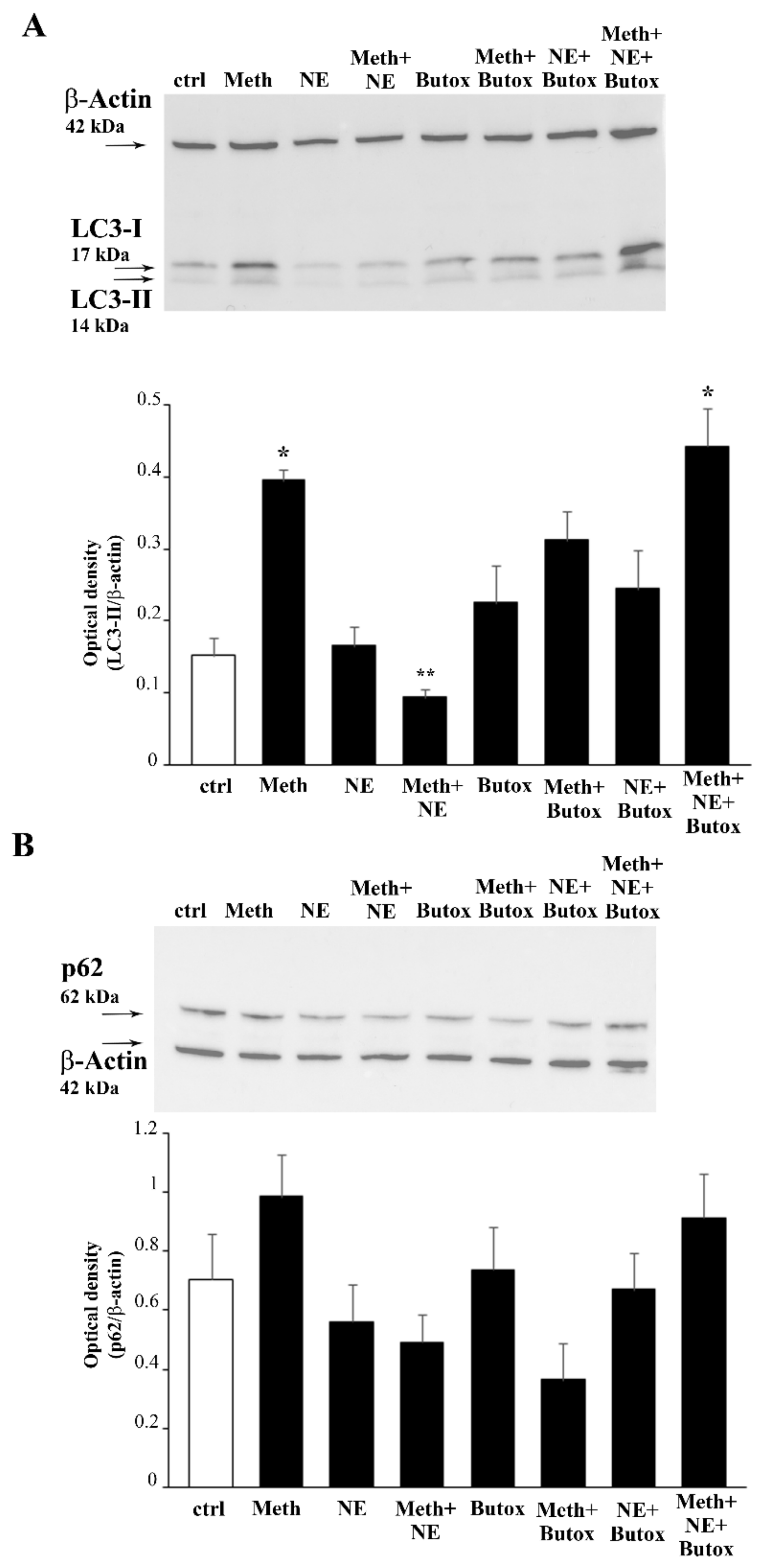
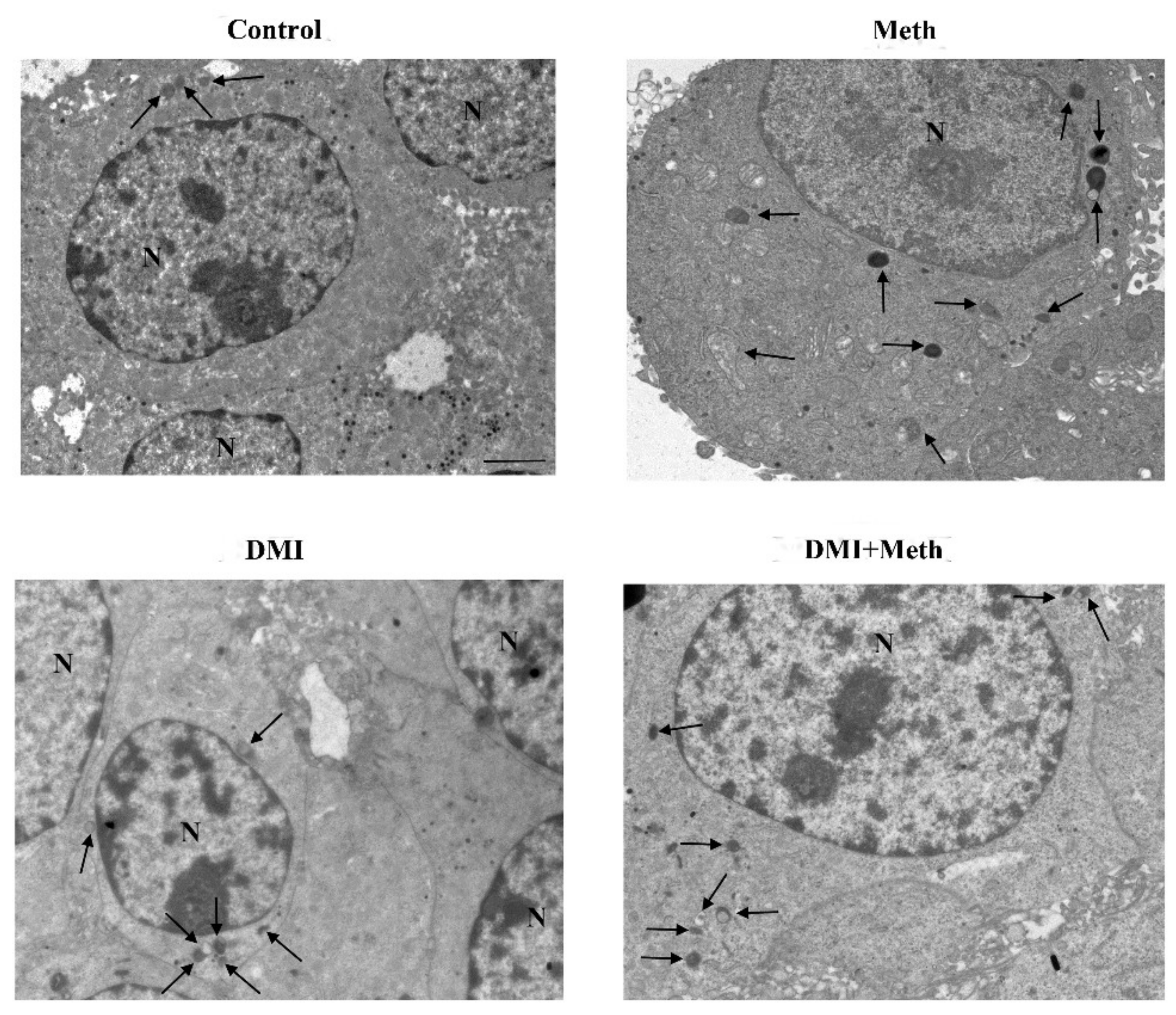
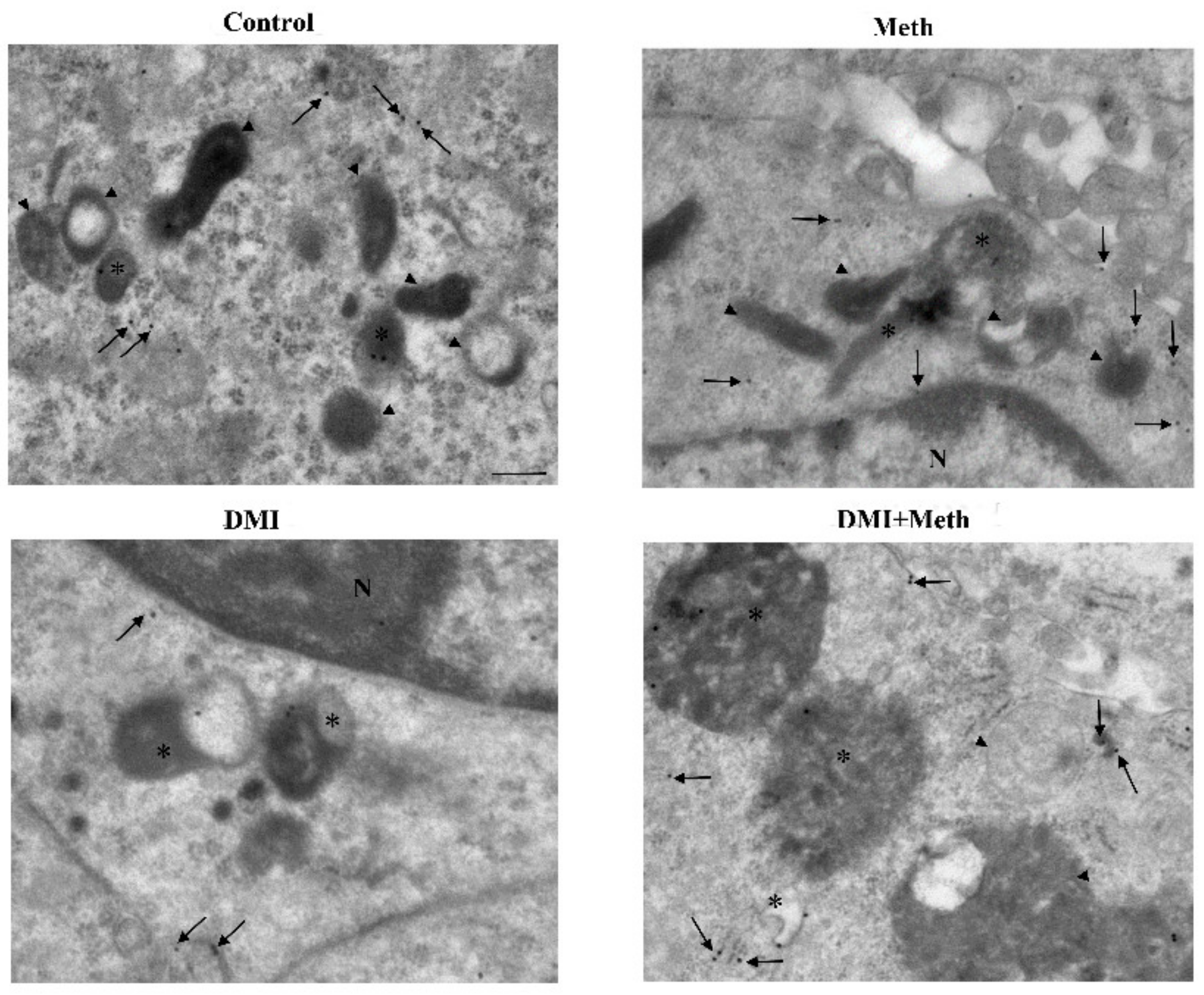
2.7. The Effects of DMI on Meth-Induced LC3 Dissipation
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Experimental Design
4.2. Cell Cycle Analysis by Flow Cytometry
4.3. Silencing RNA Transfection
- (i)
- For Western blot and TEM experiments, cells were seeded onto 6-well plates at a density of 1 × 106 cells per well;
- (ii)
- For light microscopy, cells were seeded onto 24-well plates at a density of 4 × 105 cells per well;
- (iii)
- For WST-1 assay, cells were seeded onto 96-well plates at a density of 1 × 104 cell per well.
