
Calcium and Heart Failure: How Did We Get Here and Where Are We Going?

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Calcium Cycling in Pressure Overload Hypertrophy, Dysthyroidism, and Human Heart Failure
Inotropes vs. Beta Blockers in the Treatment of Heart Failure: Paradigm Shift
Myofilaments and Ca2+ Responsiveness
Antioxidants and CaMKII Inhibitors in the Treatment of Heart Disease
Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fabiato, A. Myoplasmic free calcium concentration reached during the twitch of an intact isolated cardiac cell and during calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned cardiac cell from the adult rat or rabbit ventricle. J. Gen. Physiol. 1981, 78, 457–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef]
- Fabiato, A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 1985, 85, 247–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabiato, A.; Fabiato, F. Calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cells from adult human, dog, cat, rabbit, rat, and frog hearts and from fetal and new-born rat ventricles. Ann. N. Y. Acad. Sci. 1978, 307, 491–522. [Google Scholar] [CrossRef]
- Fabiato, A.; Fabiato, F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J. Physiol. 1975, 249, 469–495. [Google Scholar] [CrossRef] [Green Version]
- Fabiato, A.; Fabiato, F. Effects of magnesium on contractile activation of skinned cardiac cells. J. Physiol. 1975, 249, 497–517. [Google Scholar] [CrossRef] [Green Version]
- Fabiato, A.; Fabiato, F. Dependence of calcium release, tension generation and restoring forces on sarcomere length in skinned cardiac cells. Eur. J. Cardiol. 1976, 4, 13–27. [Google Scholar]
- Fabiato, A.; Fabiato, F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiace and skeletal muscles. J. Physiol. 1978, 276, 233–255. [Google Scholar] [CrossRef]
- Fabiato, A.; Fabiato, F. Use of chlorotetracycline fluorescence to demonstrate Ca2+-induced release of Ca2+ from the sarcoplasmic reticulum of skinned cardiac cells. Nature 1979, 281, 146–148. [Google Scholar] [CrossRef] [PubMed]
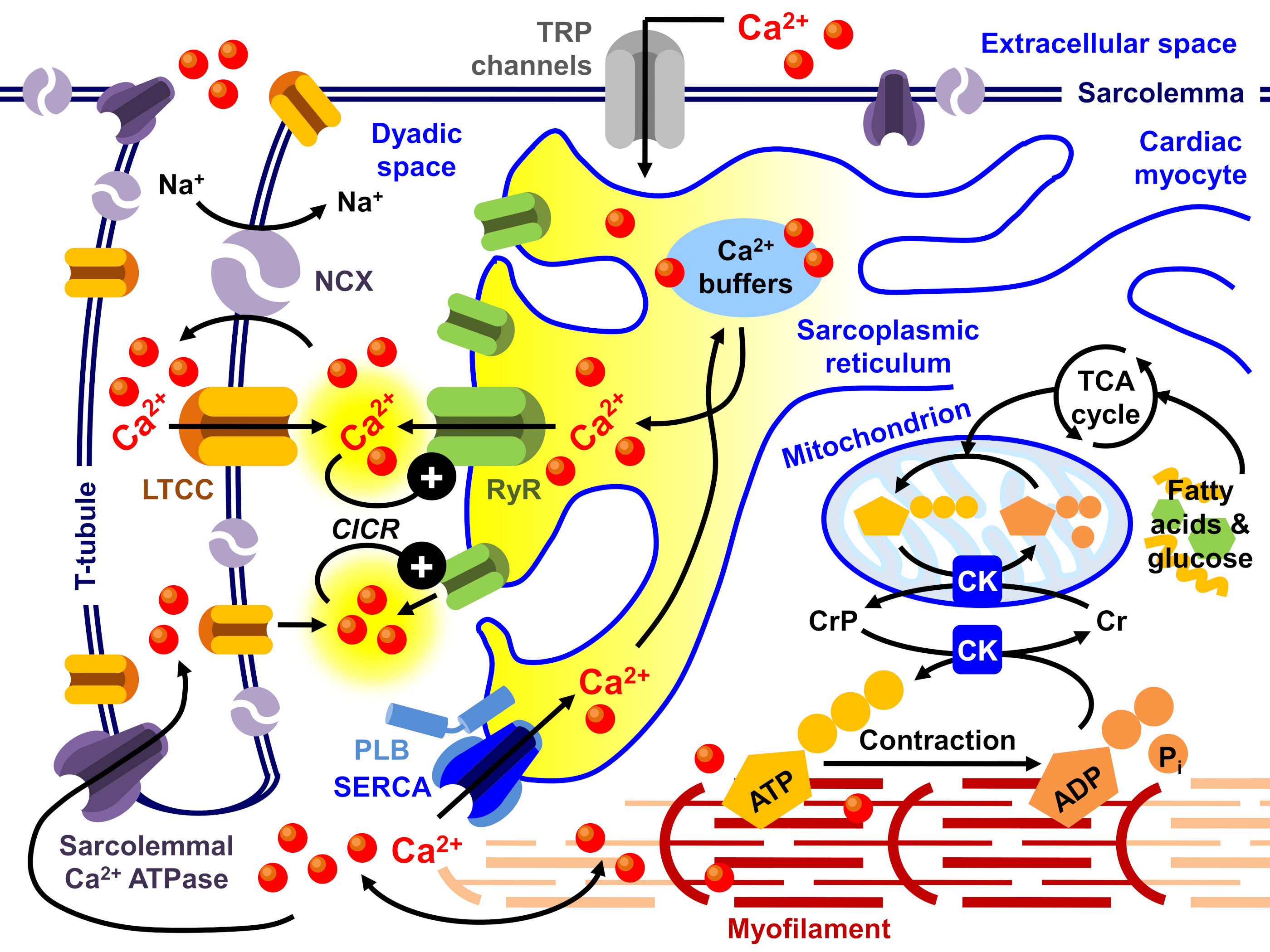
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M.; Guo, T. Calcium signaling in cardiac ventricular myocytes. Ann. N. Y. Acad. Sci. 2005, 1047, 86–98. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Eisner, D.A. Calcium Buffering in the Heart in Health and Disease. Circulation 2019, 139, 2358–2371. [Google Scholar] [CrossRef]
- Falsetti, H.L.; Mates, R.E.; Greene, D.G.; Bunnell, I.L. Vmax as an index of contractile state in man. Circulation 1971, 43, 467–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, M.J.; Levinson, G.E. An index of the contractile state of the myocardium in man. J. Clin. Investig. 1968, 47, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Pollack, G.H. Maximum velocity as an index of contractility in cardiac muscle. A critical evaluation. Circ. Res. 1970, 26, 111–127. [Google Scholar] [CrossRef] [Green Version]
- Pollack, G.H. Is Vmax a valid contractile index? Am. Heart J. 1971, 81, 572–573. [Google Scholar] [CrossRef]
- Siegel, J.H.; Sonnenblick, E.H. Isometric time-tension relationships as an index of myocardial contractility. Circ. Res. 1963, 12, 597–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penefsky, Z.J. Effects of hypothermia and stretch on contraction and relaxation of cardiac muscle. Am. J. Physiol. 1968, 214, 730–736. [Google Scholar] [CrossRef]
- Penefsky, Z.J. Ultrastructural studies of the site of action of ryanodine on heart muscle. Pflugers Arch. 1974, 347, 185–198. [Google Scholar] [CrossRef]
- Penefsky, Z.J. The determinants of contractility in the heart. Comp. Biochem. Physiol. Physiol. 1994, 109, 1–22. [Google Scholar] [CrossRef]
- Penefsky, Z.J.; Kahn, M. Mechanical and electrical effects of ryanodine on mammalian heart muscle. Am. J. Physiol. 1970, 218, 1682–1686. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Briggs, G.M.; Allen, P.D. Heart Failure: Basic Research and Clinical Aspects; Marcel Dekker: New York, NY, USA, 1993. [Google Scholar]
- Gwathmey, J.K.; Ingwall, J.S. Basic Pathophysiology of Congestive Heart Failure. Cardiol. Rev. 1995, 3, 282–291. [Google Scholar] [CrossRef]
- Ingwall, J.S.; Nascimben, L.; Gwathmey, J.K. Heart failure: Is the pathology due to calcium overload or to mismatch in energy supply and demand? In Heart Failure: Basic Science and Clinical Aspects; Gwathmey, J.K., Briggs, G.M., Allen, P.D., Eds.; Marcel Dekker: New York, NY, USA, 1993; pp. 667–700. [Google Scholar]
- Bakayan, A.; Domingo, B.; Vaquero, C.F.; Peyriéras, N.; Llopis, J. Fluorescent Protein-photoprotein Fusions and Their Applications in Calcium Imaging. Photochem. Photobiol. 2017, 93, 448–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blinks, J.R. Use of photoproteins as intracellular calcium indicators. Environ. Health Perspect. 1990, 84, 75–81. [Google Scholar] [CrossRef]
- Blinks, J.R.; Moore, E.D. Practical aspects of the use of photoproteins as biological calcium indicators. Soc. Gen. Physiol. Ser. 1986, 40, 229–238. [Google Scholar] [PubMed]
- Miller, A.L.; Karplus, E.; Jaffe, L.F. Imaging [Ca2+]i with aequorin using a photon imaging detector. Methods Cell Biol. 1994, 40, 305–338. [Google Scholar] [CrossRef] [PubMed]
- Sharifian, S.; Homaei, A.; Hemmati, R.; Luwor, R.B.; Khajeh, K. The emerging use of bioluminescence in medical research. Biomed. Pharmacother. 2018, 101, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, O. Bioluminescence in the sea: Photoprotein systems. Symp. Soc. Exp. Biol. 1985, 39, 351–372. [Google Scholar]
- Shimomura, O. Preparation and handling of aequorin solutions for the measurement of cellular Ca2+. Cell Calcium 1991, 12, 635–643. [Google Scholar] [CrossRef]
- Yoshimoto, Y.; Hiramoto, Y. Observation of intracellular Ca2+ with aequorin luminescence. Int. Rev. Cytol. 1991, 129, 45–73. [Google Scholar] [CrossRef]
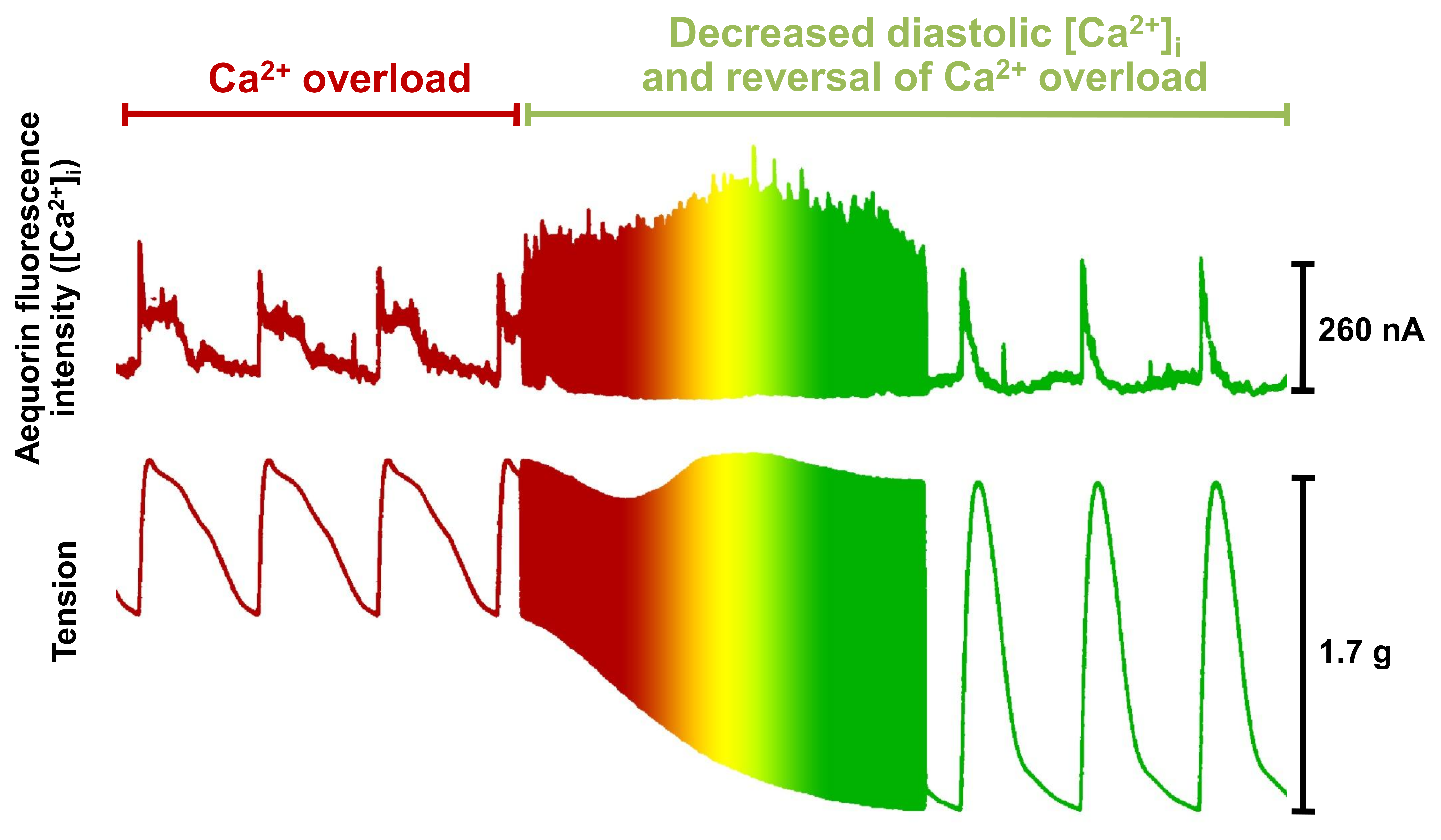
- Allen, D.G.; Blinks, J.R. Calcium transients in aequorin-injected frog cardiac muscle. Nature 1978, 273, 509–513. [Google Scholar] [CrossRef]
- Blinks, J.R. On the suitability of aequorin as an intracellular calcium detector. Nihon Seirigaku Zasshi 1972, 34, 95–96. [Google Scholar]
- Taylor, S.R.; Rüdel, R.; Blinks, J.R. Calcium transients in amphibian muscle. Fed. Proc. 1975, 34, 1379–1381. [Google Scholar]
