
The Mode of SN38 Derivatives Interacting with Nicked DNA Mimics Biological Targeting of Topo I Poisons

 , , , , , , and
, , , , , , and
Abstract
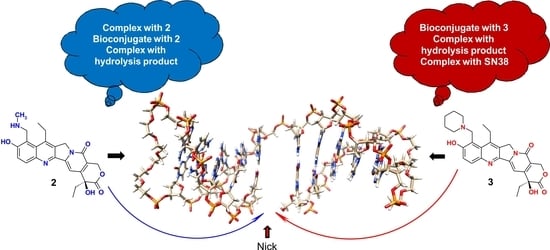
:
1. Introduction
2. Results and Discussion
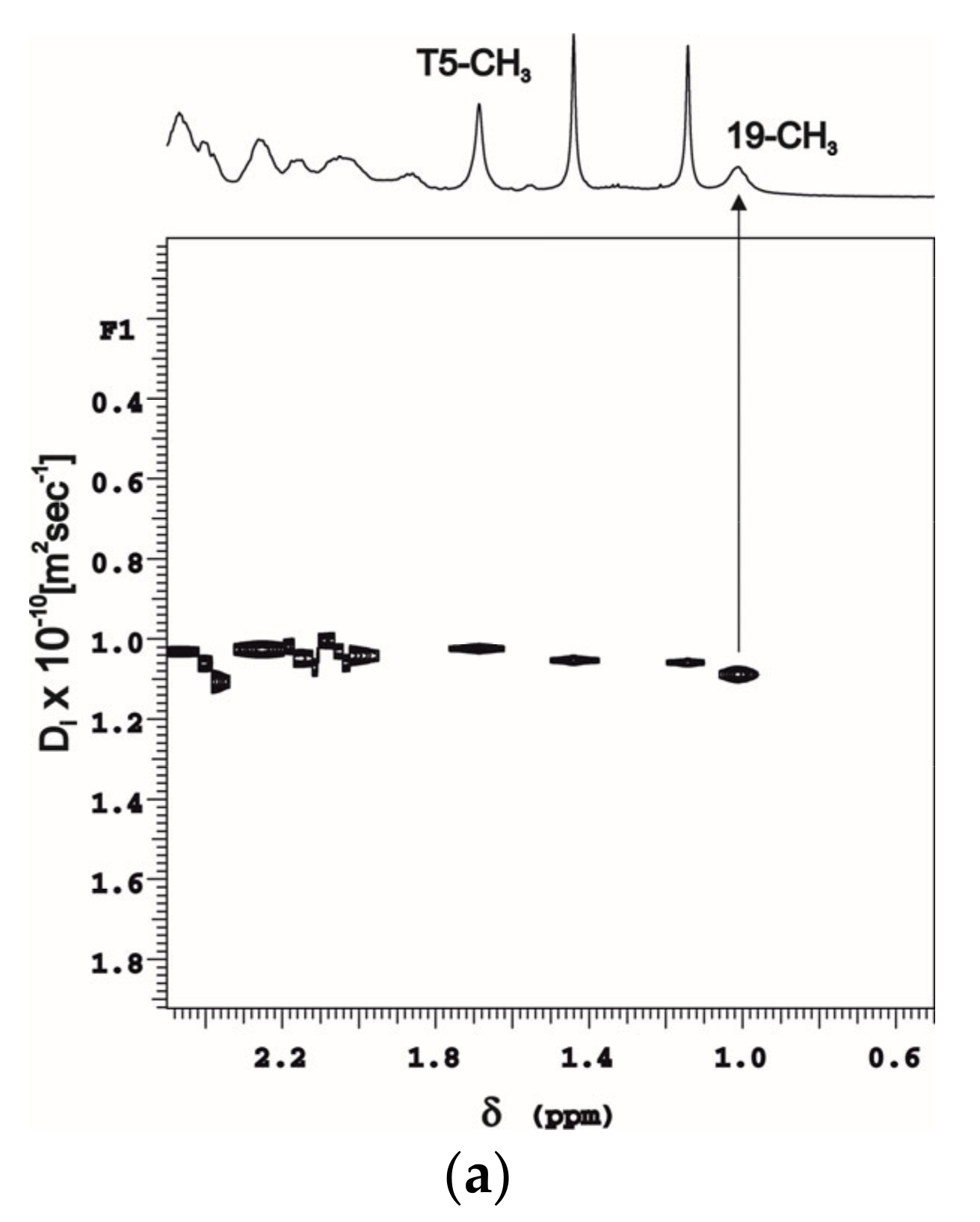
2.1. DOSY Experiments of Molecular Complexes
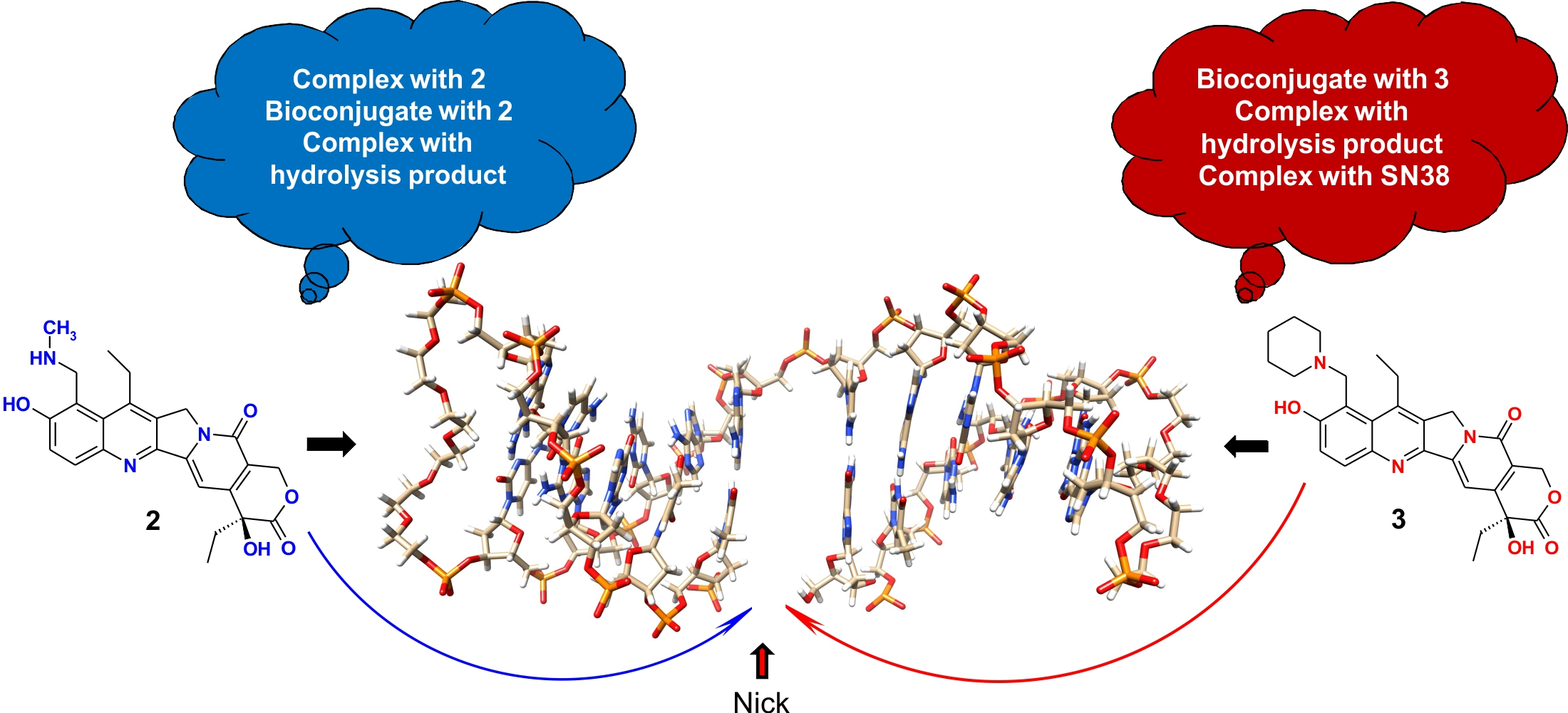
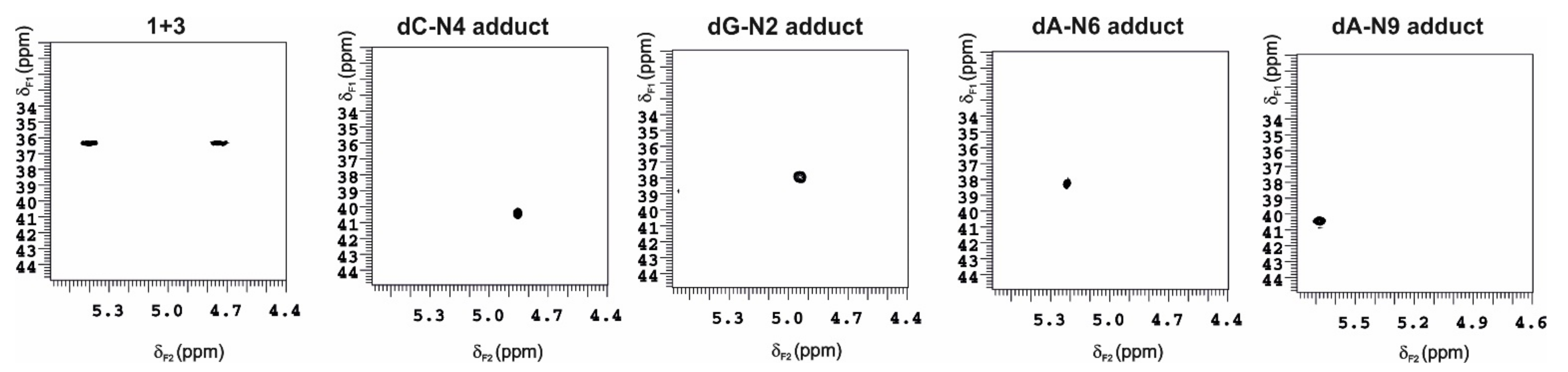
2.2. NOESY Experiment of the Molecular C Complex
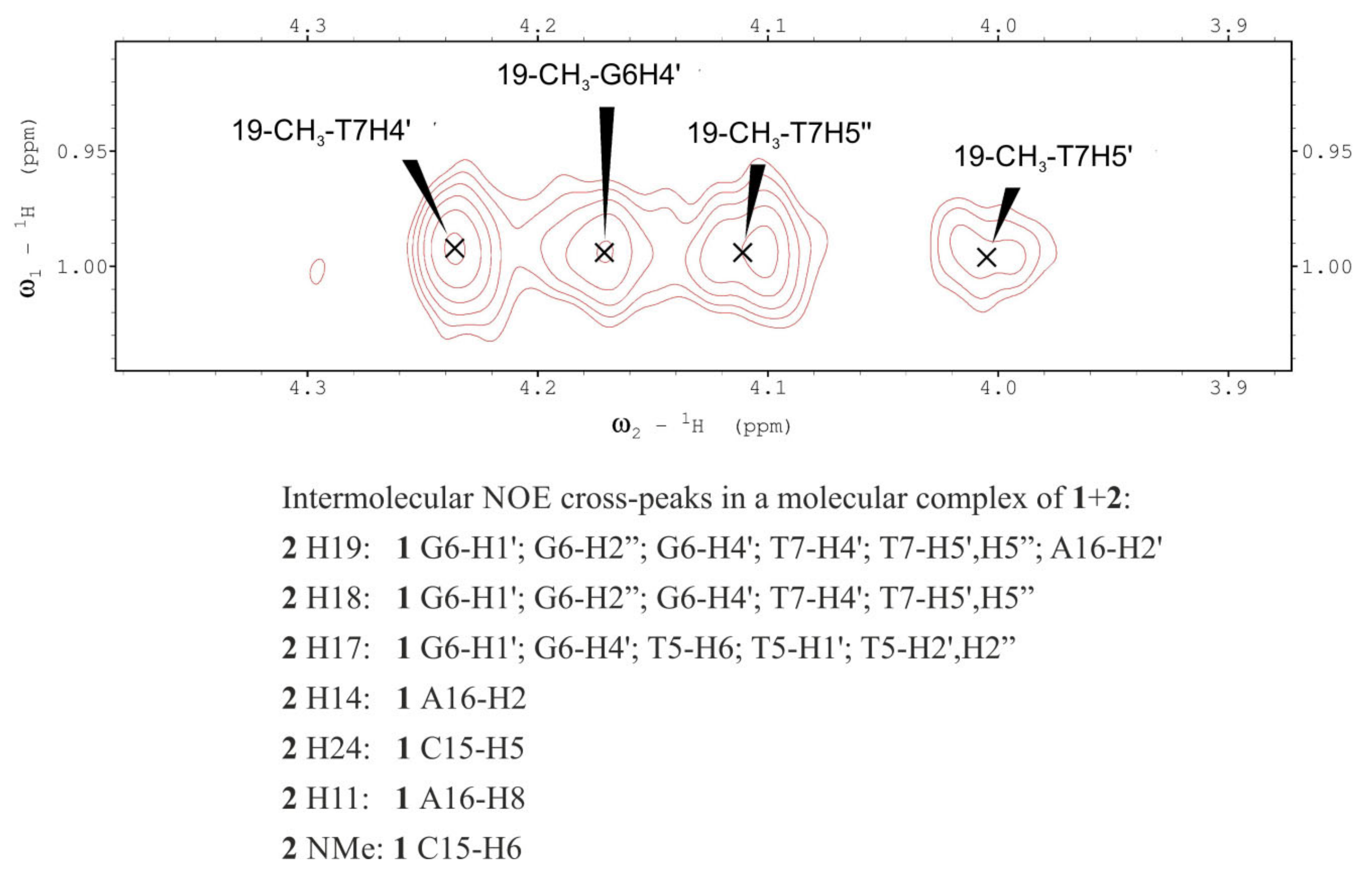
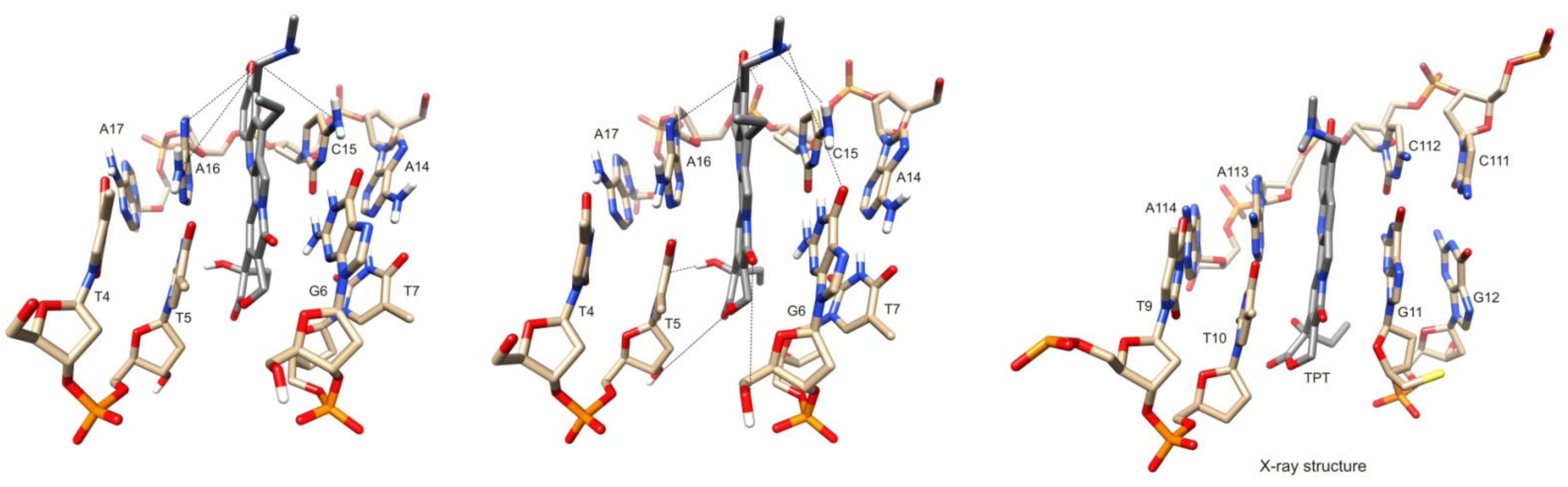
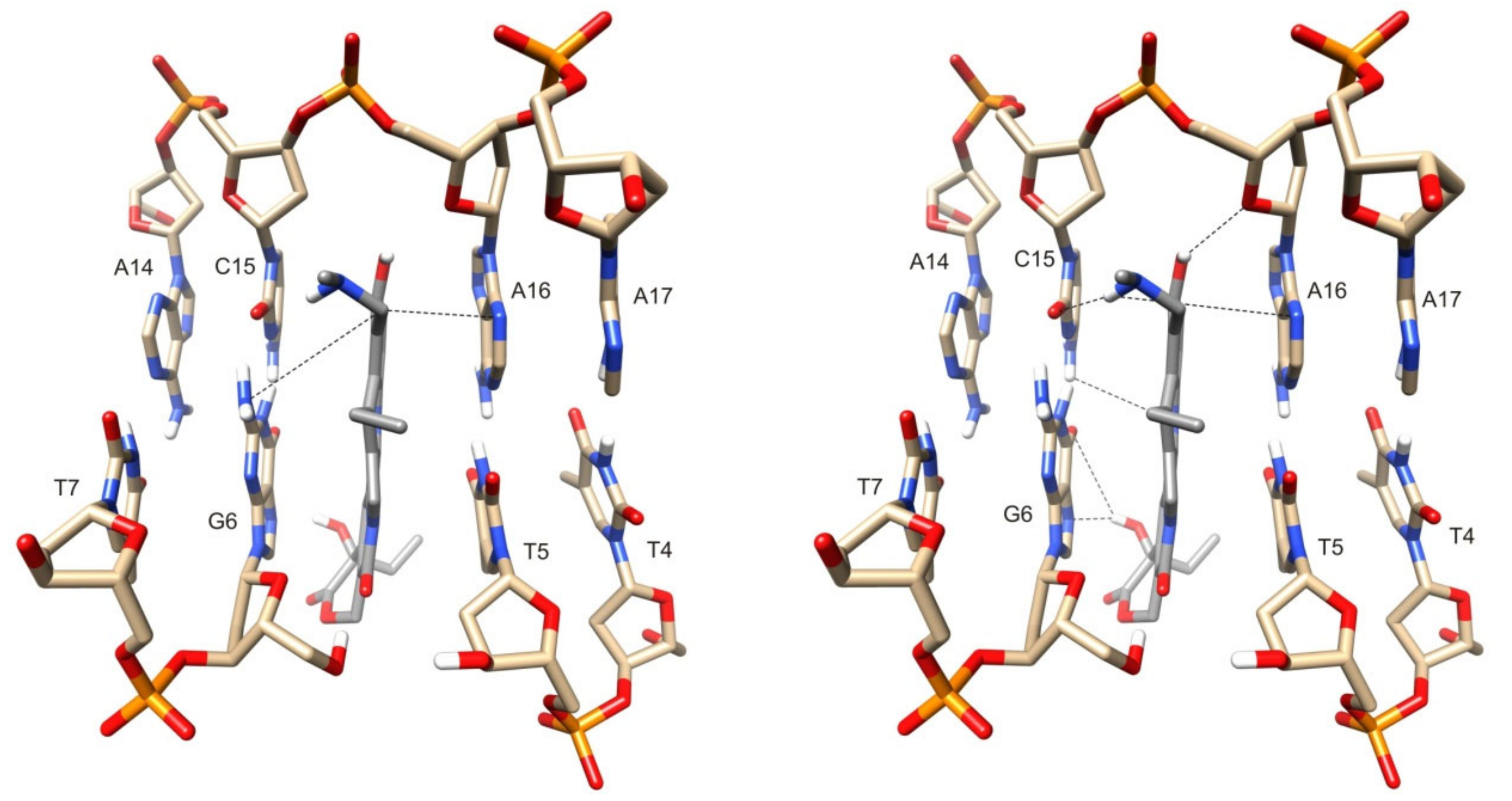
2.3. Molecular Modeling
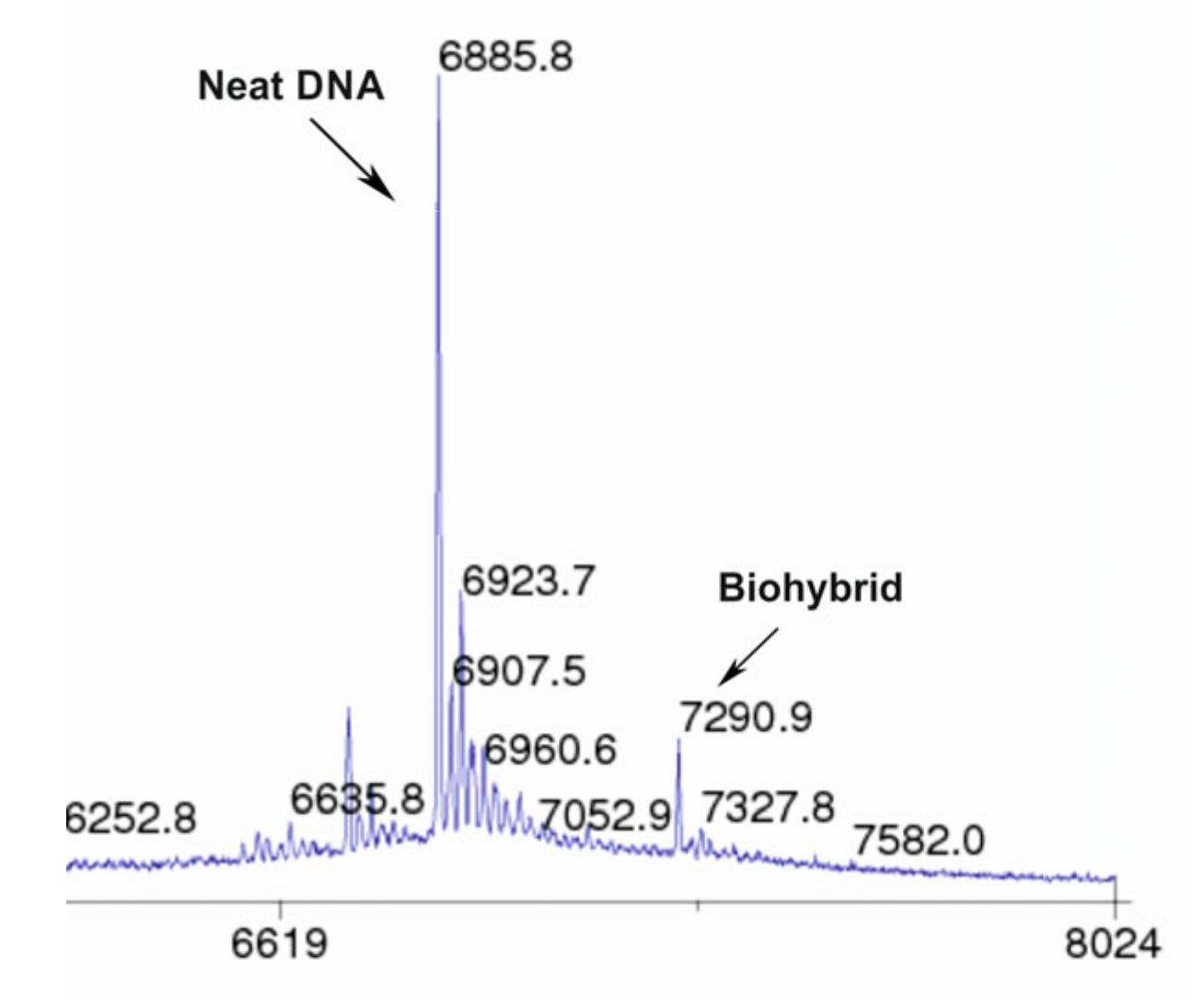
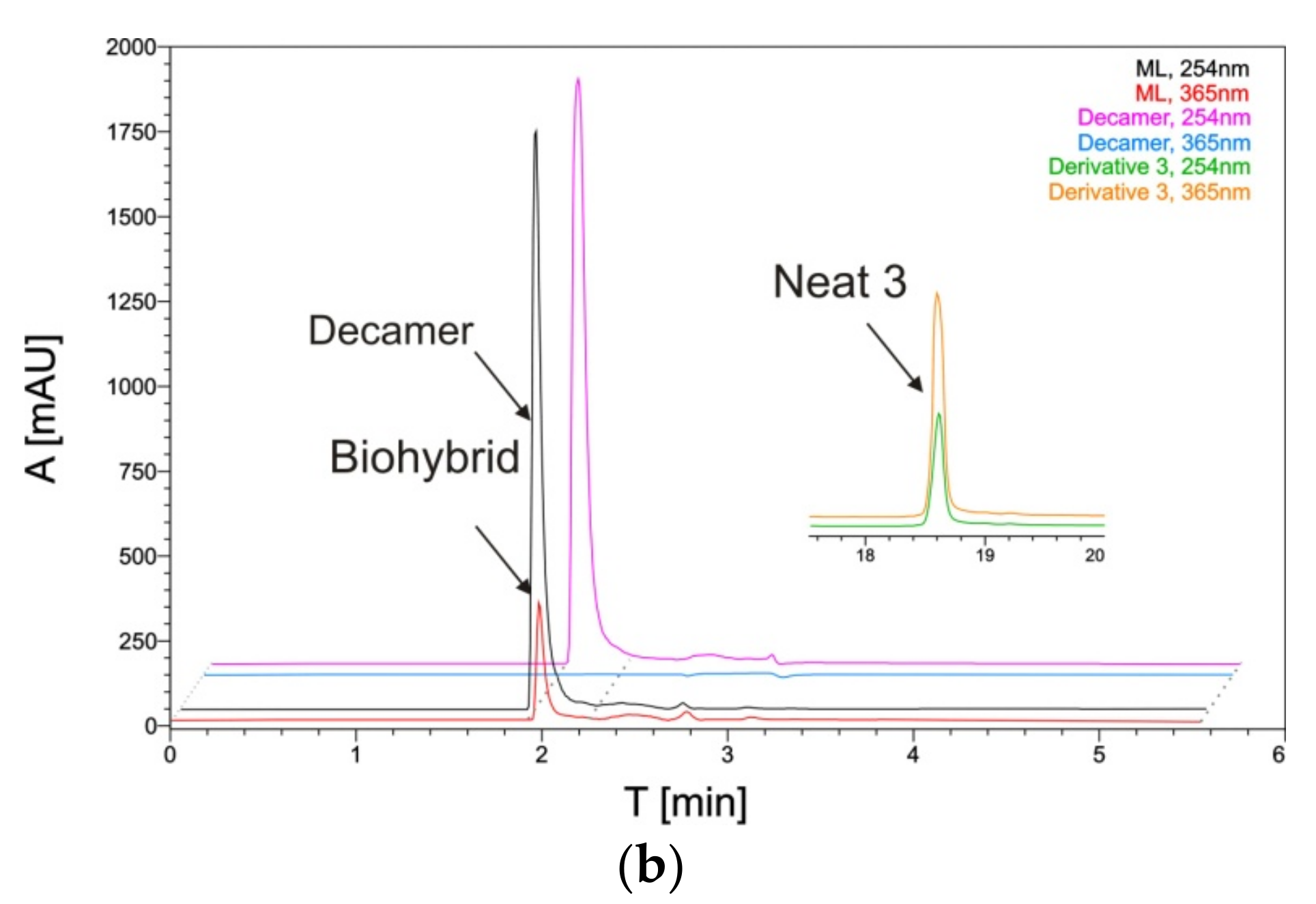
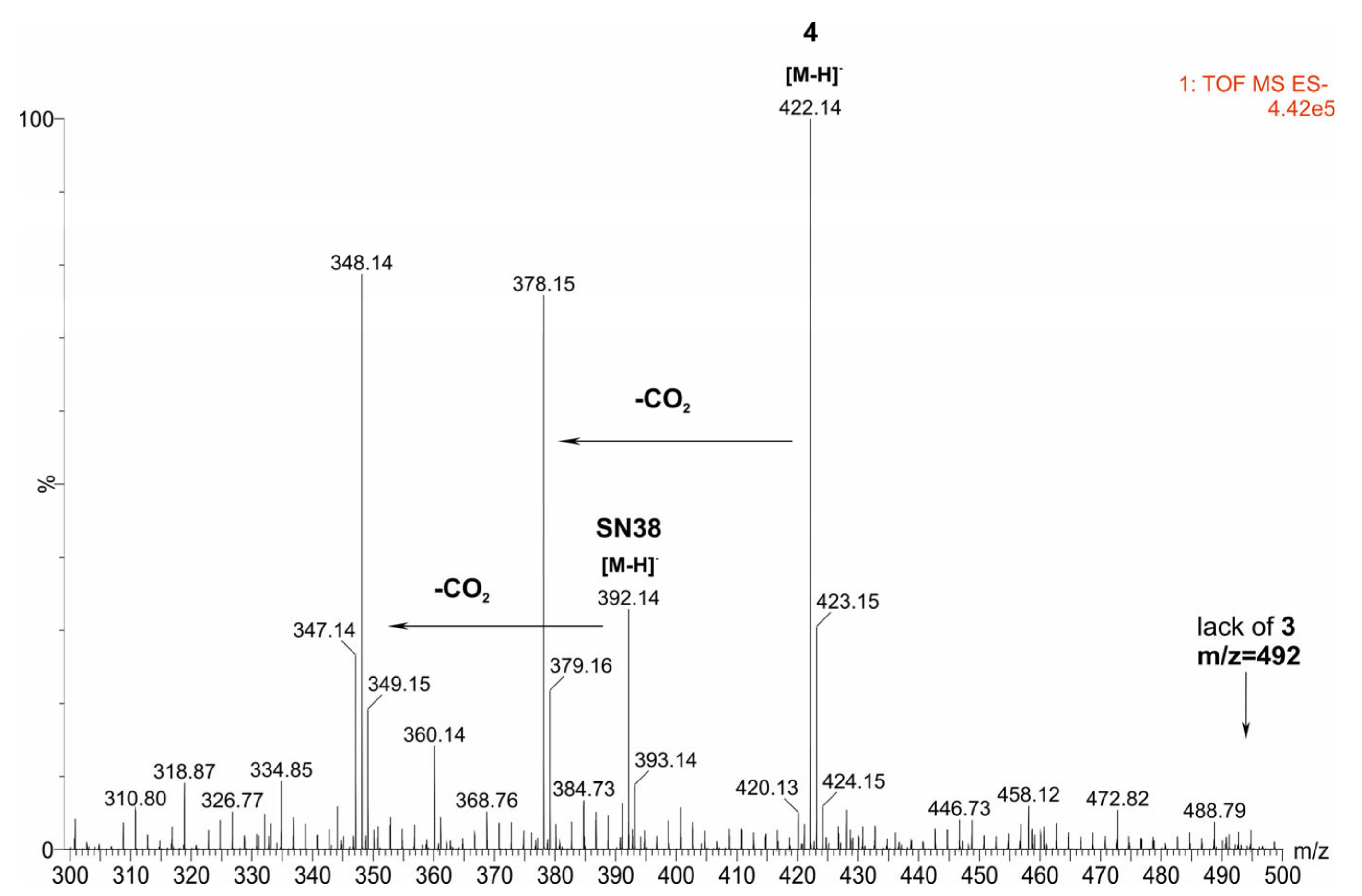
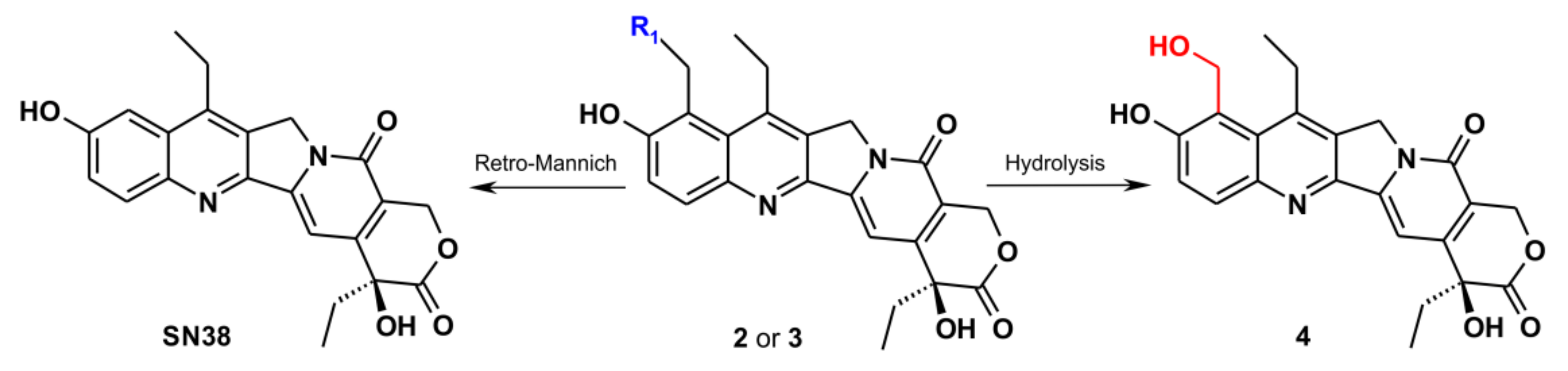
2.4. The Reaction Products Analysis
2.5. The Topo I Relaxation Activity Test
3. Materials and Methods
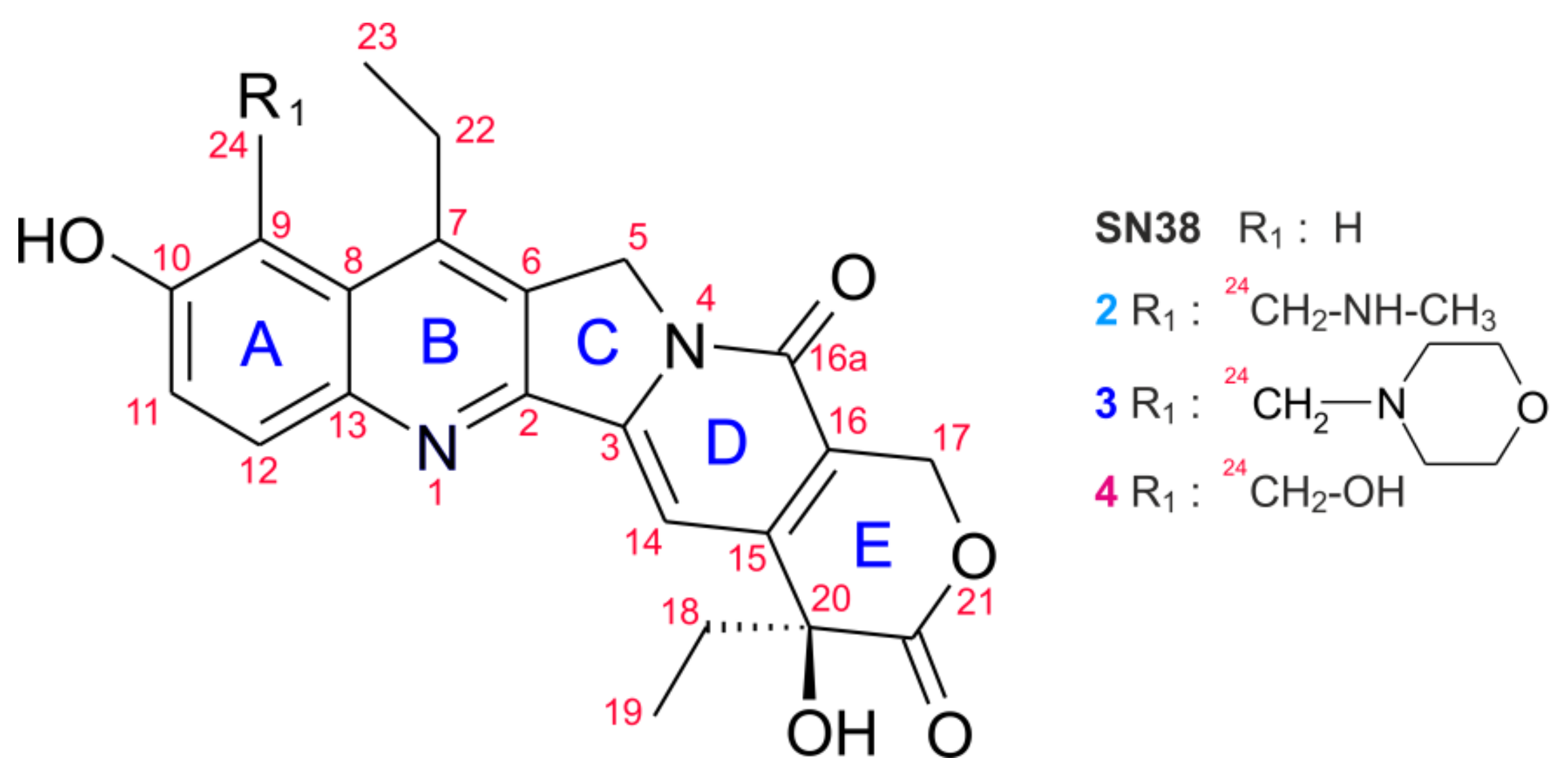
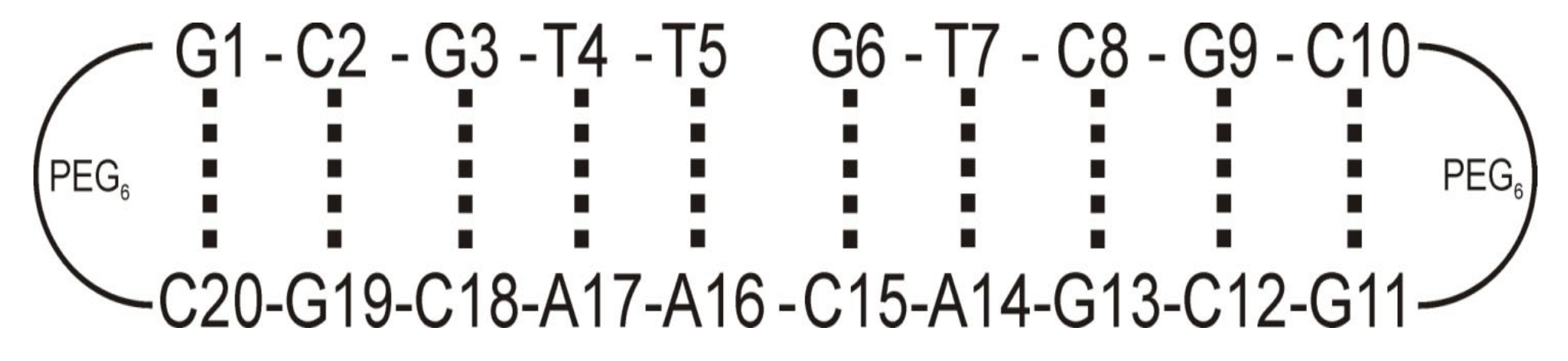
3.1. Chemical Substrates
3.2. Sample Preparation
3.3. HPLC Analysis
3.4. NMR Experiments
3.5. ESI–MS Experiments
3.6. MALDI–MS Experiments
3.7. MALDI Sample Preparation
3.8. Calculating Binding Constants from the Diffusion Coefficients
3.9. Molecular Dynamics Calculations
3.9.1. Calculating Binding Free Energies (Enthalpies) Using MM-PBSA and MM-GBSA Methods
3.9.2. PM7 Semi-Empirical Calculations
3.9.3. Cluster Analysis
3.10. The Topo I Relaxation Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pourquier, P.; Pommier, Y. Topoisomerase I-mediated DNA damage. Adv. Cancer Res. 2001, 80, 189–216. [Google Scholar]
- Stewart, L.; Redinbo, M.R.; Qiu, X.; Hol, W.G.; Champoux, J.J. A model for the mechanism of human topoisomerase I. Science 1998, 279, 1534–1541. [Google Scholar] [CrossRef]
- Pommier, Y. DNA topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [Green Version]
