
Anti-Inflammatory Influences of Cystic Fibrosis Transmembrane Conductance Regulator Drugs on Lung Inflammation in Cystic Fibrosis


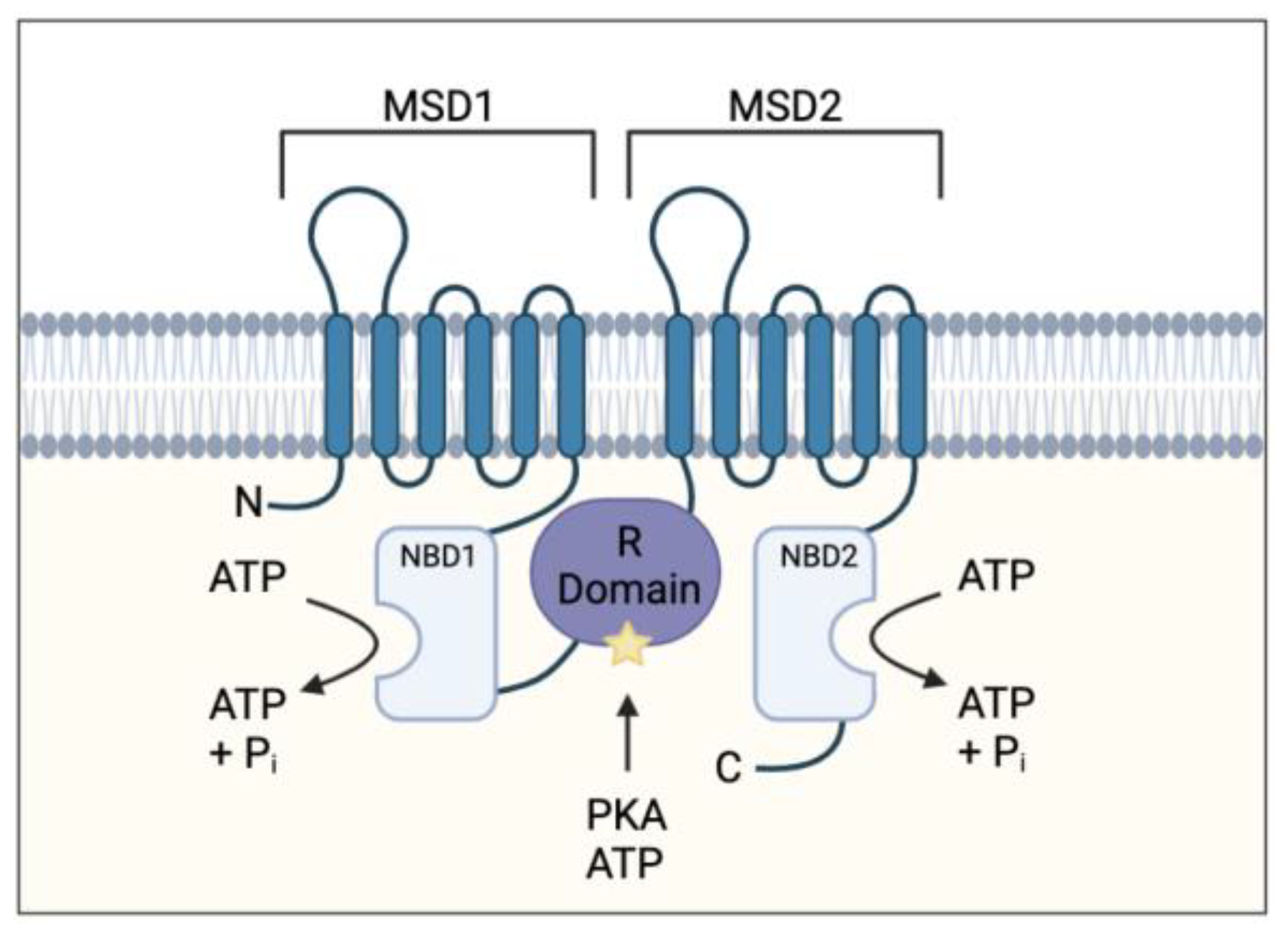{kind=link}
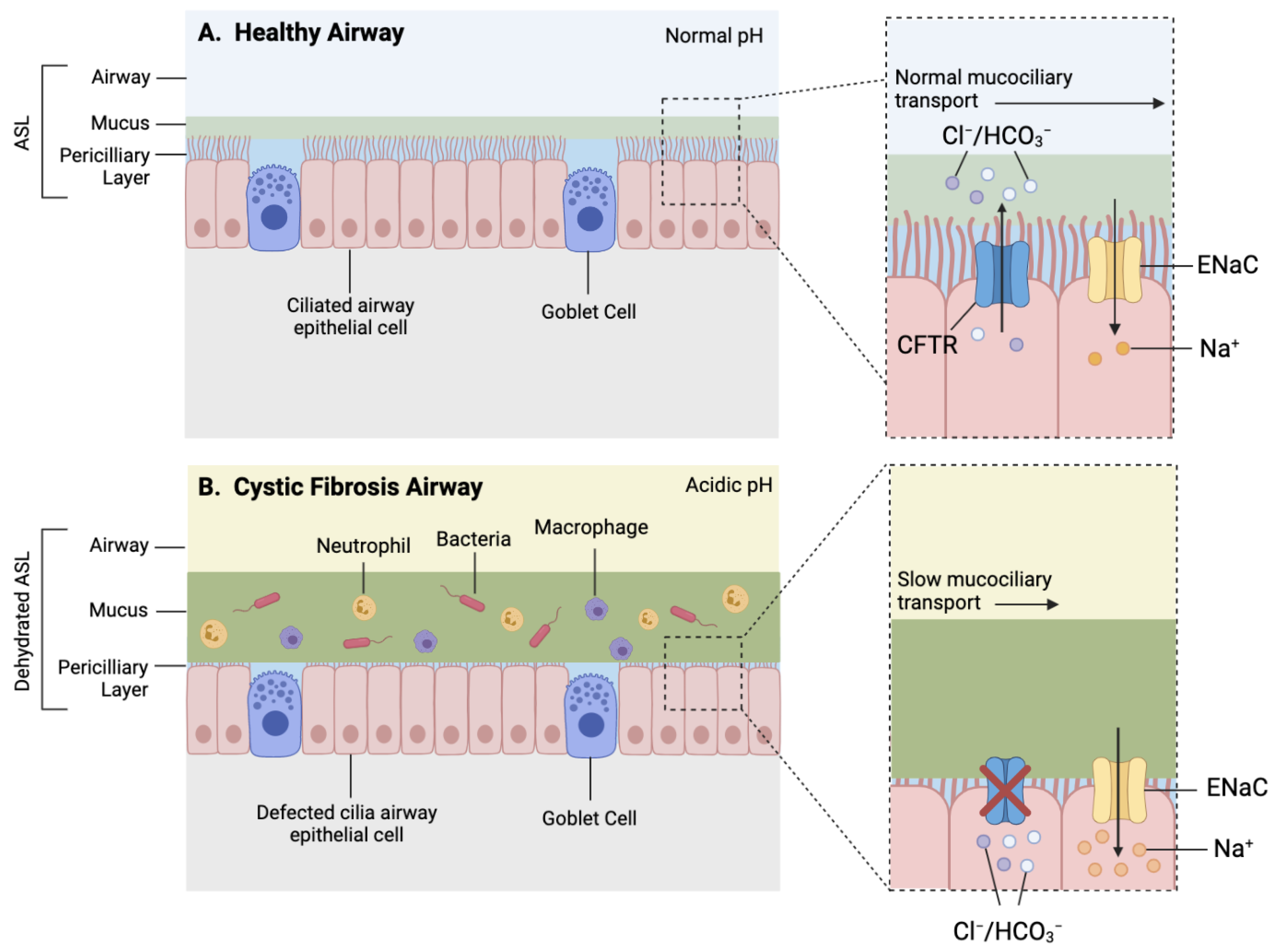{kind=link}
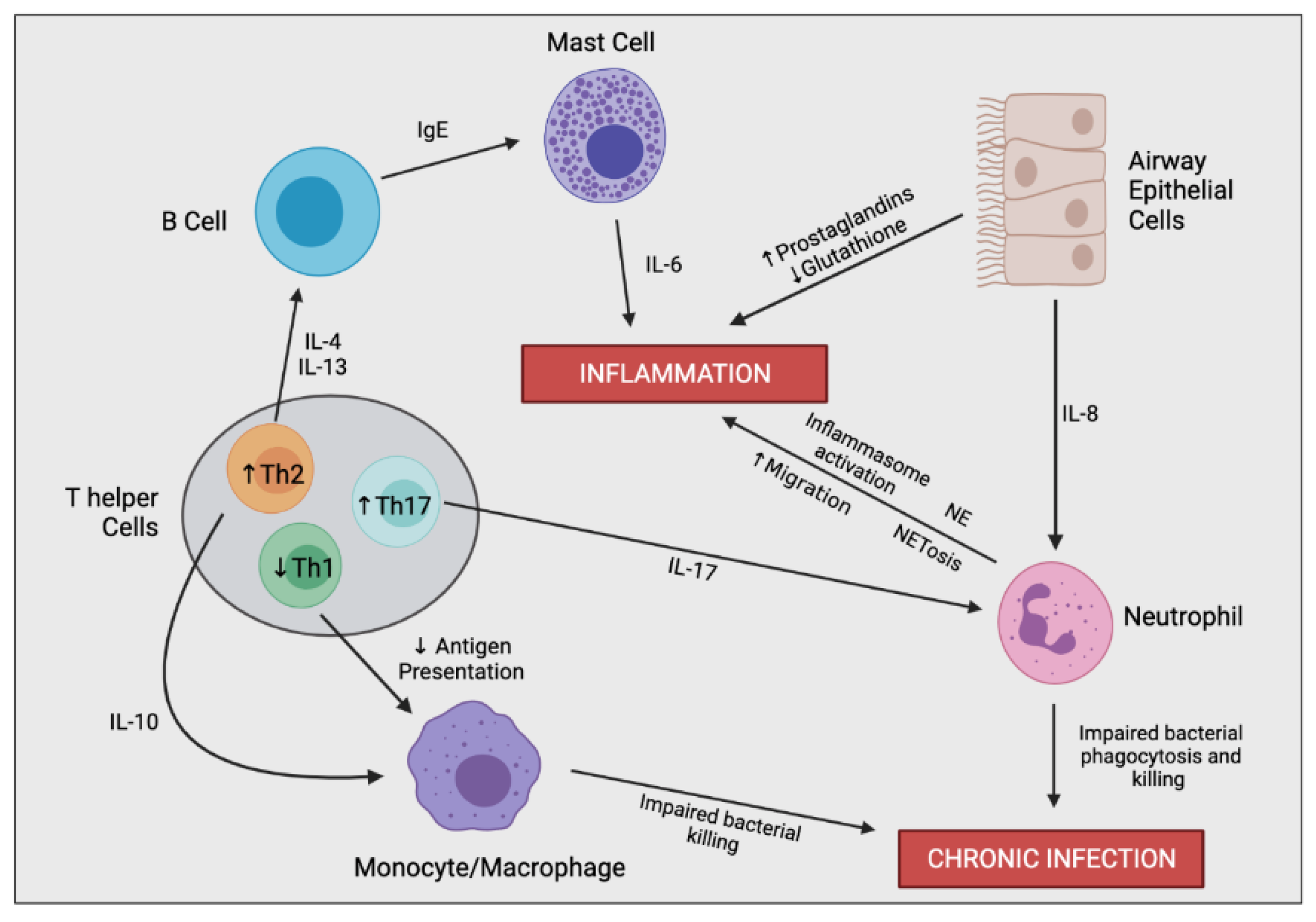{kind=link}
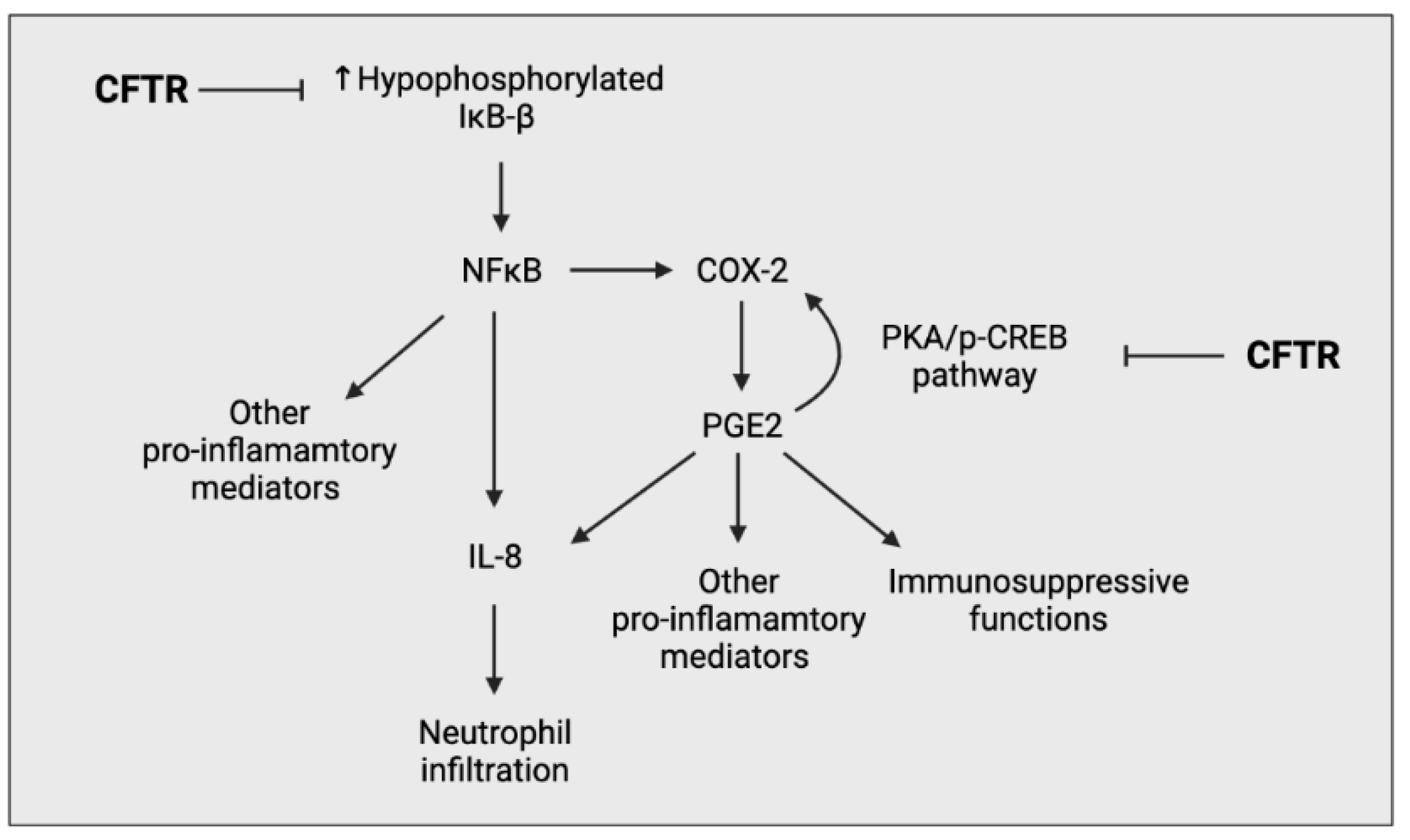{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CF-Induced