4.4. Light Microscopy
4.4.1. Trypan Blue in Suspended Cells
4.4.2. Histochemistry and Histofluorescence in Layered Cell Pellets
4.4.3. Immunofluorescence
4.5. WST-1 Assay
4.6. Western Blot Assay
4.7. TEM and Immunocytochemistry
4.7.1. Post-Embedding Immunocytochemistry
4.7.2. Ultrastructural Morphometry
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marien, M.; Briley, M.; Colpaert, F. Noradrenaline depletion exacerbates MPTP-induced striatal dopamine loss in mice. Eur. J. Pharmacol. 1993, 236, 487–489. [Google Scholar] [CrossRef]
- Fornai, F.; Bassi, L.; Torracca, M.T.; Scalori, V.; Corsini, G.U. Norepinephrine loss exacerbates methamphetamine-induced striatal dopamine depletion in mice. Eur. J. Pharmacol. 1995, 283, 99–102. [Google Scholar] [CrossRef]
- Fornai, F.; Alessandrì, M.G.; Torracca, M.T.; Bassi, L.; Corsini, G.U. Effects of Noradrenergic Lesions on MPTP/MPP+Kinetics and MPTP-Induced Nigrostriatal Dopamine Depletions. J. Pharmacol. Exp. Ther. 1997, 283, 100–107. [Google Scholar] [PubMed]
- Weinshenker, D.; Ferrucci, M.; Busceti, C.L.; Biagioni, F.; Lazzeri, G.; Liles, L.C.; Lenzi, P.; Pasquali, L.; Murri, L.; Paparelli, A.; et al. Genetic or pharmacological blockade of noradrenaline synthesis enhances the neurochemical, behavioral, and neurotoxic effects of methamphetamine. J. Neurochem. 2008, 105, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Fornai, F.; Giorgi, F.S.; Gesi, M.; Chen, K.; Alessrì, M.G.; Shih, J.C. Biochemical effects of the monoamine neurotoxins DSP-4 and MDMA in specific brain regions of MAO-B-deficient mice. Synapse 2001, 39, 213–221. [Google Scholar] [CrossRef]
- Fornai, F.; Torracca, M.T.; Bassi, L.; D’Errigo, D.A.; Scalori, V.; Corsini, G.U. Norepinephrine loss selectively enhances chronic nigrostriatal dopamine depletion in mice and rats. Brain Res. 1996, 735, 349–353. [Google Scholar] [CrossRef]
- Weinshenker, D. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci. 2018, 41, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Cirelli, C.; Pompeiano, M.; Tononi, G. Neuronal gene expression in the waking state: A role for the locus coeruleus. Science 1996, 274, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Hornykiewicz, O.; Kish, S.J. Biochemical pathophysiology of Parkinson’s disease. Adv. Neurol. 1987, 45, 19–34. [Google Scholar]
- Gesi, M.; Soldani, P.; Giorgi, F.S.; Santinami, A.; Bonaccorsi, I.; Fornai, F. The role of the locus coeruleus in the development of Parkinson’s disease. Neurosci. Biobehav. Rev. 2000, 24, 655–668. [Google Scholar] [CrossRef]
- Mann, D.M.A.; Lincoln, J.; Yates, P.O.; Stamp, J.E.; Toper, S. Changes in the monoamine containing neurones of the human CNS in senile dementia. Br. J. Psychiatry 1980, 136, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.C.; He, B.; Perez, S.E.; Ginsberg, S.D.; Mufson, E.J.; Counts, S.E. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Grosser, S.; Hollnagel, J.O.; Gilling, K.E.; Bartsch, J.C.; Heinemann, U.; Behr, J. Gating of hippocampal output by β-adrenergic receptor activation in the pilocarpine model of epilepsy. Neuroscience 2015, 286, 325–337. [Google Scholar] [CrossRef]
- Altman, I.M.; Corcoran, M.E. Facilitation of neocortical kindling by depletion of forebrain noradrenaline. Brain Res. 1983, 270, 174–177. [Google Scholar] [CrossRef]
- Corcoran, M.E. Characteristics of accelerated kindling after depletion of noradrenaline in adult rats. Neuropharmacology 1988, 27, 1081–1084. [Google Scholar] [CrossRef]
- Szot, P.; Weinshenker, D.; White, S.S.; Robbins, C.A.; Rust, N.C.; Schwartzkroin, P.A.; Palmiter, R.D. Norepinephrine-deficient mice have increased susceptibility to seizure-inducing stimuli. J. Neurosci. 1999, 19, 10985–10992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinshenker, D.; Szot, P. The role of catecholamines in seizure susceptibility: New results using genetically engineered mice. Pharmacol. Ther. 2002, 94, 213–233. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Ferrucci, M.; Lazzeri, G.; Pizzanelli, C.; Lenzi, P.; Alessandrl, M.G.; Murri, L.; Fornai, F. A damage to locus coeruleus neurons converts sporadic seizures into self-sustaining limbic status epilepticus. Eur. J. Neurosci. 2003, 17, 2593–2601. [Google Scholar] [CrossRef]