- Dallavalle, S.; Merlini, L. Camptothecin and Analogs: Structure and Synthetic Efforts. In Modern Alkaloids; Fattorusso, E., Taglialatela-Scafati, O., Eds.; Wiley: Hoboken, NJ, USA, 2007; pp. 503–520. [Google Scholar]
- Bocian, W.; Kawęcki, R.; Bednarek, E.; Sitkowski, J.; Williamson, M.P.; Hansen, P.E.; Kozerski, L. Binding of Topotecan to a Nicked DNA Oligomer in Solution. Chem. A Eur. J. 2008, 14, 2788–2794. [Google Scholar] [CrossRef]
- Reedijk, J. Why does Cisplatin reach Guanine-n7 with competing s-donor ligands available in the cell? Chem. Rev. 1999, 99, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Pande, P.; Shearer, J.; Yang, J.; Greenberg, W.A.; Rokita, S.E. Alkylation of Nucleic Acids by a Model Quinone Methide. J. Am. Chem. Soc. 1999, 121, 6773–6779. [Google Scholar] [CrossRef]
- Pommier, Y.; Kohlhagen, G.; Kohn, K.W.; Leteurtre, F.; Wani, M.C.; Wall, M.E. Interaction of an alkylating camptothecin derivative with a DNA base at topoisomerase I-DNA cleavage sites. Proc. Natl. Acad. Sci. USA 1995, 92, 8861–8865. [Google Scholar] [CrossRef] [Green Version]
- Veldhuyzen, W.F.; Lam, Y.F.; Rokita, S.E. 2′-Deoxyguanosine reacts with a model quinone methide at multiple sites. Chem. Res. Toxicol. 2001, 14, 1345–1351. [Google Scholar] [CrossRef]
- Wang, H.; Wahi, M.S.; Rokita, S.E. Immortalizing a transient electrophile for DNA cross-linking. Angew. Chem. Int. Ed. Engl. 2008, 47, 1291–1293. [Google Scholar] [CrossRef]
- Svoboda, J.; Konig, B. Templated photochemistry: Toward catalysts enhancing the efficiency and selectivity of photoreactions in homogeneous solutions. Chem. Rev. 2006, 106, 5413–5430. [Google Scholar] [CrossRef]
- Bocian, W.; Kawęcki, R.; Bednarek, E.; Sitkowski, J.; Pietrzyk, A.; Williamson, M.P.; Hansen, P.E.; Kozerski, L. Multiple binding modes of the camptothecin family to DNA oligomers. Chem. Eur. J. 2004, 10, 5776–5787. [Google Scholar] [CrossRef]