- Brozovich, F.V.; Walsh, M.P.; Morgan, K.G. Regulation of force in skinned, single cells of ferret aortic smooth muscle. Pflugers Arch. 1990, 416, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.J.; Morgan, K.G. Agonist-specific myosin phosphorylation and intracellular calcium during isometric contractions of arterial smooth muscle. Pflugers Arch. 1989, 413, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, J.P. The effects of digitalis on intracellular calcium transients in mammalian working myocardium as detected with aequorin. J. Mol. Cell. Cardiol. 1985, 17, 1065–1075. [Google Scholar] [CrossRef]
- Roura, S.; Gálvez-Montón, C.; Bayes-Genis, A. Bioluminescence imaging: A shining future for cardiac regeneration. J. Cell. Mol. Med. 2013, 17, 693–703. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Morgan, J.P. The effects of milrinone and piroximone on intracellular calcium handling in working myocardium from the ferret. Br. J. Pharmacol. 1985, 85, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.P.; Gwathmey, J.K.; DeFeo, T.T.; Morgan, K.G. The effects of amrinone and related drugs on intracellular calcium in isolated mammalian cardiac and vascular smooth muscle. Circulation 1986, 73, iii65–iii77. [Google Scholar]
- Gwathmey, J.K.; Liao, R.; Ingwall, J.S. Comparison of twitch force and calcium handling in papillary muscles from right ventricular pressure overload hypertrophy in weanling and juvenile ferrets. Cardiovasc. Res. 1995, 29, 475–481. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Morgan, J.P. Altered calcium handling in experimental pressure-overload hypertrophy in the ferret. Circ. Res. 1985, 57, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, J.P.; MacKinnon, R.; Briggs, G.M.; Gwathmey, J.K. Calcium and cardiac relaxation. In The Physiology of Diastole in Health and Disease; Grossman, W., Lorell, B., Eds.; Martinus Nijhoff: Boston, MA, USA, 1987; pp. 17–26. [Google Scholar]
- Morgan, J.P.; MacKinnon, R.; Feldman, M.D.; Grossman, W.; Gwathmey, J.K. The effects of cardiac hypertrophy on intracellular Ca2+ handling. In The Physiology of Diastole in Health and D; Grossman, W., Lorell, B., Eds.; Martinus Nijhoff: Boston, MA, USA, 1987; pp. 97–107. [Google Scholar]
- Morgan, J.P.; Bentivegna, L.A.; Perreault, C.L.; Meuse, A.J.; Allen, P.D.; Ransil, B.J.; Grossman, W.; Gwathmey, J.K. Abnormal intracellular calcium handling in hypertrophy and failure of human working myocardium. In Molecular Biology of the Cardiovascular System; Grossman, W., Lorell, B., Eds.; Alan R. Liss, Inc.: London, UK, 1990; pp. 241–248. [Google Scholar]
- MacKinnon, R.; Gwathmey, J.K.; Allen, P.D.; Briggs, G.M.; Morgan, J.P. Modulation by the thyroid state of intracellular calcium and contractility in ferret ventricular muscle. Circ. Res. 1988, 63, 1080–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwathmey, J.K.; Copelas, L.; MacKinnon, R.; Schoen, F.J.; Feldman, M.D.; Grossman, W.; Morgan, J.P. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. 1987, 61, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, R.; Kistamás, K.; Greensmith, D.J.; Venetucci, L.A.; Eisner, D.A. Systolic [Ca(2+) ](i) regulates diastolic levels in rat ventricular myocytes. J. Physiol. 2017, 595, 5545–5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okafor, C.; Liao, R.; Perreault-Micale, C.; Li, X.; Ito, T.; Stepanek, A.; Doye, A.; de Tombe, P.; Gwathmey, J.K. Mg-ATPase and Ca+ activated myosin ATPase activity in ventricular myofibrils from non-failing and diseased human hearts--effects of calcium sensitizing agents MCI-154, DPI 201-106, and caffeine. Mol. Cell. Biochem. 2003, 245, 77–89. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Hajjar, R.J. Abnormal calcium metabolism in heart muscle dysfunction. In Idiopathic Dilated Cardiomyopathy: Cellular and Molecular Mechanisms, Clinical Consequences; Figulla, H.R., Kandolf, R., McManus, B., Eds.; Springer-Verlag: New York, NY, USA, 1993; pp. 132–144. [Google Scholar]
- Gwathmey, J.K.; Hajjar, R.J. The complexity of simplicity revisited. Pathophysiology of heart failure. Resid. Staff Physician 1993, 39, 45–59. [Google Scholar]
- Feldman, M.D.; Gwathmey, J.K.; Philips, P.; Schoen, F.; Morgan, J.P. Reversal of the force-frequency relationship in working myocardium from patients with end-stage heart failure. J. Appl. Cardiol. 1988, 3, 273–283. [Google Scholar]
- Gwathmey, J.K.; Hajjar, R.J. Relation between steady-state force and intracellular [Ca2+] in intact human myocardium. Index of myofibrillar responsiveness to Ca2+. Circulation 1990, 82, 1266–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwathmey, J.K.; Slawsky, M.T.; Hajjar, R.J.; Briggs, G.M.; Morgan, J.P. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J. Clin. Invest. 1990, 85, 1599–1613. [Google Scholar] [CrossRef] [Green Version]
- Hajjar, R.J.; DiSalvo, T.G.; Schmidt, U.; Thaiyananthan, G.; Semigran, M.J.; Dec, G.W.; Gwathmey, J.K. Clinical correlates of the myocardial force-frequency relationship in patients with end-stage heart failure. J. Heart Lung Transplant. 1997, 16, 1157–1167. [Google Scholar]
- Pieske, B.; Kretschmann, B.; Meyer, M.; Holubarsch, C.; Weirich, J.; Posival, H.; Minami, K.; Just, H.; Hasenfuss, G. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation 1995, 92, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, J.K.; Hajjar, R.J. Intracellular calcium related to force development in twitch contraction of mammalian myocardium. Cell Calcium 1990, 11, 531–538. [Google Scholar] [CrossRef]
- Phillips, P.J.; Gwathmey, J.K.; Feldman, M.D.; Schoen, F.J.; Grossman, W.; Morgan, J.P. Post-extrasystolic potentiation and the force-frequency relationship: Differential augmentation of myocardial contractility in working myocardium from patients with end-stage heart failure. J. Mol. Cell. Cardiol. 1990, 22, 99–110. [Google Scholar] [CrossRef]
- Schmidt, U.; Hajjar, R.J.; Gwathmey, J.K. The force-interval relationship in human myocardium. J. Card. Fail. 1995, 1, 311–321. [Google Scholar] [CrossRef]
- Xie, L.H.; Sato, D.; Garfinkel, A.; Qu, Z.; Weiss, J.N. Intracellular Ca alternans: Coordinated regulation by sarcoplasmic reticulum release, uptake, and leak. Biophys. J. 2008, 95, 3100–3110. [Google Scholar] [CrossRef] [Green Version]
- Falcón, D.; Galeano-Otero, I.; Calderón-Sánchez, E.; Del Toro, R.; Martín-Bórnez, M.; Rosado, J.A.; Hmadcha, A.; Smani, T. TRP Channels: Current Perspectives in the Adverse Cardiac Remodeling. Front. Physiol. 2019, 10, 159. [Google Scholar] [CrossRef]
- Falcón, D.; Galeano-Otero, I.; Martín-Bórnez, M.; Fernández-Velasco, M.; Gallardo-Castillo, I.; Rosado, J.A.; Ordóñez, A.; Smani, T. TRPC Channels: Dysregulation and Ca(2+) Mishandling in Ischemic Heart Disease. Cells 2020, 9, 173. [Google Scholar] [CrossRef] [Green Version]