- Jahan, K.; Pillai, K.K.; Vohora, D. DSP-4 induced depletion of brain noradrenaline and increased 6-hertz psychomotor seizure susceptibility in mice is prevented by sodium valproate. Brain Res. Bull. 2018, 142, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Lin, C.S.; Morse, J.K.; Davis, J.N. DSP4 treatment worsens hippocampal pyramidal cell damage after transient ischemia. Neuroscience 1991, 43, 361–367. [Google Scholar] [CrossRef]
- Sternberg, Z.; Schaller, B. Central Noradrenergic Agonists in the Treatment of Ischemic Stroke-an Overview. Transl. Stroke Res. 2020, 11, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Toshimitsu, M.; Kamei, Y.; Ichinose, M.; Seyama, T.; Imada, S.; Iriyama, T.; Fujii, T. Atomoxetine, a selective norepinephrine reuptake inhibitor, improves short-term histological outcomes after hypoxic-ischemic brain injury in the neonatal male rat. Int. J. Dev. Neurosci. 2018, 70, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Gesi, M.; Lenzi, P.; Soldani, P.; Ruffoli, R.; Pellegrini, A.; Ruggieri, S.; Paparelli, A.; Fornai, F. Noradrenergic loss enhances MDMA toxicity and induces ubiquitin-positive striatal whorls. Neurol. Sci. 2002, 23, S75–S76. [Google Scholar] [CrossRef]
- Fornai, F.; Puglisi-Allegra, S. Autophagy status as a gateway for stress-induced catecholamine interplay in neurodegeneration. Neurosci. Biobehav. Rev. 2021, 123, 238–256. [Google Scholar] [CrossRef] [PubMed]
- Bucci, D.; Busceti, C.L.; Calierno, M.T.; Di Pietro, P.; Madonna, M.; Biagioni, F.; Ryskalin, L.; Limanaqi, F.; Nicoletti, F.; Fornai, F. Systematic Morphometry of Catecholamine Nuclei in the Brainstem. Front. Neuroanat. 2017, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuxe, K.; Dahlström, A.B.; Jonsson, G.; Marcellino, D.; Guescini, M.; Dam, M.; Manger, P.; Agnati, L. The discovery of central monoamine neurons gave volume transmission to the wired brain. Prog. Neurobiol. 2010, 90, 82–100. [Google Scholar] [CrossRef] [PubMed]
- Hom, D.G.; Jiang, D.; Hong, E.J.; Mo, J.Q.; Andersen, J.K. Elevated expression of glutathione peroxidase in PC12 cells results in protection against methamphetamine but not MPTP toxicity. Brain Res. Mol. Brain Res. 1997, 46, 154–160. [Google Scholar] [CrossRef]
- Imam, S.Z.; Ali, S.F. Selenium, an antioxidant, attenuates methamphetamine-induced dopaminergic toxicity and peroxynitrite generation. Brain Res. 2000, 855, 186–191. [Google Scholar] [CrossRef]
- Fornai, F.; Lenzi, P.; Gesi, M.; Soldani, P.; Ferrucci, M.; Lazzeri, G.; Capobianco, L.; Battaglia, G.; de Blasi, A.; Nicoletti, F.; et al. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J. Neurochem. 2004, 88, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell. Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef] [Green Version]
- Ryskalin, L.; Puglisi-Allegra, S.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Balestrini, L.; Fornasiero, A.; Leone, S.; Pompili, E.; Ferrucci, M.; et al. Neuroprotective Effects of Curcumin in Methamphetamine-Induced Toxicity. Molecules 2021, 26, 2493. [Google Scholar] [CrossRef]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Eisenberg, T.; Kroemer, G. Autophagy for the avoidance of neurodegeneration. Genes Dev. 2009, 23, 2253–2259. [Google Scholar] [CrossRef] [Green Version]
- Cubells, J.F.; Rayport, S.; Rajendran, G.; Sulzer, D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J. Neurosci. 1994, 14, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [CrossRef]
- Pasquali, L.; Lazzeri, G.; Isidoro, C.; Ruggieri, S.; Paparelli, A.; Fornai, F. Role of autophagy during methamphetamine neurotoxicity. Ann. N. Y. Acad. Sci. 2008, 1139, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.J.; Hanson, G.R.; Fleckenstein, A.E. The role of the plasmalemmal dopamine and vesicular monoamine transporters in methamphetamine-induced dopaminergic deficits. J. Neurochem. 2007, 101, 883–888. [Google Scholar] [CrossRef]