- Kozerski, L.; Mazurek, A.P.; Kawęcki, R.; Bocian, W.; Krajewski, P.; Bednarek, E.; Sitkowski, J.; Williamson, M.P.; Moir, A.J.; Hansen, P.E. A nicked duplex decamer DNA with a PEG(6) tether. Nucleic Acids Res 2001, 29, 1132–1143. [Google Scholar] [CrossRef] [Green Version]
- Naumczuk, B.; Hyz, K.; Kawęcki, R.; Bocian, W.; Bednarek, E.; Sitkowski, J.; Wielgus, E.; Kozerski, L. DOSY NMR and MALDI-TOF evidence of covalent binding the DNA duplex by trimethylammonium salts of topotecan upon near UV irradiation. Magn. Reson. Chem. 2015, 53, 565–571. [Google Scholar] [CrossRef]
- Kozerski, L.; Kawęcki, R.; Naumczuk, B.; Hyz, K.; Bocian, W.; Bednarek, E.; Sitkowski, J.; Wiktorska, K.; Lubelska, K. Derivatives of Camptothecin, A Method of Producing Them and Their Use. WO 2014/064654 A1, 1 May 2014. [Google Scholar]
- Naumczuk, B.; Kawęcki, R.; Bocian, W.; Bednarek, E.; Sitkowski, J.; Kozerski, L. Preliminary study of mechanism of action of SN38 derivatives. Physicochemical data, evidence of interaction and alkylation of DNA octamer d(GCGATCGC)2. Magn. Reson. Chem. 2017, 55, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Naumczuk, B.; Wiktorska, K.; Lubelska, K.; Kawęcki, R.; Bocian, W.; Bednarek, E.; Sitkowski, J.; Chilmonczyk, Z.; Kozerski, L. New generation of camptothecin derivatives spontaneously alkylating DNA. New J. Chem. 2016, 40, 7978–7985. [Google Scholar] [CrossRef]
- Naumczuk, B.; Kawęcki, R.; Sitkowski, J.; Bocian, W.; Bednarek, E.; Kozerski, L. Spontaneous 2′-deoxyguanosine alkylation by a new generation of topoisomerase I inhibitors of the camptothecin family. New J. Chem. 2016, 40, 3010–3013. [Google Scholar] [CrossRef]
- Naumczuk, B.; Kawęcki, R.; Bocian, W.; Bednarek, E.; Sitkowski, J.; Kozerski, L. Regioselective alkylation reaction of the 2′deoxycitidine with 9-aminomethyl derivatives of SN38. J. Mol. Struct. 2019, 1176, 298–302. [Google Scholar] [CrossRef]
