
Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas

 , ,
, ,  ,
,
Abstract
:1. Introduction
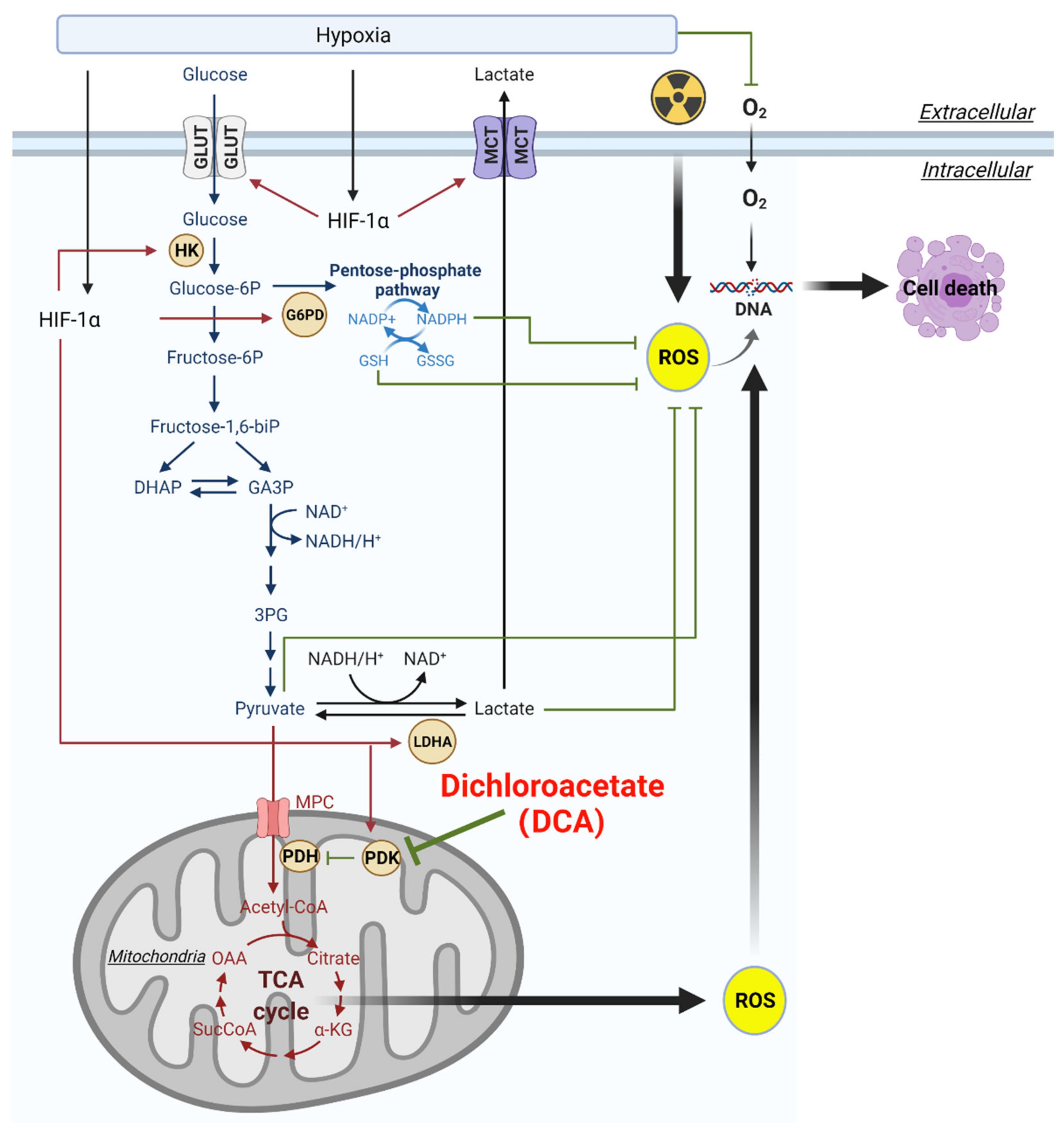
2. Aberrant Glucose Metabolism and Warburg Effect in HGGs
3. Hypoxia and Activation of HIF Can Alter Glucose Metabolism in HGGs
4. Radiotherapy and Radioresistance in HGGs
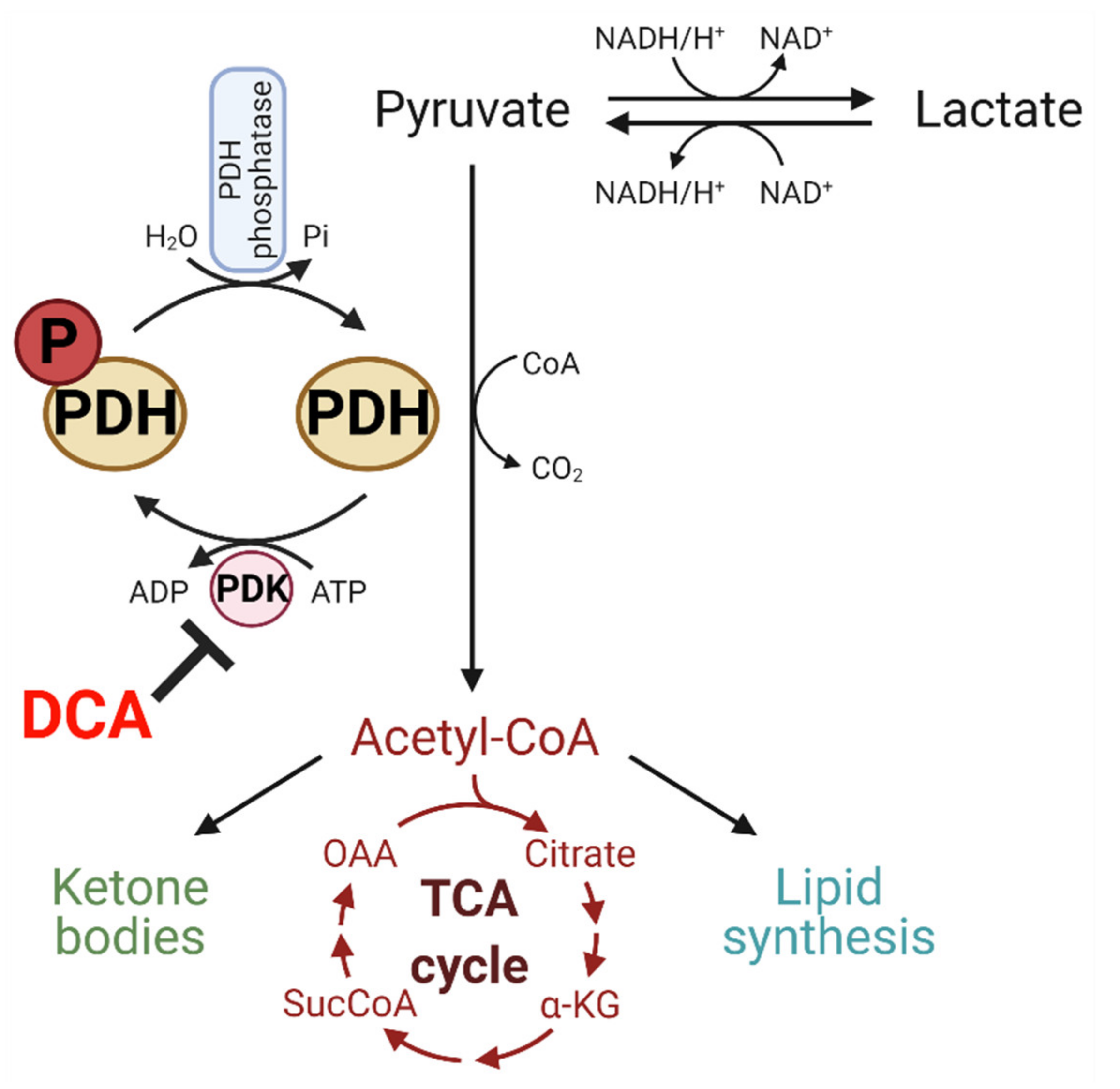
5. DCA Inhibits PDK and Has Potential to Modulate Glucose Metabolism
6. Pre-Clinical Evidence of DCA as a Radiosensitizer in HGG Treatment
6.1. DCA and Glioma Stem Cells (GSCs)
6.2. Efficacy of DCA Combined with Radiotherapy
6.3. Efficacy of DCA/Radiotherapy Combined with Chemotherapy or Metabolic Drugs
6.4. Radiosensitivity Induced by DCA Varies Depending on In Vitro and In Vivo Models
6.5. DCA Efficacy and microRNAs
7. Clinical Trials of DCA in Cancer and as an Anti-HGG Drug
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quick-Weller, J.; Lescher, S.; Forster, M.T.; Konczalla, J.; Seifert, V.; Senft, C. Combination of 5-ALA and iMRI in re-resection of recurrent glioblastoma. Br. J. Neurosurg. 2016, 30, 313–317. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Meijer, T.W.; Kaanders, J.H.; Span, P.N.; Bussink, J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin. Cancer Res. 2012, 18, 5585–5594. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Yacoub, S.; Shiverick, K.T.; Namiki, K.; Sakai, Y.; Porvasnik, S.; Urbanek, C.; Rosser, C.J. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008, 68, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Delaney, L.M.; Ho, N.; Morrison, J.; Farias, N.R.; Mosser, D.D.; Coomber, B.L. Dichloroacetate affects proliferation but not survival of human colorectal cancer cells. Apoptosis Int. J. Program. Cell Death 2015, 20, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.E.; Jin, H.O.; Kim, H.A.; Seong, M.K.; Kim, E.K.; Ye, S.K.; Choe, T.B.; Lee, J.K.; Kim, J.I.; Park, I.C.; et al. Targeting HIF-1alpha is a prerequisite for cell sensitivity to dichloroacetate (DCA) and metformin. Biochem. Biophys. Res. Commun. 2016, 469, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Decollogne, S.; Dilda, P.J.; Hau, E.; Chung, S.A.; Luk, P.P.; Hogg, P.J.; McDonald, K.L. Dual-targeting of aberrant glucose metabolism in glioblastoma. J. Exp. Clin. Cancer Res. 2015, 34, 14. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer 2015, 14, 1794–1804. [Google Scholar] [CrossRef] [Green Version]