- Kadota, T.; Yamaai, T.; Saito, Y.; Akita, Y.; Kawashima, S.; Moroi, K.; Inagaki, N.; Kadota, K. Expression of dopamine transporter at the tips of growing neurites of PC12 cells. J. Histochem. Cytochem. 1996, 44, 989–996. [Google Scholar] [CrossRef]
- Loder, M.K.; Melikian, H.E. The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J. Biol. Chem. 2003, 278, 22168–22174. [Google Scholar] [CrossRef] [Green Version]
- Müller, H.K.; Kragballe, M.; Fjorback, A.W.; Wiborg, O. Differential regulation of the serotonin transporter by vesicle-associated membrane protein 2 in cells of neuronal versus non-neuronal origin. PLoS ONE 2014, 9, e97540. [Google Scholar] [CrossRef]
- Tellez, R.; Rocha, L.; Castillo, C.; Meneses, A. Autoradiographic study of serotonin transporter during memory formation. Behav. Brain Res. 2010, 212, 12–26. [Google Scholar] [CrossRef]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Lencesova, L.; Sirova, M.; Csaderova, L.; Laukova, M.; Sulova, Z.; Kvetnansky, R.; Krizanova, O. Changes and role of adrenoceptors in PC12 cells after phenylephrine administration and apoptosis induction. Neurochem. Int. 2010, 57, 884–892. [Google Scholar] [CrossRef]
- Yun, J.; Gaivin, R.J.; McCune, D.F.; Boongird, A.; Papay, R.S.; Ying, Z.; Gonzalez-Cabrera, P.J.; Najm, I.; Perez, D.M. Gene expression profile of neurodegeneration induced by alpha1B-adrenergic receptor overactivity: nMDA/GABAA dysregulation and apoptosis. Brain 2003, 126, 2667–2681. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Fornai, F.; Busceti, C.L.; Lembo, G.; Nicoletti, F.; De Blasi, A. Alpha-1B adrenergic receptor knockout mice are protected against methamphetamine toxicity. J. Neurochem. 2003, 86, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paladini, C.A.; Williams, J.T. Noradrenergic inhibition of midbrain dopamine neurons. J. Neurosci. 2004, 24, 4568–4575. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.G. The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br. J. Pharmacol. 2010, 160, 1048–1061. [Google Scholar] [CrossRef] [Green Version]
- Ippolito, M.; Benovic, J.L. Biased agonism at β-adrenergic receptors. Cell. Signal. 2021, 80, 109905. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Alessandrì, M.G.; Torracca, M.T.; Bassi, L.; Scalori, V.; Corsini, G.U. Noradrenergic Modulation of Methamphetamine-Induced Striatal Dopamine Depletion. Ann. N. Y. Acad. Sci. 1998, 844, 166–177. [Google Scholar] [CrossRef]
- Fornai, F.; Giorgi, F.S.; Alessandrí, M.G.; Giusiani, M.; Corsini, G.U. Effects of pretreatment with N-(2-chloroethyl)-N-ethyl-2- bromobenzylamine (DSP-4) on methamphetamine pharmacokinetics and striatal dopamine losses. J. Neurochem. 1999, 72, 777–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aránguiz-Urroz, P.; Canales, J.; Copaja, M.; Troncoso, R.; Vicencio, J.M.; Carrillo, C.; Lara, H.; Lavandero, S.; Díaz-Araya, G. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2011, 1812, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Zaglia, T.; Milan, G.; Franzoso, M.; Bertaggia, E.; Pianca, N.; Piasentini, E.; Voltarelli, V.A.; Chiavegato, D.; Brum, P.C.; Glass, D.J.; et al. Cardiac sympathetic neurons provide trophic signal to the heart via β2-adrenoceptor-dependent regulation of proteolysis. Cardiovasc. Res. 2013, 97, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Farah, B.L.; Sinha, R.A.; Wu, Y.; Singh, B.K.; Zhou, J.; Bay, B.H.; Yen, P.M. β-Adrenergic agonist and antagonist regulation of autophagy in HepG2 cells, primary mouse hepatocytes, and mouse liver. PLoS ONE 2014, 9, e98155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhi, X.; Li, B.; Li, Z.; Zhang, J.; Yu, J.; Zhang, L.; Xu, Z. Adrenergic modulation of AMPK-dependent autophagy by chronic stress enhances cell proliferation and survival in gastric cancer. Int. J. Oncol. 2019, 54, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Jiang, P.; Yang, T.; Huang, M.; Xie, J.; Luo, C.; Qi, W.; Zhou, T.; Yang, Z.; Zou, Y.; et al. β2 adrenergic receptor signaling promotes neuroblastoma cell proliferation by activating autophagy. Oncol. Rep. 2019, 42, 1295–1306. [Google Scholar] [CrossRef]