- Naumczuk, B.; Bocian, W.; Sitkowski, J.; Kawęcki, R.; Kozerski, L. Solvent-dependent regioselectivity of 2′-deoxyadenosine alkylation by 9-aminomethyl derivatives of SN38. New J. Chem. 2019, 43, 18975–18978. [Google Scholar] [CrossRef]
- Rivory, L.P.; Robert, J. Pharmacology of camptothecin and its derivatives. Bull. Cancer 1995, 82, 265–285. [Google Scholar]
- Pommier, Y.; Marchand, C. Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discov. 2011, 11, 25–36. [Google Scholar] [CrossRef]
- Coletta, A.; Desideri, A. Role of the protein in the DNA sequence specificity of the cleavage site stabilized by the camptothecin topoisomerase IB inhibitor: A metadynamics study. Nucleic Acids Res. 2013, 41, 9977–9986. [Google Scholar] [CrossRef]
- Antałek, B. Accounting for spin relaxation in quantitative pulse gradient spin echo NMR mixture analysis. J. Am. Chem. Soc. 2006, 128, 8402–8403. [Google Scholar] [CrossRef]
- Antałek, B. Using Pulsed Gradient Spin Echo NMR for Chemical Mixture Analysis: How to Obtain Optimum Results. Concepts Magn. Reson. 2002, 14, 225–258. [Google Scholar] [CrossRef]
- Staker, B.L.; Hjerrild, K.; Feese, M.D.; Behnke, C.A.; Burgin, A.B., Jr.; Stewart, L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 2002, 99, 15387–15392. [Google Scholar] [CrossRef] [Green Version]
- Laco, G.S.; Collins, J.R.; Luke, B.T.; Kroth, H.; Sayer, J.M.; Jerina, D.M.; Pommier, Y. Human topoisomerase I inhibition: Docking camptothecin and derivatives into a structure-based active site model. Biochemistry 2002, 41, 1428–1435. [Google Scholar] [CrossRef]
- Fan, Y.; Weinstein, J.N.; Kohn, K.W.; Shi, L.M.; Pommier, Y. Molecular modeling studies of the DNA-topoisomerase I ternary cleavable complex with camptothecin. J. Med. Chem. 1998, 41, 2216–2226. [Google Scholar] [CrossRef]