- Hof, T.; Chaigne, S.; Récalde, A.; Sallé, L.; Brette, F.; Guinamard, R. Transient receptor potential channels in cardiac health and disease. Nat. Rev. Cardiol. 2019, 16, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gwathmey, J.K.; Xie, L.H. Role of Transient Receptor Potential Canonical Channels in Heart Physiology and Pathophysiology. Front. Cardiovasc. Med. 2020, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Xie, J.; Yu, A.S.; Stock, J.; Du, J.; Yue, L. Role of TRP channels in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H157–H182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Ahmad, A.A.; Seidel, T.; Hunter, C.; Streiff, M.; Nikolova, L.; Spitzer, K.W.; Sachse, F.B. Location and function of transient receptor potential canonical channel 1 in ventricular myocytes. J. Mol. Cell. Cardiol. 2020, 139, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.A.; Streiff, M.; Hunter, C.; Hu, Q.; Sachse, F.B. Physiological and pathophysiological role of transient receptor potential canonical channels in cardiac myocytes. Prog. Biophys. Mol. Biol. 2017, 130, 254–263. [Google Scholar] [CrossRef]
- Wen, H.; Zhao, Z.; Fefelova, N.; Xie, L.H. Potential Arrhythmogenic Role of TRPC Channels and Store-Operated Calcium Entry Mechanism in Mouse Ventricular Myocytes. Front. Physiol. 2018, 9, 1785. [Google Scholar] [CrossRef] [PubMed]
- Nikolova-Krstevski, V.; Wagner, S.; Yu, Z.Y.; Cox, C.D.; Cvetkovska, J.; Hill, A.P.; Huttner, I.G.; Benson, V.; Werdich, A.A.; MacRae, C.; et al. Endocardial TRPC-6 Channels Act as Atrial Mechanosensors and Load-Dependent Modulators of Endocardial/Myocardial Cross-Talk. JACC Basic Transl. Sci. 2017, 2, 575–590. [Google Scholar] [CrossRef]
- Eder, P.; Molkentin, J.D. TRPC channels as effectors of cardiac hypertrophy. Circ. Res. 2011, 108, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Invest. 2006, 116, 3114–3126. [Google Scholar] [CrossRef]
- Sabourin, J.; Robin, E.; Raddatz, E. A key role of TRPC channels in the regulation of electromechanical activity of the developing heart. Cardiovasc. Res. 2011, 92, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Doleschal, B.; Primessnig, U.; Wölkart, G.; Wolf, S.; Schernthaner, M.; Lichtenegger, M.; Glasnov, T.N.; Kappe, C.O.; Mayer, B.; Antoons, G.; et al. TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1. Cardiovasc. Res. 2015, 106, 163–173. [Google Scholar] [CrossRef]
- Camacho Londoño, J.E.; Tian, Q.; Hammer, K.; Schröder, L.; Camacho Londoño, J.; Reil, J.C.; He, T.; Oberhofer, M.; Mannebach, S.; Mathar, I.; et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur. Heart J. 2015, 36, 2257–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, M.; Zhang, Z.S.; Mao, L.; Graham, V.; Burch, J.; Stiber, J.; Tsiokas, L.; Winn, M.; Abramowitz, J.; Rockman, H.A.; et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res. 2009, 105, 1023–1030. [Google Scholar] [CrossRef]
- Makarewich, C.A.; Zhang, H.; Davis, J.; Correll, R.N.; Trappanese, D.M.; Hoffman, N.E.; Troupes, C.D.; Berretta, R.M.; Kubo, H.; Madesh, M.; et al. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ. Res. 2014, 115, 567–580. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Li, S.; Liu, B.; Susperreguy, S.; Formoso, K.; Yao, J.; Kang, J.; Shi, A.; Birnbaumer, L.; Liao, Y. Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc. Natl. Acad. Sci. USA 2017, 114, E4582–E4591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Hang, P.; Zhu, W.; Su, Z.; Liang, H.; Du, Z. Whole genome network analysis of ion channels and connexins in myocardial infarction. Cell. Physiol. Biochem. 2011, 27, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Gené, G.G.; Tomás, M.; Plata, C.; Selent, J.; Pastor, M.; Fandos, C.; Senti, M.; Lucas, G.; Elosua, R.; et al. A gain-of-function SNP in TRPC4 cation channel protects against myocardial infarction. Cardiovasc. Res. 2011, 91, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Shan, D.; Marchase, R.B.; Chatham, J.C. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia-reperfusion in adult mouse cardiomyocytes. Am. J. Physiol. Cell Physiol. 2008, 294, C833–C841. [Google Scholar] [CrossRef] [Green Version]
- Kojima, A.; Fukushima, Y.; Ito, Y.; Ding, W.G.; Kitagawa, H.; Matsuura, H. Transient Receptor Potential Canonical Channel Blockers Improve Ventricular Contractile Functions After Ischemia/Reperfusion in a Langendorff-perfused Mouse Heart Model. J. Cardiovasc. Pharmacol. 2018, 71, 248–255. [Google Scholar] [CrossRef]
- Zhu, Y.; Gao, M.; Zhou, T.; Xie, M.; Mao, A.; Feng, L.; Yao, X.; Wong, W.T.; Ma, X. The TRPC5 channel regulates angiogenesis and promotes recovery from ischemic injury in mice. J. Biol. Chem. 2019, 294, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Li, J. Canonical transient receptor potential 3 channels in atrial fibrillation. Eur. J. Pharmacol. 2018, 837, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wu, W.Y.; Li, G.; Zhang, Y.H.; Sun, Y.; Qiu, F.; Yang, Q.; Xiao, G.S.; Li, G.R.; Wang, Y. Regulation of the TRPC1 channel by endothelin-1 in human atrial myocytes. Heart Rhythm 2019, 16, 1575–1583. [Google Scholar] [CrossRef]
- Harada, M.; Luo, X.; Qi, X.Y.; Tadevosyan, A.; Maguy, A.; Ordog, B.; Ledoux, J.; Kato, T.; Naud, P.; Voigt, N.; et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 2012, 126, 2051–2064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Wu, H.J.; Che, H.; Sun, H.Y.; Cheng, L.C.; Li, X.; Au, W.K.; Tse, H.F.; Li, G.R. Functional transient receptor potential canonical type 1 channels in human atrial myocytes. Pflugers Arch. 2013, 465, 1439–1449. [Google Scholar] [CrossRef]
- Siri-Angkul, N.; Song, Z.; Fefelova, N.; Gwathmey, J.K.; Chattipakorn, S.C.; Qu, Z.; Chattipakorn, N.; Xie, L.H. Activation of TRPC (Transient Receptor Potential Canonical) Channel Currents in Iron Overloaded Cardiac Myocytes. Circ. Arrhythm. Electrophysiol. 2021, 14, e009291. [Google Scholar] [CrossRef]
- Montell, C. The TRP superfamily of cation channels. Sci. STKE 2005, 2005, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Voets, T.; Peters, J. TRP channels in disease. Sci. STKE 2005, 2005, re8. [Google Scholar] [CrossRef]
- Rohacs, T. Teaching resources. TRP channels. Sci. STKE 2005, 2005, tr14. [Google Scholar] [CrossRef]
- Almanaitytė, M.; Jurevičius, J.; Mačianskienė, R. Effect of Carvacrol, TRP Channels Modulator, on Cardiac Electrical Activity. BioMed Res. Int. 2020, 2020, 6456805. [Google Scholar] [CrossRef]