- Lee, J.H.; Wolfe, D.M.; Darji, S.; McBrayer, M.K.; Colacurcio, D.J.; Kumar, A.; Stavrides, P.; Mohan, P.S.; Nixon, R.A. β2-adrenergic Agonists Rescue Lysosome Acidification and Function in PSEN1 Deficiency by Reversing Defective ER-to-lysosome Delivery of ClC-7. J. Mol. Biol. 2020, 432, 2633–2650. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Du, Y.; Zhang, L.; Chen, X.; He, D. Chloride channel 7 protects from redox status impairment-induced renal tubular epithelial cell apoptosis by activating autophagy. Life Sci. 2020, 261, 118484. [Google Scholar] [CrossRef] [PubMed]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/β-catenin signaling. Biochim. Biophys. Acta Mol. Cell. Res. 2021, 1868, 118926. [Google Scholar] [CrossRef]
- Woo, J.A.; Liu, T.; Fang, C.C.; Castaño, M.A.; Kee, T.; Yrigoin, K.; Yan, Y.; Cazzaro, S.; Matlack, J.; Wang, X.; et al. β-Arrestin2 oligomers impair the clearance of pathological tau and increase tau aggregates. Proc. Natl. Acad. Sci. USA 2020, 117, 5006–5015. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Jia, M.Q.; Xie, Z.H.; Liu, X.; Yang, H.; Zheng, X.; Yuan, H.; Bi, J. Arrestins contribute to amyloid beta-induced cell death via modulation of autophagy and the α7nAch receptor in SH-SY5Y cells. Sci. Rep. 2017, 7, 3446. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Qu, L.; Wan, C.; Xiao, M.; Ni, W.; Jiang, F.; Fan, Y.; Lu, C.; Ni, R. A novel β2-AR/YB-1/β-catenin axis mediates chronic stress-associated metastasis in hepatocellular carcinoma. Oncogenesis 2020, 9, 84. [Google Scholar] [CrossRef]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavridis, M.; Degryse, A.D.; Lategan, A.J.; Marien, M.R.; Colpaert, F.C. Effects of locus coeruleus lesions on parkinsonian signs, striatal dopamine and substantia nigra cell loss after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in monkeys: A possible role for the locus coeruleus in the progression of Parkinson’s disease. Neuroscience 1991, 41, 507–523. [Google Scholar] [CrossRef]
- German, D.C.; Manaye, K.F.; White, C.L.; Woodward, D.J.; McIntire, D.D.; Smith, W.K.; Kalaria, R.N.; Mann, D.M.A. Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol. 1992, 32, 667–676. [Google Scholar] [CrossRef]
- Pifl, C.; Kish, S.J.; Hornykiewicz, O. Thalamic noradrenaline in Parkinson’s disease: Deficits suggest role in motor and non-motor symptoms. Mov. Disord. 2012, 27, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories From 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Ryskalin, L.; Ruffoli, R.; Biagioni, F.; Limanaqi, F.; Ferrucci, M.; Busceti, C.L.; Bonuccelli, U.; Fornai, F. The Neuroanatomy of the Reticular Nucleus Locus Coeruleus in Alzheimer’s Disease. Front. Neuroanat. 2017, 11, 80. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, M.; Giorgi, F.S.; Bartalucci, A.; Busceti, C.L.; Fornai, F. The Effects of Locus Coeruleus and Norepinephrine in Methamphetamine Toxicity. Curr. Neuropharmacol. 2013, 11, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Violin, J.D.; Seidler, F.J.; Slotkin, T.A. Modeling the developmental neurotoxicity of chlorpyrifos in vitro: Macromolecule synthesis in PC12 cells. Toxicol. Appl. Pharmacol. 1998, 151, 182–191. [Google Scholar] [CrossRef]
- Qiao, D.; Seidler, F.J.; Slotkin, T.A. Developmental neurotoxicity of chlorpyrifos modeled in vitro: Comparative effects of metabolites and other cholinesterase inhibitors on DNA synthesis in PC12 and C6 cells. Environ. Health Perspect. 2001, 109, 909–913. [Google Scholar] [CrossRef]
- Balle, T.; Perregaard, J.; Ramirez, M.T.; Larsen, A.K.; Søby, K.K.; Liljefors, T.; Andersen, K. Synthesis and structure-affinity relationship investigations of 5-heteroaryl- substituted analogues of the antipsychotic sertindole. A new class of highly selective alpha(1) adrenoceptor antagonists. J. Med. Chem. 2003, 46, 265–283. [Google Scholar] [CrossRef]
- Tsuchihashi, H.; Nakashima, Y.; Kinami, J.; Nagatomo, T. Characteristics of 125I-iodocyanopindolol binding to beta-adrenergic and serotonin-1B receptors of rat brain: Selectivity of beta-adrenergic agents. Jpn. J. Pharmacol. 1990, 52, 195–200. [Google Scholar] [CrossRef]