- Pelta, M.D.; Morris, G.A.; Stchedroff, M.J.; Hammond, S.J. A one-shot sequence for high-resolution diffusion-ordered spectroscopy. Magn. Reson. Chem. 2002, 40, S147–S152. [Google Scholar] [CrossRef]
- Waldeck, A.R.; Kuchel, P.W.; Lennon, A.J.; Chapman, B.E. NMR diffusion measurements to characterise membrane transport and solute binding. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 30, 39–68. [Google Scholar] [CrossRef]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; MGaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Luo, R.; David, L.; Gilson, M.K. Accelerated Poisson-Boltzmann calculations for static and dynamic systems. J. Comput. Chem. 2002, 23, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J. MOPAC2016, Version: 17181L. Available online: http://OpenMOPAC.net (accessed on 5 April 2021).
- Klamt, A.; Schuurmann, G. Cosmo—A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Maxwell, A.; Burton, N.P.; O’Hagan, N. High-throughput assays for DNA gyrase and other topoisomerases. Nucleic Acids Res. 2006, 34, e104. [Google Scholar] [CrossRef] [Green Version]
- Seol, Y.; Zhang, H.; Agama, K.; Lorence, N.; Pommier, Y.; Neuman, K.C. Single-Molecule Supercoil Relaxation Assay as a Screening Tool to Determine the Mechanism and Efficacy of Human Topoisomerase IB Inhibitors. Mol. Cancer 2015, 14, 2552–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathijssen, R.H.; van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Concentration (mM) | 1 (T7-CH3) Integral | 2 (19-CH3) Integral | Di of 1 (10−10 m2 s−1) | Di of 2 (10−10 m2 s−−1) | Ka (mM−1) | % of Complex | |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | |||||||
| Complex of 1 + 2 after 5 days | 1.5 a | 3.25 b | 3 | 6.5 | 1.147 | 1.475 | 4.07 | 41% c 89% d |