- McKelvey, K.J.; Wilson, E.B.; Short, S.; Melcher, A.A.; Biggs, M.; Diakos, C.I.; Howell, V.M. Glycolysis and Fatty Acid Oxidation Inhibition Improves Survival in Glioblastoma. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Abdelmalak, M.; Lew, A.; Ramezani, R.; Shroads, A.L.; Coats, B.S.; Langaee, T.; Shankar, M.N.; Neiberger, R.E.; Subramony, S.H.; Stacpoole, P.W. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol. Genet. Metab. 2013, 109, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudard, S.; Arvelo, F.; Miccoli, L.; Apiou, F.; Dutrillaux, A.M.; Poisson, M.; Dutrillaux, B.; Poupon, M.F. High glycolysis in gliomas despite low hexokinase transcription and activity correlated to chromosome 10 loss. Br. J. Cancer 1996, 74, 839–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13C] glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From Molecular Pathology to Targeted Treatment. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.-L.; Rajagopalan Kartik, N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of Tumor Metabolism Reveals Mitochondrial Glucose Oxidation in Genetically Diverse Human Glioblastomas in the Mouse Brain In Vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324. [Google Scholar] [CrossRef] [Green Version]
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic Targeting of Lactate Efflux by Malignant Glioma Inhibits Invasiveness and Induces Necrosis: An In Vivo Study. Neoplasia 2011, 13, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Crane, C.A.; Austgen, K.; Haberthur, K.; Hofmann, C.; Moyes, K.W.; Avanesyan, L.; Fong, L.; Campbell, M.J.; Cooper, S.; Oakes, S.A.; et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc. Natl. Acad. Sci. USA 2014, 111, 12823–12828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Griguer, C.E.; Oliva, C.R. Bioenergetics pathways and therapeutic resistance in gliomas: Emerging role of mitochondria. Curr. Pharm. Des. 2011, 17, 2421–2427. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.R.; Hudson, A.L.; Khong, P.; Parkinson, J.F.; Dwight, T.; Ikin, R.J.; Zhu, Y.; Cheng, Z.J.; Vafaee, F.; Chen, J.; et al. Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci. Rep. 2016, 6, 22477. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Hudson, A.L.; Prasanna Kumar, R.; Wilmott, J.S.; Attrill, G.H.; Long, G.V.; Scolyer, R.A.; Clarke, S.J.; Wheeler, H.R.; Diakos, C.I.; et al. Temporal and spatial modulation of the tumor and systemic immune response in the murine Gl261 glioma model. PLoS ONE 2020, 15, e0226444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Duraj, T.; García-Romero, N.; Carrión-Navarro, J.; Madurga, R.; Mendivil, A.O.; Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef]
- Prabhu, A.H.; Kant, S.; Kesarwani, P.; Ahmed, K.; Forsyth, P.; Nakano, I.; Chinnaiyan, P. Integrative cross-platform analyses identify enhanced heterotrophy as a metabolic hallmark in glioblastoma. Neuro-Oncology 2019, 21, 337–347. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Yu, M.; Tsoli, M.; Chang, C.; Joshi, S.; Liu, J.; Ryall, S.; Chornenkyy, Y.; Siddaway, R.; Hawkins, C.; et al. Targeting reduced mitochondrial DNA quantity as a therapeutic approach in pediatric high-grade gliomas. Neuro-Oncology 2020, 22, 139–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, A.; Schneider, F.; Vaupel, P.; Sommer, C.; Schmidberger, H. Differential expression of HIF-1 in glioblastoma multiforme and anaplastic astrocytoma. Int. J. Oncol. 