- Soriano-Ursúa, M.A.; Valencia-Hernández, I.; Arellano-Mendoza, M.G.; Correa-Basurto, J.; Trujillo-Ferrara, J.G. Synthesis, pharmacological and in silico evaluation of 1-(4-di-hydroxy-3,5-dioxa-4-borabicyclo[4.4.0]deca-7,9,11-trien-9-yl)-2-(tert- butylamino)ethanol, a compound designed to act as a beta2 adrenoceptor agonist. Eur. J. Med. Chem. 2009, 44, 2840–2846. [Google Scholar] [CrossRef]
- Huang, Q.; Lv, J.; Dong, T.; Liu, H.; Xu, L.; Wu, M. Cryptochrome 1 Alleviates the Antiproliferative Effect of Isoproterenol on Human Gastric Cancer Cells. Dose Response 2020, 18, 1559325820939022. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.J.; Zeng, X.S.; Yang, L.H.; Bai, J. The epinephrine increases tyrosine hydroxylase expression through upregulating thioredoxin-1 in PC12 cells. Biochimie 2015, 115, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Rezmann-Vitti, L.A.; Nero, T.L.; Jackman, G.P.; Machida, C.A.; Duke, B.J.; Louis, W.J.; Louis, S.N. Role of Tyr(356(7.43)) and Ser(190(4.57)) in antagonist binding in the rat beta1-adrenergic receptor. J. Med. Chem. 2006, 49, 3467–3477. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.N.; Loxley, R.A.; Barron, A.; Cleary, S.; Phillips, J.K. Comparative studies of PC12 and mouse pheochromocytoma-derived rodent cell lines as models for the study of neuroendocrine systems. In Vitro Cell. Dev. Biol. Anim. 2005, 41, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.Y.; Ordway, G.A. Down-regulation of norepinephrine transporters on PC12 cells by transporter inhibitors. J. Neurochem. 1997, 68, 134–141. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Fabrizi, C.; Pompili, E.; De Vito, S.; Somma, F.; Catizone, A.; Ricci, G.; Lenzi, P.; Fornai, F.; Fumagalli, L. Impairment of the autophagic flux in astrocytes intoxicated by trimethyltin. Neurotoxicology 2016, 52, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Bartek, J. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Bendayan, M.; Zollinger, M. Ultrastructural localization of antigenic sites on osmium-fixed tissues applying the protein A-gold technique. J. Histochem. Cytochem. 1983, 31, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Lenzi, P.; Marongiu, R.; Falleni, A.; Gelmetti, V.; Busceti, C.L.; Michiorri, S.; Valente, E.M.; Fornai, F. A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 2012, 150, 194–217. [Google Scholar] [CrossRef]
- Lucocq, J.M.; Habermann, A.; Watt, S.; Backer, J.M.; Mayhew, T.M.; Griffiths, G. A rapid method for assessing the distribution of gold labeling on thin sections. J. Histochem. Cytochem. 2004, 52, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome a Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef] [Green Version]






















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazzeri, G.; Busceti, C.L.; Biagioni, F.; Fabrizi, C.; Morucci, G.; Giorgi, F.S.; Ferrucci, M.; Lenzi, P.; Puglisi-Allegra, S.; Fornai, F. Norepinephrine Protects against Methamphetamine Toxicity through β2-Adrenergic Receptors Promoting LC3 Compartmentalization. Int. J. Mol. Sci. 2021, 22, 7232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137232
Lazzeri G, Busceti CL, Biagioni F, Fabrizi C, Morucci G, Giorgi FS, Ferrucci M, Lenzi P, Puglisi-Allegra S, Fornai F. Norepinephrine Protects against Methamphetamine Toxicity through β2-Adrenergic Receptors Promoting LC3 Compartmentalization. International Journal of Molecular Sciences. 2021; 22(13):7232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137232
Chicago/Turabian StyleLazzeri, Gloria, Carla L. Busceti, Francesca Biagioni, Cinzia Fabrizi, Gabriele Morucci, Filippo S. Giorgi, Michela Ferrucci, Paola Lenzi, Stefano Puglisi-Allegra, and Francesco Fornai. 2021. "Norepinephrine Protects against Methamphetamine Toxicity through β2-Adrenergic Receptors Promoting LC3 Compartmentalization" International Journal of Molecular Sciences 22, no. 13: 7232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137232