| Compound 2 | - | 2.0 | - | - | - | 3.07 | - | - |
| Decamer 1 | 0.96 | - | - | - | 1.13 | - | - | - |
| Compound 2 | Free; δ (ppm) | Complex 1 + 2; δ (ppm) | Δ (ppm) | Δ (Hz) |
|---|---|---|---|---|
| 19-CH3 | 0.96 | 0.993 | 0.033 | 16.5 |
| 23-CH3 | 1.50 | 1.377 | −0.123 | −61.5 |
| 18-CH2 | 1.95 | 1.908 | −0.042 | −21 |
| N–CH3 | 2.82 | 2.894 | 0.074 | 37 |
| 22-CH2 | 3.09 | 2.911 | −0.179 | −89.5 |
| 24-CH2 | 4.54 | 4.512 | −0.028 | −14 |
| 24-CH2 | 4.62 | 4.512 | −0.108 | −54 |
| 5-CH2 | 5.02 | 4.65 | −0.370 | −185 |
| 17-CH2 | 5.36 | 5.259 | −0.101 | −50.5 |
| 17-CH2 | 5.51 | 5.259 | −0.251 | −125.5 |
| 14-CH | 7.18 | 7.041 | −0.139 | −69.5 |
| 11-CH | 7.32 | 7.081 | −0.239 | −119.5 |
| 12-CH | 7.71 | 7.333 | −0.377 | −188.5 |
| Base | H1’ | H2’ | H2” | H3’ | H4’ | H5’, H5” | H6/8 | H2/5/Me |
|---|---|---|---|---|---|---|---|---|
| T4 | −0.008 | −0.026 | −0.048 | 0.014 | −0.035 | -/−0.003 | −0.005 | −0.023 |
| T5 | 0.020 | 0.052 | −0.046 | −0.006 | −0.009 | −0.001/- | 0.067 | −0.012 |
| nick | ----------- | ---------- | ---------- | ----------- | ---------- | ------------------ | ----------- | ----------- |
| G6 | −0.342 | −0.284 | −0.068 | 0.017 | −0.037 | −0.076/−0.222 | 0.020 | - |
| T7 | −0.045 | −0.022 | −0.037 | −0.018 | −0.082 | -/0.013 | −0.204 | −0.025 |
| A14 | −0.044 | −0.035 | −0.128 | −0.118 | −0.057 | -/- | −0.093 | −0.058 |
| C15 | 0.170 | 0.010 | −0.199 | - | 0.021 | -/0.000 | 0.027 | −0.245 |
| A16 | −0.230 | 0.024 | −0.093 | −0.043 | 0.029 | -/- | −0.224 | −0.204 |
| A17 | −0.040 | −0.048 | −0.031 | −0.026 | −0.015 | -/0.073 | −0.069 | −0.081 |
| Deriv. 2 | DNA 1 | NOE (a) | Structure 1 (b) | Structure 2 (b) | Structure 3 (b) | Structure 4 (b) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <6Å | <5Å | <4Å | <3Å | <6Å | <5Å | <4Å | <3Å | <6Å | <5Å | <4Å | <3Å | <6Å | <5Å | <4Å | <3Å | |||