2012, 41, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; Van Meir, E.G. Pseudopalisades in Glioblastoma Are Hypoxic, Express Extracellular Matrix Proteases, and Are Formed by an Actively Migrating Cell Population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeom, K.W.; Lober, R.M.; Nelson, M.D.; Panigrahy, A.; Blüml, S. Citrate concentrations increase with hypoperfusion in pediatric diffuse intrinsic pontine glioma. J. Neuro-Oncol. 2015, 122, 383–389. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-w.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigfield, S.M.; Winter, S.C.; Giatromanolaki, A.; Taylor, J.; Koukourakis, M.L.; Harris, A.L. PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br. J. Cancer 2008, 98, 1975–1984. [Google Scholar] [CrossRef]
- Kaynar, M.Y.; Sanus, G.Z.; Hnimoglu, H.; Kacira, T.; Kemerdere, R.; Atukeren, P.; Gumustas, K.; Canbaz, B.; Tanriverdi, T. Expression of hypoxia inducible factor-1α in tumors of patients with glioblastoma multiforme and transitional meningioma. J. Clin. Neurosci. 2008, 15, 1036–1042. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.L.; Zani, B.M.; Popov, V.M.; Fratticci, A.; Cerasani, M.; Di Genova, D.; Mancini, M.; Ciccarelli, C.; Ficorella, C.; et al. Hypoxia sustains glioblastoma radioresistance through ERKs/DNA-PKcs/HIF-1alpha functional interplay. Int. J. Oncol. 2014, 44, 2121–2131. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Squatrito, M.; Holland, E.C. Radiation resistance and stem-like cells in brain tumors. Cancer Cell 2006, 10, 454–456. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef] [Green Version]
- Schaue, D.; McBride, W.H. Counteracting tumor radioresistance by targeting DNA repair. Mol. Cancer 2005, 4, 1548–1550. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 2018, 37, 87. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Cook, K.; Gee, H.E.; Hau, E. Hypoxia, metabolism, and the circadian clock: New links to overcome radiation resistance in high-grade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 129. [Google Scholar] [CrossRef] [PubMed]
- Chédeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef] [PubMed]
- Pena-Rico, M.A.; Calvo-Vidal, M.N.; Villalonga-Planells, R.; Martinez-Soler, F.; Gimenez-Bonafe, P.; Navarro-Sabate, A.; Tortosa, A.; Bartrons, R.; Manzano, A. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother. Oncol. 2011, 101, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Werbrouck, C.; Evangelista, C.C.S.; Lobón-Iglesias, M.