- Feng, J.; Zong, P.; Yan, J.; Yue, Z.; Li, X.; Smith, C.; Ai, X.; Yue, L. Upregulation of transient receptor potential melastatin 4 (TRPM4) in ventricular fibroblasts from heart failure patients. Pflugers Arch. 2021, 473, 521–531. [Google Scholar] [CrossRef]
- Frede, W.; Medert, R.; Poth, T.; Gorenflo, M.; Vennekens, R.; Freichel, M.; Uhl, S. TRPM4 Modulates Right Ventricular Remodeling Under Pressure Load Accompanied With Decreased Expression Level. J. Card. Fail. 2020, 26, 599–609. [Google Scholar] [CrossRef]
- Hedon, C.; Lambert, K.; Chakouri, N.; Thireau, J.; Aimond, F.; Cassan, C.; Bideaux, P.; Richard, S.; Faucherre, A.; Le Guennec, J.Y.; et al. New role of TRPM4 channel in the cardiac excitation-contraction coupling in response to physiological and pathological hypertrophy in mouse. Prog. Biophys. Mol. Biol. 2021, 159, 105–117. [Google Scholar] [CrossRef]
- Amarouch, M.Y.; El Hilaly, J. Inherited Cardiac Arrhythmia Syndromes: Focus on Molecular Mechanisms Underlying TRPM4 Channelopathies. Cardiovasc. Ther. 2020, 2020, 6615038. [Google Scholar] [CrossRef]
- Guinamard, R.; Bouvagnet, P.; Hof, T.; Liu, H.; Simard, C.; Sallé, L. TRPM4 in cardiac electrical activity. Cardiovasc. Res. 2015, 108, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, N.E.; Miller, B.A.; Wang, J.; Elrod, J.W.; Rajan, S.; Gao, E.; Song, J.; Zhang, X.Q.; Hirschler-Laszkiewicz, I.; Shanmughapriya, S.; et al. Ca2+ entry via Trpm2 is essential for cardiac myocyte bioenergetics maintenance. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H637–H650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Guo, W.; Hao, B.; Shi, X.; Lu, Y.; Wong, C.W.; Ma, V.W.; Yip, T.T.; Au, J.S.; Hao, Q.; et al. Mechanistic study of TRPM2-Ca(2+)-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy 2016, 12, 1340–1354. [Google Scholar] [CrossRef] [Green Version]
- Xian, W.; Wang, H.; Moretti, A.; Laugwitz, K.L.; Flockerzi, V.; Lipp, P. Domain zipping and unzipping modulates TRPM4’s properties in human cardiac conduction disease. FASEB J. 2020, 34, 12114–12126. [Google Scholar] [CrossRef]
- Chaigne, S.; Cardouat, G.; Louradour, J.; Vaillant, F.; Charron, S.; Sacher, F.; Ducret, T.; Guinamard, R.; Vigmond, E.; Hof, T. Transient receptor potential vanilloid 4 channel participates in mouse ventricular electrical activity. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1156–H1169. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Lisco, A.M.; Rudebush, T.L.; Yu, L.; Gao, L.; Kitzerow, O.; Zucker, I.H.; Wang, H.J. Identification of Cardiac Expression Pattern of Transient Receptor Potential Vanilloid Type 1 (TRPV1) Receptor using a Transgenic Reporter Mouse Model. Neurosci. Lett. 2020, 737, 135320. [Google Scholar] [CrossRef] [PubMed]
- Peana, D.; Polo-Parada, L.; Domeier, T.L. Arrhythmogenesis in the aged heart following ischaemia-reperfusion: Role of Transient Receptor Potential Vanilloid 4. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Entin-Meer, M.; Keren, G. Potential roles in cardiac physiology and pathology of the cation channel TRPV2 expressed in cardiac cells and cardiac macrophages: A mini-review. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H181–H188. [Google Scholar] [CrossRef]
- Iwata, Y.; Katayama, Y.; Okuno, Y.; Wakabayashi, S. Novel inhibitor candidates of TRPV2 prevent damage of dystrophic myocytes and ameliorate against dilated cardiomyopathy in a hamster model. Oncotarget 2018, 9, 14042–14057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, Y.; Ohtake, H.; Suzuki, O.; Matsuda, J.; Komamura, K.; Wakabayashi, S. Blockade of sarcolemmal TRPV2 accumulation inhibits progression of dilated cardiomyopathy. Cardiovasc. Res. 2013, 99, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Yu, T.; Xiao, C.; Sheng, D.; Yang, M.; Cheng, Q.; Wu, J.; Lian, T.; Zhao, Y.; Zhang, S. Expression of transient receptor potential vanilloid genes and proteins in diabetic rat heart. Mol. Biol. Rep. 2021, 48, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.E.; Mann, A.; Jones, S.; Robbins, N.; Alkhattabi, A.; Worley, M.C.; Gao, X.; Lasko-Roiniotis, V.M.; Karani, R.; Fulford, L.; et al. Transient receptor potential vanilloid 2 function regulates cardiac hypertrophy via stretch-induced activation. J. Hypertens. 2017, 35, 602–611. [Google Scholar] [CrossRef]
- Liao, J.; Wu, Q.; Qian, C.; Zhao, N.; Zhao, Z.; Lu, K.; Zhang, S.; Dong, Q.; Chen, L.; Li, Q.; et al. TRPV4 blockade suppresses atrial fibrillation in sterile pericarditis rats. JCI insight 2020, 5, e137528. [Google Scholar] [CrossRef]
- Matsumura, T.; Matsui, M.; Iwata, Y.; Asakura, M.; Saito, T.; Fujimura, H.; Sakoda, S. A Pilot Study of Tranilast for Cardiomyopathy of Muscular Dystrophy. Intern. Med. 2018, 57, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Obata, K.; Morita, H.; Takaki, M. Mechanism underlying the negative inotropic effect in rat left ventricle in hyperthermia: The role of TRPV1. J. Physiol. Sci. 2020, 70, 4. [Google Scholar] [CrossRef]
- O’Connor, B.; Robbins, N.; Koch, S.E.; Rubinstein, J. TRPV2 channel-based therapies in the cardiovascular field. Molecular underpinnings of clinically relevant therapies. Prog. Biophys. Mol. Biol. 2021, 159, 118–125. [Google Scholar] [CrossRef]
- Schmidt, U.; Hajjar, R.J.; Helm, P.A.; Kim, C.S.; Doye, A.A.; Gwathmey, J.K. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J. Mol. Cell. Cardiol. 1998, 30, 1929–1937. [Google Scholar] [CrossRef]
- Schmidt, U.; Hajjar, R.J.; Kim, C.S.; Lebeche, D.; Doye, A.A.; Gwathmey, J.K. Human heart failure: cAMP stimulation of SR Ca(2+)-ATPase activity and phosphorylation level of phospholamban. Am. J. Physiol. 1999, 277, H474–H480. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Gwathmey, J.K. Cross-bridge dynamics in human ventricular myocardium. Regulation of contractility in the failing heart. Circulation 1992, 86, 1819–1826. [Google Scholar] [CrossRef] [Green Version]
- Gwathmey, J.K.; Liao, R.; Helm, P.A.; Thaiyananthan, G.; Hajjar, R.J. Is contractility depressed in the failing human heart? Cardiovasc. Drugs Ther. 1995, 9, 581–587. [Google Scholar] [CrossRef]
- Liao, R.; Helm, P.A.; Hajjar, R.J.; Saha, C.; Gwathmey, J.K. [Ca2+]i in human heart failure: A review and discussion of current areas of controversy. Yale J. Biol. Med. 1994, 67, 247–264. [Google Scholar] [PubMed]
- Feldman, M.D.; Copelas, L.; Gwathmey, J.K.; Phillips, P.; Warren, S.E.; Schoen, F.J.; Grossman, W.; Morgan, J.P. Deficient production of cyclic AMP: Pharmacologic evidence of an important cause of contractile dysfunction in patients with end-stage heart failure. Circulation 1987, 75, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Carver, J.R.; Rodeheffer, R.J.; Ivanhoe, R.J.; DiBianco, R.; Zeldis, S.M.; Hendrix, G.H.; Bommer, W.J.; Elkayam, U.; Kukin, M.L.; et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Effect of phosphodiesterase inhibitors on survival of patients with chronic congestive heart failure. Am. J. Cardiol. 1989, 63, 41a–45a. [Google Scholar] [CrossRef]