Lung Inflammation
2.1. Persistent Lung Infections
2.2. Dysregulated Lung Inflammatory Response in CF
2.2.1. Epithelial Cells
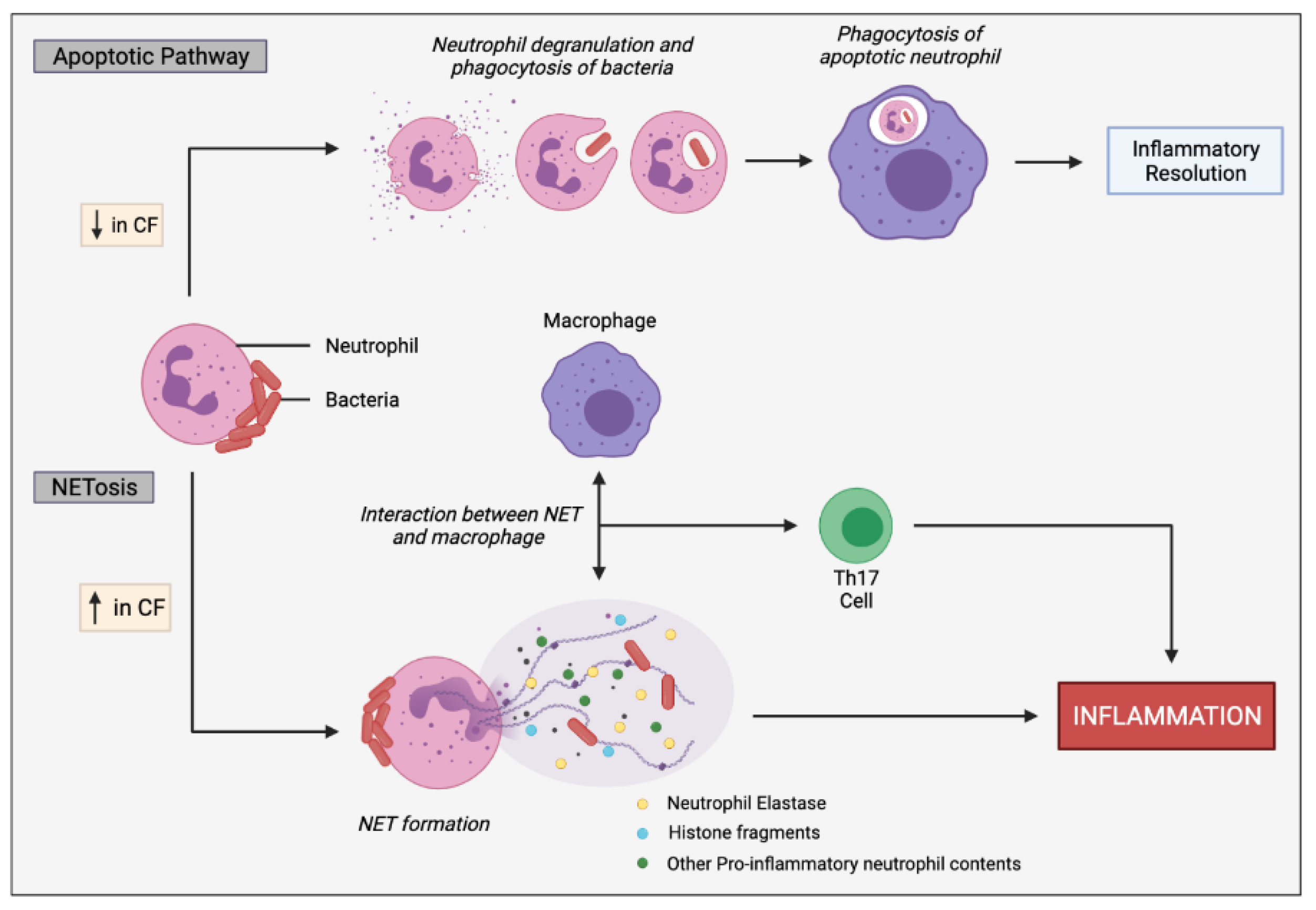
2.2.2. Neutrophils
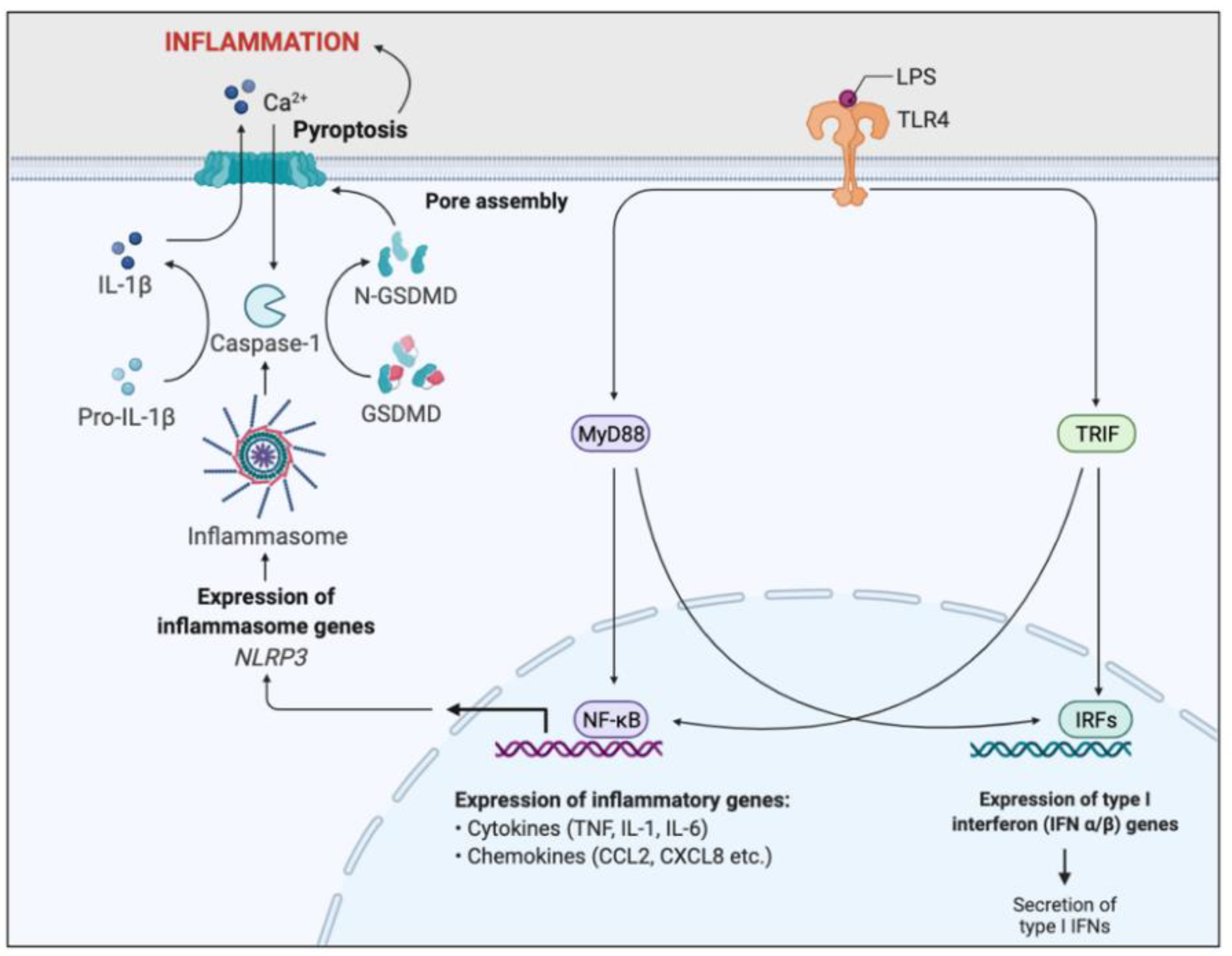
2.2.3. Macrophages
2.2.4. Lymphocytes
3. CFTR Modulators and Inflammation