-J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, G.A.; Perkins, N.D. Transcriptional cross talk between NF-kappaB and p53. Mol. Cell Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.; Vuckovic, I.; Zhang, S.; Xiong, Y.; Carlson, B.L.; Jacobs, J.; Olson, I.; Petterson, X.-M.; Macura, S.I.; Sarkaria, J.; et al. Radiation Induced Metabolic Alterations Associate With Tumor Aggressiveness and Poor Outcome in Glioblastoma. Front. Oncol. 2020, 10, 535. [Google Scholar] [CrossRef]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. CMLS 2014, 71, 2577–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacpoole, P.W.; Felts, J.M. Diisopropylammonium dichloroacetate (DIPA) and sodium dichloracetate (DCA): Effect on glucose and fat metabolism in normal and diabetic tissue. Metab. Clin. Exp. 1970, 19, 71–78. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Nagaraja, N.V.; Hutson, A.D. Efficacy of dichloroacetate as a lactate-lowering drug. J. Clin. Pharm. 2003, 43, 683–691. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Jeoung, N.H.; Park, K.G.; Lee, I.K. Transcriptional regulation of pyruvate dehydrogenase kinase. Diabetes Metab. J. 2012, 36, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, M.O.; Jahn, S.C.; Zhong, G.; Smeltz, M.G.; Hu, Z.; Stacpoole, P.W. Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1. Pharm. Ther. 2017, 170, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.I.; Ganswindt, U. The Role of Cancer Stem Cells in Radiation Resistance. Front. Oncol. 2020, 10, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef]
- Wicks, R.T.; Azadi, J.; Mangraviti, A.; Zhang, I.; Hwang, L.; Joshi, A.; Bow, H.; Hutt-Cabezas, M.; Martin, K.L.; Rudek, M.A.; et al. Local delivery of cancer-cell glycolytic inhibitors in high-grade glioma. Neuro-Oncology 2015, 17, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Zwicker, F.; Kirsner, A.; Peschke, P.; Roeder, F.; Debus, J.; Huber, P.E.; Weber, K.J. Dichloroacetate induces tumor-specific radiosensitivity in vitro but attenuates radiation-induced tumor growth delay in vivo. Strahlenther. Onkol. 2013, 189, 684–692. [Google Scholar] [CrossRef]
- Yang, S.-B.; Gao, K.-D.; Jiang, T.; Cheng, S.-J.; Li, W.-B. Bevacizumab combined with chemotherapy for glioblastoma: A meta-analysis of randomized controlled trials. Oncotarget 2017, 8, 57337–57344. [Google Scholar] [CrossRef] [Green Version]
- Niyazi, M.; Harter, P.N.; Hattingen, E.; Rottler, M.; von Baumgarten, L.; Proescholdt, M.; Belka, C.; Lauber, K.; Mittelbronn, M. Bevacizumab and radiotherapy for the treatment of glioblastoma: Brothers in arms or unholy alliance? Oncotarget 2016, 7, 2313–2328. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, K.; Wigfield, S.; Gee, H.E.; Devlin, C.M.; Singleton, D.; Li, J.L.; Buffa, F.; Huffman, M.; Sinn, A.L.; Silver, J.; et al. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J. Mol. Med. 2013, 91, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albayrak, G.; Konac, E.; Akin Dere, U.; Emmez, H. Targeting Cancer Cell Metabolism with Metformin, Dichloroacetate and Memantine in Glioblastoma (GBM). Turk. Neurosurg. 2020. [Google Scholar] [CrossRef]