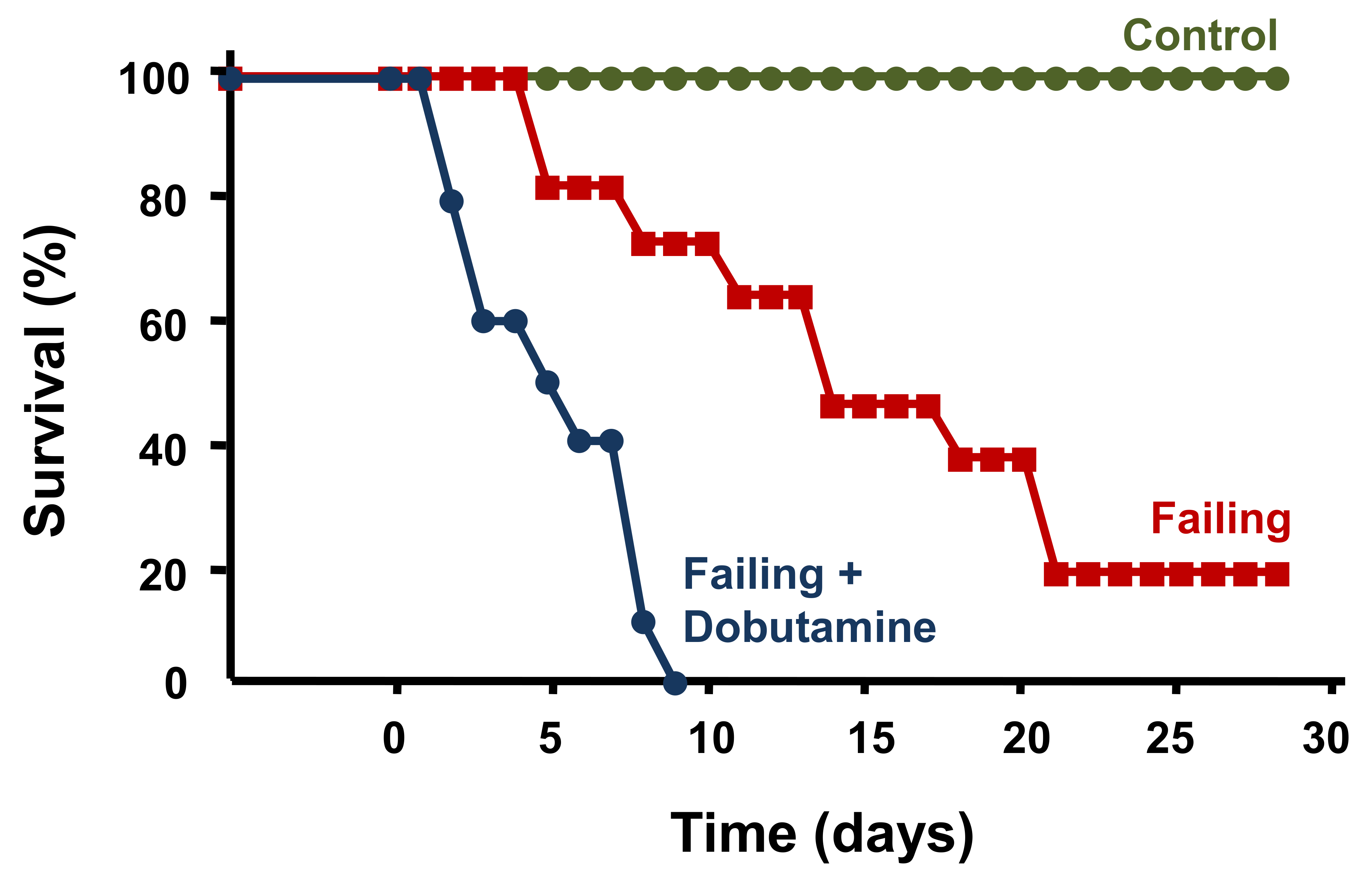
- O’Connor, C.M.; Gattis, W.A.; Uretsky, B.F.; Adams, K.F., Jr.; McNulty, S.E.; Grossman, S.H.; McKenna, W.J.; Zannad, F.; Swedberg, K.; Gheorghiade, M.; et al. Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: Insights from the Flolan International Randomized Survival Trial (FIRST). Am. Heart J. 1999, 138, 78–86. [Google Scholar] [CrossRef]
- Stevenson, L.W. Clinical use of inotropic therapy for heart failure: Looking backward or forward? Part I: Inotropic infusions during hospitalization. Circulation 2003, 108, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Gwathmey, J.K.; Slawsky, M.T.; Briggs, G.M.; Morgan, J.P. Role of intracellular sodium in the regulation of intracellular calcium and contractility. Effects of DPI 201-106 on excitation-contraction coupling in human ventricular myocardium. J. Clin. Invest. 1988, 82, 1592–1605. [Google Scholar] [CrossRef] [Green Version]
- Hajjar, R.J.; Gwathmey, J.K. Modulation of calcium-activation in control and pressure--Overload hypertrophied ferret hearts: Effect of DPI 201-106 on myofilament calcium responsiveness. J. Mol. Cell. Cardiol. 1991, 23, 65–75. [Google Scholar] [CrossRef]
- Kihara, Y.; Gwathmey, J.K.; Grossman, W.; Morgan, J.P. Mechanisms of positive inotropic effects and delayed relaxation produced by DPI 201-106 in mammalian working myocardium: Effects on intracellular calcium handling. Br. J. Pharmacol. 1989, 96, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, R.; Gwathmey, J.K. Effects of MCI-154 and caffeine on Ca(++)-regulated interactions between troponin subunits from bovine heart. J. Pharmacol. Exp. Ther. 1994, 270, 831–839. [Google Scholar] [PubMed]
- Warren, S.E.; Gwathmey, J.K.; Feldman, M.D.; Phillips, P.J.; Grossman, W.; Morgan, J.P. Inotropic and lusitropic effects of DPI 201-106 on human myocardium. J. Appl. Cardiol. 1989, 4, 177–188. [Google Scholar]
- Warren, S.E.; Kihara, Y.; Pesaturo, J.; Gwathmey, J.K.; Phillips, P.; Morgan, J.P. Inotropic and lusitropic effects of MCI-154 (6-[4-(4- pyridyl)aminophenyl]-4,5-dihydro-3(2H)-pyridazinone) on human myocardium. J. Mol. Cell. Cardiol. 1989, 21, 1037–1045. [Google Scholar] [CrossRef]
- Davidoff, A.J.; Gwathmey, J.K. Pathophysiology of cardiomyopathies: Part I. Animal models and humans. Curr. Opin. Cardiol. 1994, 9, 357–368. [Google Scholar] [CrossRef]
- Gwathmey, J.K. Experimental cardiomyopathies. In Cardiomyopathies, myocarditis, and pericardial disease; Braunwald, E., Ed.; Atlas of Heart Diseases; Current Science: Philadelphia, PA, USA, 1995; Volume 2, pp. 11.10–11.21. [Google Scholar]
- Gwathmey, J.K.; Davidoff, A.J. Pathophysiology of cardiomyopathies: Part II. Drug-induced and other interventions. Curr. Opin. Cardiol. 1994, 9, 369–378. [Google Scholar] [CrossRef]
- Genao, A.; Seth, K.; Schmidt, U.; Carles, M.; Gwathmey, J.K. Dilated cardiomyopathy in turkeys: An animal model for the study of human heart failure. Lab. Anim. Sci. 1996, 46, 399–404. [Google Scholar]
- Glass, M.G.; Fuleihan, F.; Liao, R.; Lincoff, A.M.; Chapados, R.; Hamlin, R.; Apstein, C.S.; Allen, P.D.; Ingwall, J.S.; Hajjar, R.J.; et al. Differences in cardioprotective efficacy of adrenergic receptor antagonists and Ca2+ channel antagonists in an animal model of dilated cardiomyopathy. Effects on gross morphology, global cardiac function, and twitch force. Circ. Res. 1993, 73, 1077–1089. [Google Scholar] [CrossRef] [Green Version]
- Gruver, E.J.; Glass, M.G.; Marsh, J.D.; Gwathmey, J.K. An animal model of dilated cardiomyopathy: Characterization of dihydropyridine receptors and contractile performance. Am. J. Physiol. 1993, 265, H1704–H1711. [Google Scholar] [CrossRef]
- Gwathmey, J.K. Morphological changes associated with furazolidone-induced cardiomyopathy: Effects of digoxin and propranolol. J. Comp. Pathol. 1991, 104, 33–45. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Hajjar, R.J. Calcium-activated force in a turkey model of spontaneous dilated cardiomyopathy: Adaptive changes in thin myofilament Ca2+ regulation with resultant implications on contractile performance. J. Mol. Cell. Cardiol. 1992, 24, 1459–1470. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Hamlin, R.L. Protection of turkeys against furazolidone-induced cardiomyopathy. Am. J. Cardiol. 1983, 52, 626–628. [Google Scholar] [CrossRef]
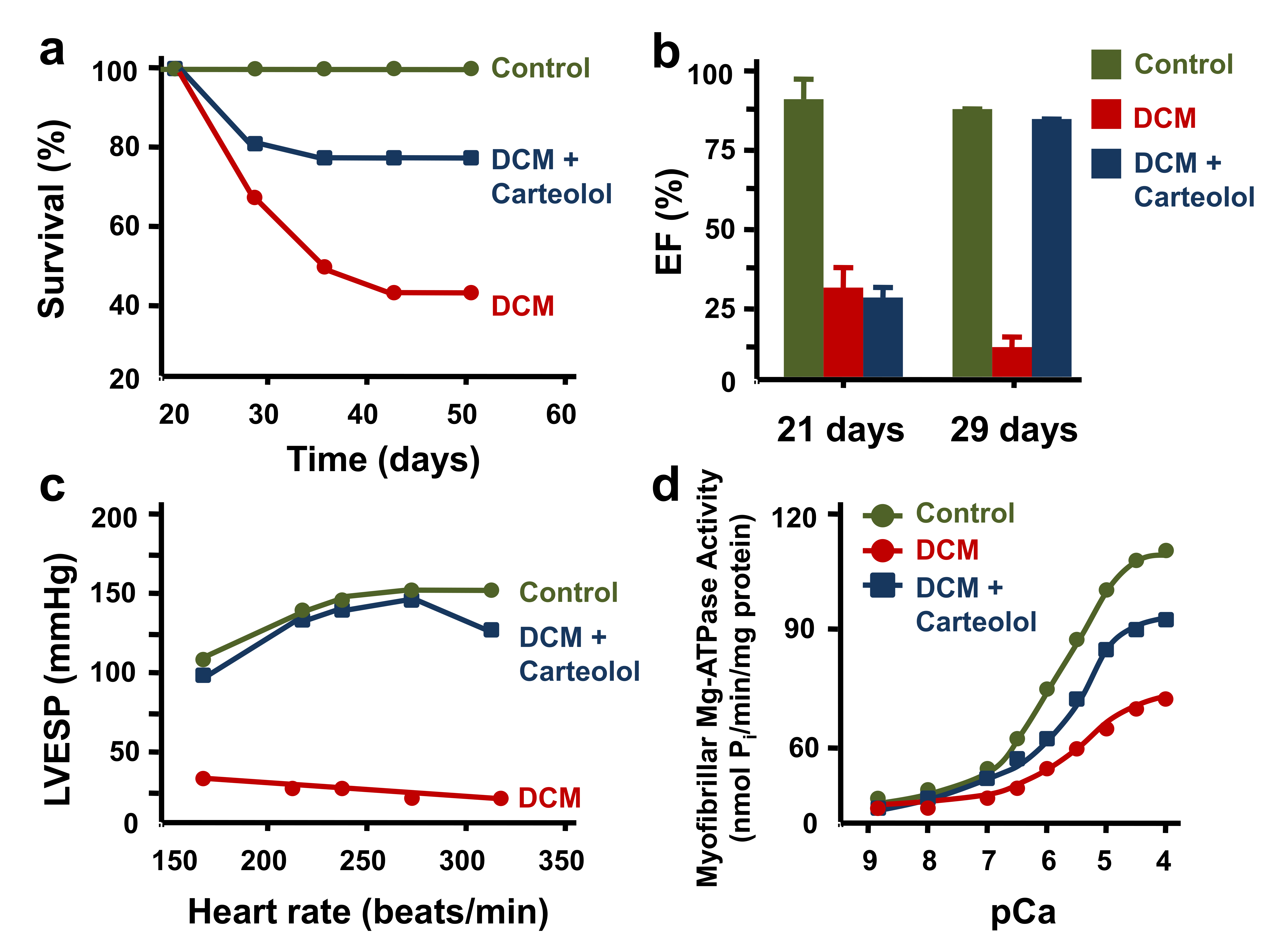
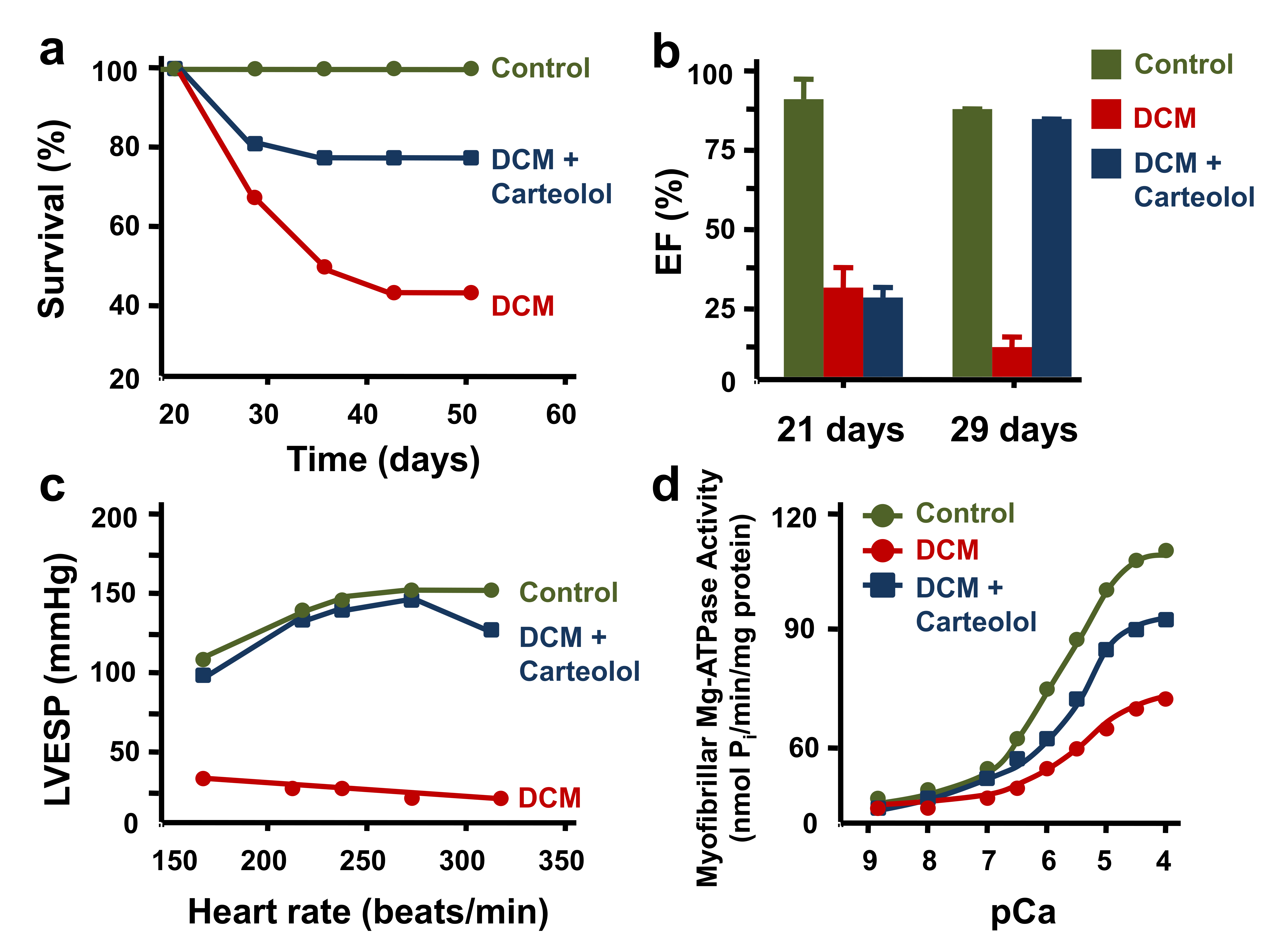
- Gwathmey, J.K.; Kim, C.S.; Hajjar, R.J.; Khan, F.; DiSalvo, T.G.; Matsumori, A.; Bristow, M.R. Cellular and molecular remodeling in a heart failure model treated with the beta-blocker carteolol. Am. J. Physiol. 1999, 276, H1678–H1690. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Morgan, J.P. Calcium handling in myocardium from amphibian, avian, and mammalian species: The search for two components. J. Comp. Physiol. B 1991, 161, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, R.J.; Liao, R.; Young, J.B.; Fuleihan, F.; Glass, M.G.; Gwathmey, J.K. Pathophysiological and biochemical characterisation of an avian model of dilated cardiomyopathy: Comparison to findings in human dilated cardiomyopathy. Cardiovasc. Res. 1993, 27, 2212–2221. [Google Scholar] [CrossRef]
- Kim, C.S.; Davidoff, A.J.; Maki, T.M.; Doye, A.A.; Gwathmey, J.K. Intracellular calcium and the relationship to contractility in an avian model of heart failure. J. Comp. Physiol. B 2000, 170, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.S.; Matsumori, A.; Goldberg, L.; Doye, A.A.; McCoy, Q.; Gwathmey, J.K. Effects of pranidipine, a calcium channel antagonist, in an avian model of heart failure. Cardiovasc. Drugs Ther. 1999, 13, 455–463. [Google Scholar] [CrossRef]
- Liao, R.; Carles, M.; Gwathmey, J.K. Animal models of cardiovascular disease for pharmacologic drug development and testing: Appropriateness of comparison to the human disease state and pharmacotherapeutics. Am. J. Ther. 1997, 4, 149–158. [Google Scholar] [CrossRef]
- Liao, R.; Nascimben, L.; Friedrich, J.; Gwathmey, J.K.; Ingwall, J.S. Decreased energy reserve in an animal model of dilated cardiomyopathy. Relationship to contractile performance. Circ. Res. 1996, 78, 893–902. [Google Scholar] [CrossRef]