| 11 | C15H2” | 8.0% | ++++ | ++++ | ++++ | ++ | ++++ | +++ | + | * | ||||||||
| 12 | A16H8 | 8.4% | ++++ | ++++ | +++ | +++ | + | |||||||||||
| 14 | A16H2 | 23.5% | ++++ | ++++ | ++++ | +++ | ++ | ++++ | ++++ | ++++ | + | |||||||
| 17a | G6H1′ | 11.0% | ++++ | ++++ | ++++ | +++ | + | |||||||||||
| 17b | ++++ | ++++ | +++ | . | ++++ | ++ | * | |||||||||||
| 17a | G6H4′ | 16.2% | ++++ | ++++ | ++++ | +++ | ++ | . | ||||||||||
| 17b | ++++ | ++++ | ++++ | + | ++++ | ++ | ++ | * | ||||||||||
| 17a | T5H1′ | 6.9% | ++++ | +++ | * | + | * | |||||||||||
| 17b | ++++ | ++++ | +++ | + | + | |||||||||||||
| 17a | T5H2” | 14.8% | ++++ | ++++ | +++ | +++ | ++ | + | * | |||||||||
| 17b | ++++ | ++++ | ++++ | +++ | ++++ | +++ | + | |||||||||||
| 17a | T5H2′ | 9.4% | ++++ | +++ | + | ++++ | +++ | ++ | * | |||||||||
| 17b | ++++ | ++++ | ++++ | ++ | ++++ | ++++ | + | |||||||||||
| 17a | T5H6 | 4.7% | ++++ | + | ++++ | ++++ | ++ | |||||||||||
| 17b | ++++ | ++++ | +++ | ++++ | ++++ | * | ||||||||||||
| 18 | G6H1’ | 12.3% | ++++ | ++++ | +++ | ++ | * | + | * | |||||||||
| 18 | G6H2” | 9.1% | +++ | ++ | * | |||||||||||||
| 18 | G6H4′ | 11.7% | ++++ | +++ | ++ | + | * | |||||||||||
| 18 | T7H4′ | 5.7% | ++++ | +++ | ++ | * | * | * | ||||||||||
| 18 | T7H5” | 4.7% | ++++ | +++ | + | * | ||||||||||||
| 18 | T7H5′ | 11.5% | ++++ | ++++ | +++ | ++ | * | * | ||||||||||
| 19 | A16H2 | 2.3% | +++ | * | ++++ | ++++ | ++ | * | ||||||||||
| 19 | G6H1′ | 5.4% | ++++ | +++ | ++ | * | * | |||||||||||
| 19 | G6H2” | 4.1% | +++ | ++ | * | |||||||||||||
| 19 | G6H4′ | 7.9% | ++++ | +++ | ++ | * | * | |||||||||||
| 19 | T7H4′ | 12.1% | ++++ | ++++ | ++++ | + | * | * | ||||||||||
| 19 | T7H5” | 5.7% | ++++ | +++ | ++ | * | * | |||||||||||
| 19 | T7H5′ | 10.9% | ++++ | ++++ | +++ | ++ | * | * | ||||||||||
| 24 | C15H5 | 5.7% | ++++ | ++++ | ++ | + | ||||||||||||