- Jiang, W.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, Z.; Mikkelsen, T.; Poisson, L.; Shackelford, D.B.; Brodie, C. Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget 2016, 7, 56456–56470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korsakova, L.; Krasko, J.A.; Stankevicius, E. Metabolic-targeted Combination Therapy with Dichloroacetate and Metformin Suppresses Glioblastoma Cell Line Growth In Vitro and In Vivo. In Vivo 2021, 35, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Dwarakanath, B.S. Modulation of Immuno-biome during Radio-sensitization of Tumors by Glycolytic Inhibitors. Curr. Med. Chem. 2020, 27, 4002–4015. [Google Scholar] [CrossRef]
- Fleming, A.B.; Saltzman, W.M. Pharmacokinetics of the carmustine implant. Clin. Pharm. 2002, 41, 403–419. [Google Scholar] [CrossRef]
- Cardoso, A.M.S.; Sousa, M.; Morais, C.M.; Oancea-Castillo, L.R.; Régnier-Vigouroux, A.; Rebelo, O.; Tão, H.; Barbosa, M.; Pedroso, M.C.L.; Jurado, A.S. MiR-144 overexpression as a promising therapeutic strategy to overcome glioblastoma cell invasiveness and resistance to chemotherapy. Hum. Mol. Genet. 2019, 28, 2738–2751. [Google Scholar] [CrossRef] [PubMed]
- Lhakhang, T.W.; Chaudhry, M.A. Interactome of Radiation-Induced microRNA-Predicted Target Genes. Comp. Funct. Genom. 2012, 2012, 569731. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garon, E.B.; Christofk, H.R.; Hosmer, W.; Britten, C.D.; Bahng, A.; Crabtree, M.J.; Hong, C.S.; Kamranpour, N.; Pitts, S.; Kabbinavar, F.; et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 443–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.D.; Bennett, S.K.; Coupland, L.A.; Forwood, K.; Lwin, Y.; Pooryousef, N.; Tea, I.; Truong, T.T.; Neeman, T.; Crispin, P.; et al. GSTZ1 genotypes correlate with dichloroacetate pharmacokinetics and chronic side effects in multiple myeloma patients in a pilot phase 2 clinical trial. Pharm. Res. Perspect. 2019, 7, e00526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Q.S.; Sangha, R.; Spratlin, J.; Vos, L.J.; Mackey, J.R.; McEwan, A.J.; Venner, P.; Michelakis, E.D. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 603–610. [Google Scholar] [CrossRef]
- Bowker-Kinley, M.M.; Davis, W.I.; Wu, P.; Harris, R.A.; Popov, K.M. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998, 329 Pt 1, 191–196. [Google Scholar] [CrossRef]
- Shroads, A.L.; Guo, X.; Dixit, V.; Liu, H.P.; James, M.O.; Stacpoole, P.W. Age-dependent kinetics and metabolism of dichloroacetate: Possible relevance to toxicity. J. Pharm. Exp. 2008, 324, 1163–1171. [Google Scholar] [CrossRef]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxid. Med. Cell. Longev. 2019, 2019, 8201079. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Trial | Description | Population Description | Main Conclusions | Ref. |