- Okafor, C.C.; Perreault-Micale, C.; Hajjar, R.J.; Lebeche, D.; Skiroman, K.; Jabbour, G.; Doye, A.A.; Lee, M.X.; Laste, N.; Gwathmey, J.K. Chronic treatment with carvedilol improves ventricular function and reduces myocyte apoptosis in an animal model of heart failure. BMC Physiol. 2003, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Okafor, C.C.; Saunders, L.; Li, X.; Ito, T.; Dixon, M.; Stepenek, A.; Hajjar, R.J.; Wood, J.R.; Doye, A.A.; Gwathmey, J.K. Myofibrillar responsiveness to cAMP, PKA, and caffeine in an animal model of heart failure. Biochem. Biophys. Res. Commun. 2003, 300, 592–599. [Google Scholar] [CrossRef]
- Washington, B.; Butler, K.; Doye, A.A.; Jang, M.; Hajjar, R.J.; Gwathmey, J.K. Heart function challenged with beta-receptor agonism or antagonism in a heart failure model. Cardiovasc. Drugs Ther. 2001, 15, 479–486. [Google Scholar] [CrossRef]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; et al. 2020 International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2018, 71, e127–e248. [Google Scholar] [CrossRef]
- Ferri, C. The role of nebivolol in the management of hypertensive patients: From pharmacological profile to treatment guidelines. Future Cardiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Giles, T.D.; Weber, M.A.; Basile, J.; Gradman, A.H.; Bharucha, D.B.; Chen, W.; Pattathil, M. Efficacy and safety of nebivolol and valsartan as fixed-dose combination in hypertension: A randomised, multicentre study. Lancet 2014, 383, 1889–1898. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.V.; Bhatt, D.L.; Barsness, G.W.; Beatty, A.L.; Deedwania, P.C.; Inzucchi, S.E.; Kosiborod, M.; Leiter, L.A.; Lipska, K.J.; Newman, J.D.; et al. Clinical Management of Stable Coronary Artery Disease in Patients With Type 2 Diabetes Mellitus: A Scientific Statement From the American Heart Association. Circulation 2020, 141, e779–e806. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Han, J.K.; Yang, H.M.; Park, K.W.; Kang, H.J.; Koo, B.K.; Jeong, M.H.; Kim, H.S. Long-term efficacy of vasodilating β-blocker in patients with acute myocardial infarction: Nationwide multicenter prospective registry. Korean J. Intern. Med. 2021, 36, S62–S71. [Google Scholar] [CrossRef]
- Verma, S.; Peterson, E.L.; Liu, B.; Sabbah, H.N.; Williams, L.K.; Lanfear, D.E. Effectiveness of beta blockers in patients with and without a history of myocardial infarction. Eur. J. Clin. Pharmacol. 2020, 76, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Ziff, O.J.; Samra, M.; Howard, J.P.; Bromage, D.I.; Ruschitzka, F.; Francis, D.P.; Kotecha, D. Beta-blocker efficacy across different cardiovascular indications: An umbrella review and meta-analytic assessment. BMC Med. 2020, 18, 103. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Circulation 2018, 138, e272–e391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, S.J.; Dorian, P.; Roberts, R.S.; Gent, M.; Bailin, S.; Fain, E.S.; Thorpe, K.; Champagne, J.; Talajic, M.; Coutu, B.; et al. Comparison of beta-blockers, amiodarone plus beta-blockers, or sotalol for prevention of shocks from implantable cardioverter defibrillators: The OPTIC Study: A randomized trial. JAMA 2006, 295, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadeh, K.; Shivkumar, K.; Jeevaratnam, K. Targeting the β-adrenergic receptor in the clinical management of congenital long QT syndrome. Ann. N. Y. Acad. Sci. 2020, 1474, 27–46. [Google Scholar] [CrossRef]
- Gordan, R.; Gwathmey, J.K.; Xie, L.H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204–214. [Google Scholar] [CrossRef]
- Jasmin, G.; Proschek, L. Calcium and myocardial cell injury. An appraisal in the cardiomyopathic hamster. Can. J. Physiol. Pharmacol. 1984, 62, 891–898. [Google Scholar] [CrossRef]
- Lax, D.; Martínez-Zaguilán, R.; Gillies, R.J. Furazolidone increases thapsigargin-sensitive Ca(2+)-ATPase in chick cardiac myocytes. Am. J. Physiol. 1994, 267, H734–H741. [Google Scholar] [CrossRef]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Desai, A.S.; Barnard, D.; Bouchard, A.; Jaski, B.; Lyon, A.R.; et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016, 387, 1178–1186. [Google Scholar] [CrossRef]
- Jaski, B.E.; Jessup, M.L.; Mancini, D.M.; Cappola, T.P.; Pauly, D.F.; Greenberg, B.; Borow, K.; Dittrich, H.; Zsebo, K.M.; Hajjar, R.J. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card. Fail. 2009, 15, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Jessup, M.; Greenberg, B.; Mancini, D.; Cappola, T.; Pauly, D.F.; Jaski, B.; Yaroshinsky, A.; Zsebo, K.M.; Dittrich, H.; Hajjar, R.J. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011, 124, 304–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulot, J.S.; Ishikawa, K.; Hajjar, R.J. Gene therapy for the treatment of heart failure: Promise postponed. Eur. Heart J. 2016, 37, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, R.J.; Gwathmey, J.K. Calcium-sensitizing inotropic agents in the treatment of heart failure: A critical view. Cardiovasc. Drugs Ther. 1991, 5, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, R.J.; Gwathmey, J.K.; Briggs, G.M.; Morgan, J.P. Differential effect of DPI 201-106 on the sensitivity of the myofilaments to Ca2+ in intact and skinned trabeculae from control and myopathic human hearts. J. Clin. Invest. 1988, 82, 1578–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waagstein, F.; Bristow, M.R.; Swedberg, K.; Camerini, F.; Fowler, M.B.; Silver, M.A.; Gilbert, E.M.; Johnson, M.R.; Goss, F.G.; Hjalmarson, A. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Metoprolol in Dilated Cardiomyopathy (MDC) Trial Study Group. Lancet 1993, 342, 1441–1446. [Google Scholar] [CrossRef]
- Packer, M.; Bristow, M.R.; Cohn, J.N.; Colucci, W.S.; Fowler, M.B.; Gilbert, E.M.; Shusterman, N.H. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N. Engl. J. Med. 1996, 334, 1349–1355. [Google Scholar] [CrossRef]
- Miyata, S.; Minobe, W.; Bristow, M.R.; Leinwand, L.A. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ. Res. 2000, 86, 386–390. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.C.; Yang, H.; Yang, M.; Wang, C.K.; Shi, J.; Berg, E.A.; Pimentel, D.R.; Gwathmey, J.K.; Hajjar, R.J.; Helmes, M.; et al. A novel mutant cardiac troponin C disrupts molecular motions critical for calcium binding affinity and cardiomyocyte contractility. Biophys. J. 2008, 94, 3577–3589. [Google Scholar] [CrossRef] [Green Version]