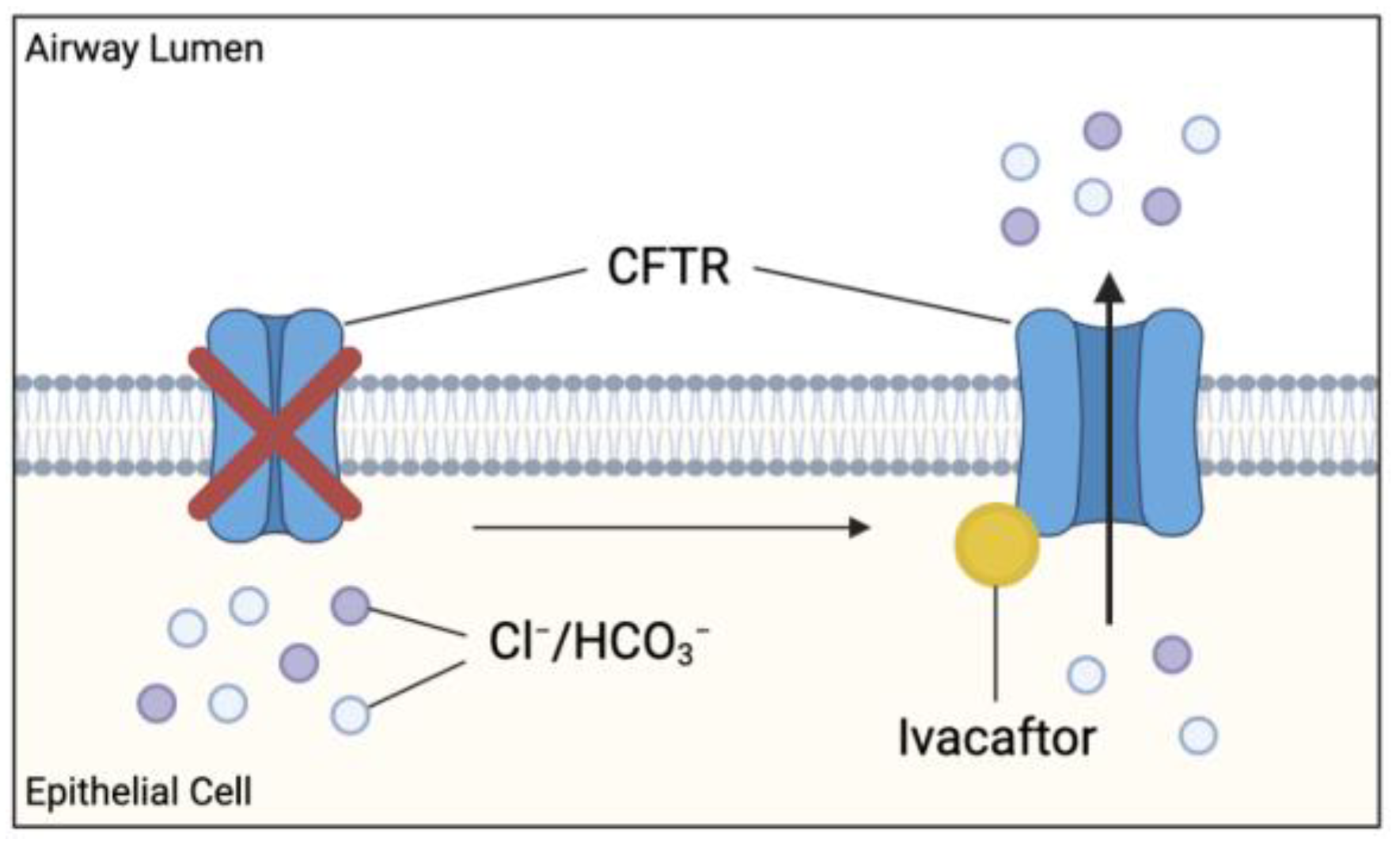
3.1. Ivacaftor
3.2. Ivacaftor Combinations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rang, C.; Keating, D.; Wilson, J.; Kotsimbos, T. Re-imagining cystic fibrosis care: Next generation thinking. Eur. Respir. J. 2020, 55, 1902443. [Google Scholar] [CrossRef]