|---|---|---|---|---|
| Michelakis et al. | Small study of 49 freshly isolated glioblastoma samples and 5 patients with glioblastoma | 5 patients with glioblastoma | Indications of clinical efficacy were present at a dose that did not cause peripheral neuropathy and at serum concentrations of DCA sufficient to inhibit the target enzyme of DCA | [75] |
| Garon et al., NCT01029925 | Open label phase II trial | 6 patients with stage IIIB/IV non-small cell lung (NSCLC) and one patient with breast cancer | Firm conclusions regarding the association between these adverse events and DCA are unclear. Further development of DCA should be in patients with longer life expectancy, in whom sustained therapeutic levels can be achieved, and potentially in combination with cisplatin. | [85] |
| Tian et al. | Open label non randomized phase II trial | 7 myeloma patients | Promoter GSTZ1 polymorphisms may be important determinants of DCA concentrations and neuropathy during chronic treatment. Novel dosing regimens may be necessary to achieve effective DCA concentrations in cancer patients while avoiding neuropathy. | [86] |
| Dunbar et al., NCT01111097 | Open-label single-arm phase 1 study | 15 adults with recurrent WHO grade III–IV gliomas or brain metastases from a primary cancer outside the central nervous system | Chronic, oral DCA is feasible and well-tolerated in patients with recurrent malignant gliomas and other tumors metastatic to the brain. Genetic-based dosing is confirmed and should be incorporated into future trials of chronic DCA administration. | [84] |
| Chu et al. | Open-label phase 1 study | 24 patients with advanced solid malignancies | Progressive increase in DCA trough levels and a trend towards decreased (18) F-FDG uptake with length of DCA therapy was observed. The recommended phase II dose of DCA is 6.25 mg/kg BID. | [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cook, K.M.; Shen, H.; McKelvey, K.J.; Gee, H.E.; Hau, E. Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas. Int. J. Mol. Sci. 2021, 22, 7265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147265
Cook KM, Shen H, McKelvey KJ, Gee HE, Hau E. Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas. International Journal of Molecular Sciences. 2021; 22(14):7265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147265
Chicago/Turabian StyleCook, Kristina M., Han Shen, Kelly J. McKelvey, Harriet E. Gee, and Eric Hau. 2021. "Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas" International Journal of Molecular Sciences 22, no. 14: 7265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147265