- Hajjar, R.J.; Schwinger, R.H.; Schmidt, U.; Kim, C.S.; Lebeche, D.; Doye, A.A.; Gwathmey, J.K. Myofilament calcium regulation in human myocardium. Circulation 2000, 101, 1679–1685. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.J.; Morgan, K.G. Intracellular calcium levels in phorbol ester-induced contractions of vascular muscle. Am. J. Physiol. 1987, 253, H1365–H1371. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Hajjar, R.J. Effect of protein kinase C activation on sarcoplasmic reticulum function and apparent myofibrillar Ca2+ sensitivity in intact and skinned muscles from normal and diseased human myocardium. Circ. Res. 1990, 67, 744–752. [Google Scholar] [CrossRef] [Green Version]
- Ringvold, H.C.; Khalil, R.A. Protein Kinase C as Regulator of Vascular Smooth Muscle Function and Potential Target in Vascular Disorders. Adv. Pharmacol. 2017, 78, 203–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehlik, J.; Movsesian, M.A. Inhibitors of cyclic nucleotide phosphodiesterase 3 and 5 as therapeutic agents in heart failure. Expert Opin. Investig. Drugs 2006, 15, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Movsesian, M.A. cAMP and cGMP signaling cross-talk: Role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 2007, 100, 1569–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movsesian, M.A.; Alharethi, R. Inhibitors of cyclic nucleotide phosphodiesterase PDE3 as adjunct therapy for dilated cardiomyopathy. Expert Opin. Investig. Drugs 2002, 11, 1529–1536. [Google Scholar] [CrossRef]
- Follmann, M.; Ackerstaff, J.; Redlich, G.; Wunder, F.; Lang, D.; Kern, A.; Fey, P.; Griebenow, N.; Kroh, W.; Becker-Pelster, E.M.; et al. Discovery of the Soluble Guanylate Cyclase Stimulator Vericiguat (BAY 1021189) for the Treatment of Chronic Heart Failure. J. Med. Chem. 2017, 60, 5146–5161. [Google Scholar] [CrossRef] [Green Version]
- Pieske, B.; Butler, J.; Filippatos, G.; Lam, C.; Maggioni, A.P.; Ponikowski, P.; Shah, S.; Solomon, S.; Kraigher-Krainer, E.; Samano, E.T.; et al. Rationale and design of the SOluble guanylate Cyclase stimulatoR in heArT failurE Studies (SOCRATES). Eur. J. Heart Fail. 2014, 16, 1026–1038. [Google Scholar] [CrossRef]
- Crassous, P.A.; Shu, P.; Huang, C.; Gordan, R.; Brouckaert, P.; Lampe, P.D.; Xie, L.H.; Beuve, A. Newly Identified NO-Sensor Guanylyl Cyclase/Connexin 43 Association Is Involved in Cardiac Electrical Function. J. Am. Heart Assoc. 2017, 6, e006397. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Gwathmey, J.K. Direct evidence of changes in myofilament responsiveness to Ca2+ during hypoxia and reoxygenation in myocardium. Am. J. Physiol. 1990, 259, H784–H795. [Google Scholar] [CrossRef]
- MacKinnon, R.; Gwathmey, J.K.; Morgan, J.P. Differential effects of reoxygenation on intracellular calcium and isometric tension. Pflugers Arch. 1987, 409, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Pesaturo, J.A.; Gwathmey, J.K. The role of mitochondria and sarcoplasmic reticulum calcium handling upon reoxygenation of hypoxic myocardium. Circ. Res. 1990, 66, 696–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapados, R.A.; Gruver, E.J.; Ingwall, J.S.; Marsh, J.D.; Gwathmey, J.K. Chronic administration of cardiovascular drugs: Altered energetics and transmembrane signaling. Am. J. Physiol. 1992, 263, H1576–H1586. [Google Scholar] [CrossRef]
- Nascimben, L.; Ingwall, J.S.; Pauletto, P.; Friedrich, J.; Gwathmey, J.K.; Saks, V.; Pessina, A.C.; Allen, P.D. Creatine kinase system in failing and nonfailing human myocardium. Circulation 1996, 94, 1894–1901. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J.; Shen, H.; Gwathmey, J.K. Myocardial Ribonuclease Activity in Heart Failure. In Mechanisms of Heart Failure; Singal, P.K., Dixon, I.M.C., Dhalla, N.S., Eds.; Kluwer Academic Publishers: New York, NY, USA, 1995; pp. 9–18. [Google Scholar]
- Gwathmey, J.K.; Tsaioun, K.; Hajjar, R.J. Cardionomics: A new integrative approach for screening cardiotoxicity of drug candidates. Expert Opin. Drug Metab. Toxicol. 2009, 5, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Choudhary, S.; Singh, P.K.; Sapra, B.; Silakari, O. Structural investigation on the selective COX-2 inhibitors mediated cardiotoxicity: A review. Life Sci. 2020, 251, 117631. [Google Scholar] [CrossRef]
- Brenner, G.B.; Makkos, A.; Nagy, C.T.; Onódi, Z.; Sayour, N.V.; Gergely, T.G.; Kiss, B.; Görbe, A.; Sághy, É.; Zádori, Z.S.; et al. Hidden Cardiotoxicity of Rofecoxib Can be Revealed in Experimental Models of Ischemia/Reperfusion. Cells 2020, 9, 551. [Google Scholar] [CrossRef] [Green Version]
- Jüni, P.; Nartey, L.; Reichenbach, S.; Sterchi, R.; Dieppe, P.A.; Egger, M. Risk of cardiovascular events and rofecoxib: Cumulative meta-analysis. Lancet 2004, 364, 2021–2029. [Google Scholar] [CrossRef]
- Mason, R.P.; Walter, M.F.; Day, C.A.; Jacob, R.F. A biological rationale for the cardiotoxic effects of rofecoxib: Comparative analysis with other COX-2 selective agents and NSAids. Subcell. Biochem. 2007, 42, 175–190. [Google Scholar]
- Mason, R.P.; Walter, M.F.; McNulty, H.P.; Lockwood, S.F.; Byun, J.; Day, C.A.; Jacob, R.F. Rofecoxib increases susceptibility of human LDL and membrane lipids to oxidative damage: A mechanism of cardiotoxicity. J. Cardiovasc. Pharmacol. 2006, 47 (Suppl. 1), S7–S14. [Google Scholar] [CrossRef]
- Beckendorf, J.; van den Hoogenhof, M.M.G.; Backs, J. Physiological and unappreciated roles of CaMKII in the heart. Basic Res. Cardiol. 2018, 113, 29. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.J.; Wang, W. Research progress on the role of CaMKII in heart disease. Am. J. Transl. Res. 2020, 12, 7625–7639. [Google Scholar]
- Tanaka, S.; Fujio, Y.; Nakayama, H. Caveolae-Specific CaMKII Signaling in the Regulation of Voltage-Dependent Calcium Channel and Cardiac Hypertrophy. Front. Physiol. 2018, 9, 1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Jiang, K.; Liu, X.; Qin, M.; Xiang, Y. CaMKII in Regulation of Cell Death During Myocardial Reperfusion Injury. Front. Mol. Biosci. 2021, 8, 668129. [Google Scholar] [CrossRef]
- Erickson, J.R. Mechanisms of CaMKII Activation in the Heart. Front. Pharmacol. 2014, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Liao, Z.; Lu, X.; Katschinski, D.M.; Mercola, M.; Chen, J.; Heller Brown, J.; Molkentin, J.D.; Bossuyt, J.; Bers, D.M. Hyperglycemia Acutely Increases Cytosolic Reactive Oxygen Species via O-linked GlcNAcylation and CaMKII Activation in Mouse Ventricular Myocytes. Circ. Res. 2020, 126, e80–e96. [Google Scholar] [CrossRef] [PubMed]
- Greer-Short, A.; Musa, H.; Alsina, K.M.; Ni, L.; Word, T.A.; Reynolds, J.O.; Gratz, D.; Lane, C.; El-Refaey, M.; Unudurthi, S.; et al. Calmodulin kinase II regulates atrial myocyte late sodium current, calcium handling, and atrial arrhythmia. Heart Rhythm 2020, 17, 503–511. [Google Scholar] [CrossRef]