| N–CH3 | C15H6 | 1.7% | ++ | * | * | |||||||||||||
| Atom Names | Atom Distances in Structure 1 in (Å) | |||
| DNA | Comp. 2 | 100% Cluster a | 50% Cluster a | 25% Cluster a |
| C15-N4 | -CH2NHCH3 | 7.23 ± 12.64 | 3.90 ± 0.33 | 3.62 ± 0.21 |
| A16-N7 | -CH2NHCH3 | 7.53 ± 12.78 | 4.27 ± 0.32 | 4.01 ± 0.25 |
| A16-N6 | -CH2NHCH3 | 8.47 ± 12.79 | 5.12 ± 0.49 | 4.75 ± 0.44 |
| Atom Distances in Structure 3 in (Å) | ||||
| G6-N2 | -CH2NHCH3 | 6.43 ± 10.88 | 4.03 ± 0.32 | 3.76 ± 0.21 |
| A16-N3 | -CH2NHCH3 | 5.51 ± 10.65 | 3.37 ± 0.13 | 3.26 ± 0.09 |
| IC50 (µM) | |||
| CPT | SN38 | 2 | 3 |
| 92.9 ± 16.1 | 40.3 ± 13.17 | 100.7 ± 10.0 | 48.7 ± 8.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bocian, W.; Naumczuk, B.; Urbanowicz, M.; Sitkowski, J.; Bierczyńska-Krzysik, A.; Bednarek, E.; Wiktorska, K.; Milczarek, M.; Kozerski, L. The Mode of SN38 Derivatives Interacting with Nicked DNA Mimics Biological Targeting of Topo I Poisons. Int. J. Mol. Sci. 2021, 22, 7471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147471
Bocian W, Naumczuk B, Urbanowicz M, Sitkowski J, Bierczyńska-Krzysik A, Bednarek E, Wiktorska K, Milczarek M, Kozerski L. The Mode of SN38 Derivatives Interacting with Nicked DNA Mimics Biological Targeting of Topo I Poisons. International Journal of Molecular Sciences. 2021; 22(14):7471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147471
Chicago/Turabian StyleBocian, Wojciech, Beata Naumczuk, Magdalena Urbanowicz, Jerzy Sitkowski, Anna Bierczyńska-Krzysik, Elżbieta Bednarek, Katarzyna Wiktorska, Małgorzata Milczarek, and Lech Kozerski. 2021. "The Mode of SN38 Derivatives Interacting with Nicked DNA Mimics Biological Targeting of Topo I Poisons" International Journal of Molecular Sciences 22, no. 14: 7471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147471