- Hegyi, B.; Bers, D.M.; Bossuyt, J. CaMKII signaling in heart diseases: Emerging role in diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 127, 246–259. [Google Scholar] [CrossRef]
- Yan, J.; Zhao, W.; Thomson, J.K.; Gao, X.; DeMarco, D.M.; Carrillo, E.; Chen, B.; Wu, X.; Ginsburg, K.S.; Bakhos, M.; et al. Stress Signaling JNK2 Crosstalk With CaMKII Underlies Enhanced Atrial Arrhythmogenesis. Circ. Res. 2018, 122, 821–835. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, X.; Zimmerman, R.J.; Wang, Q.; Ross, M.A.; Granger, J.M.; Luczak, E.D.; Bedja, D.; Jiang, H.; Feng, N. CaMKII exacerbates heart failure progression by activating class I HDACs. J. Mol. Cell. Cardiol. 2020, 149, 73–81. [Google Scholar] [CrossRef]
- Zhao, Z.; Babu, G.J.; Wen, H.; Fefelova, N.; Gordan, R.; Sui, X.; Yan, L.; Vatner, D.E.; Vatner, S.F.; Xie, L.H. Overexpression of adenylyl cyclase type 5 (AC5) confers a proarrhythmic substrate to the heart. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H240–H249. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.H.; Chen, F.; Karagueuzian, H.S.; Weiss, J.N. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 2009, 104, 79–86. [Google Scholar] [CrossRef]
- Mustroph, J.; Neef, S.; Maier, L.S. CaMKII as a target for arrhythmia suppression. Pharmacol. Ther. 2017, 176, 22–31. [Google Scholar] [CrossRef]
- Nassal, D.; Gratz, D.; Hund, T.J. Challenges and Opportunities for Therapeutic Targeting of Calmodulin Kinase II in Heart. Front. Pharmacol. 2020, 11, 35. [Google Scholar] [CrossRef]
- Mustroph, J.; Drzymalski, M.; Baier, M.; Pabel, S.; Biedermann, A.; Memmel, B.; Durczok, M.; Neef, S.; Sag, C.M.; Floerchinger, B.; et al. The oral Ca/calmodulin-dependent kinase II inhibitor RA608 improves contractile function and prevents arrhythmias in heart failure. ESC heart failure 2020, 7, 2871–2883. [Google Scholar] [CrossRef]
- Sossalla, S.; Fluschnik, N.; Schotola, H.; Ort, K.R.; Neef, S.; Schulte, T.; Wittköpper, K.; Renner, A.; Schmitto, J.D.; Gummert, J.; et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ. Res. 2010, 107, 1150–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Kudej, R.K.; Wen, H.; Fefelova, N.; Yan, L.; Vatner, D.E.; Vatner, S.F.; Xie, L.H. Antioxidant defense and protection against cardiac arrhythmias: Lessons from a mammalian hibernator (the woodchuck). FASEB J. 2018, 32, 4229–4240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, A.J.; Schultz, C.; Selvanayagam, J.B.; Moir, S.; Kovacs, R.; Dib, N.; Zlotnick, D.; Al-Omary, M.; Sugito, S.; Selvarajah, A.; et al. Calcium/Calmodulin-Dependent Protein Kinase II Delta Inhibition and Ventricular Remodeling After Myocardial Infarction: A Randomized Clinical Trial. JAMA Cardiol. 2021, e210676. [Google Scholar] [CrossRef]
- Anderson, M.E. To Be or Not to Be a CaMKII Inhibitor? JAMA Cardiol. 2021, 21. [Google Scholar] [CrossRef]
- Anderson, M.E. Oxidant stress promotes disease by activating CaMKII. J. Mol. Cell. Cardiol. 2015, 89, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Quick, A.P.; Cao, S.; Reynolds, J.; Chiang, D.Y.; Beavers, D.; Li, N.; Wang, G.; Rodney, G.G.; Anderson, M.E.; et al. Oxidized CaMKII (Ca(2+)/Calmodulin-Dependent Protein Kinase II) Is Essential for Ventricular Arrhythmia in a Mouse Model of Duchenne Muscular Dystrophy. Circ. Arrhythm. Electrophysiol. 2018, 11, e005682. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Martelli, A.; Testai, L.; Colletti, A.; Cicero, A.F.G. Coenzyme Q(10): Clinical Applications in Cardiovascular Diseases. Antioxidants 2020, 9, 341. [Google Scholar] [CrossRef] [Green Version]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Taylor, C.J.; Ordóñez-Mena, J.M.; Roalfe, A.K.; Lay-Flurrie, S.; Jones, N.R.; Marshall, T.; Hobbs, F.D.R. Trends in survival after a diagnosis of heart failure in the United Kingdom 2000-2017: Population based cohort study. BMJ 2019, 364, l223. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Arrigo, M.; Jessup, M.; Mullens, W.; Reza, N.; Shah, A.M.; Sliwa, K.; Mebazaa, A. Acute heart failure. Nat. Rev. Dis. Primers 2020, 6, 16. [Google Scholar] [CrossRef]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017, 136, e137–e161. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Ishikawa, K. Introducing Genes to the Heart: All About Delivery. Circ. Res. 2017, 120, 33–35. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Hajjar, R.J. Current Methods in Cardiac Gene Therapy: Overview. Methods Mol. Biol. 2017, 1521, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Weber, T.; Hajjar, R.J. Human Cardiac Gene Therapy. Circ. Res. 2018, 123, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Gianni, D.; Chan, J.; Gwathmey, J.K.; del Monte, F.; Hajjar, R.J. SERCA2a in heart failure: Role and therapeutic prospects. J. Bioenerg. Biomembr. 2005, 37, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.; Yaroshinsky, A.; Zsebo, K.M.; Butler, J.; Felker, G.M.; Voors, A.A.; Rudy, J.J.; Wagner, K.; Hajjar, R.J. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: The CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC. Heart failure 2014, 2, 84–92. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Yerevanian, A.; Hajjar, R.J. Targeting sarcoplasmic reticulum calcium ATPase by gene therapy. Hum. Gene Ther. 2013, 24, 937–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwathmey, J.K.; Yerevanian, A.I.; Hajjar, R.J. Cardiac gene therapy with SERCA2a: From bench to bedside. J. Mol. Cell. Cardiol. 2011, 50, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y.; et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008, 51, 1112–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siri-Angkul, N.; Dadfar, B.; Jaleel, R.; Naushad, J.; Parambathazhath, J.; Doye, A.A.; Xie, L.-H.; Gwathmey, J.K. Calcium and Heart Failure: How Did We Get Here and Where Are We Going? Int. J. Mol. Sci. 2021, 22, 7392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147392
Siri-Angkul N, Dadfar B, Jaleel R, Naushad J, Parambathazhath J, Doye AA, Xie L-H, Gwathmey JK. Calcium and Heart Failure: How Did We Get Here and Where Are We Going? International Journal of Molecular Sciences. 2021; 22(14):7392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147392
Chicago/Turabian StyleSiri-Angkul, Natthaphat, Behzad Dadfar, Riya Jaleel, Jazna Naushad, Jaseela Parambathazhath, Angelia A. Doye, Lai-Hua Xie, and Judith K. Gwathmey. 2021. "Calcium and Heart Failure: How Did We Get Here and Where Are We Going?" International Journal of Molecular Sciences 22, no. 14: 7392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147392