- McElvaney, O.J.; Wade, P.; Murphy, M.; Reeves, E.P.; McElvaney, N.G. Targeting airway inflammation in cystic fibrosis. Expert Rev. Respir. Med. 2019, 13, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, C.; Cantin, A.M. Cystic Fibrosis: Pathophysiology of Lung Disease. Semin. Respir. Crit. Care Med. 2019, 40, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharm. 2019, 10, 1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, F.; Habgood, M.; Schneider-Futschik, E.K. The Balance between the Safety of Mother, Fetus, and Newborn Undergoing Cystic Fibrosis Transmembrane Conductance Regulator Treatments during Pregnancy. ACS Pharm. Transl. Sci. 2020, 3, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Ghelani, D.P.; Schneider-Futschik, E.K. Emerging Cystic Fibrosis Transmembrane Conductance Regulator Modulators as New Drugs for Cystic Fibrosis: A Portrait of in Vitro Pharmacology and Clinical Translation. ACS Pharm. Transl. Sci. 2020, 3, 4–10. [Google Scholar] [CrossRef]
- Goss, C.H.; Muhlebach, M.S. Review: Staphylococcus aureus and MRSA in cystic fibrosis. J. Cyst. Fibros. 2011, 10, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Pillarisetti, N.; Williamson, E.; Linnane, B.; Skoric, B.; Robertson, C.F.; Robinson, P.; Massie, J.; Hall, G.L.; Sly, P.; Stick, S.; et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am. J. Respir. Crit. Care. Med. 2011, 184, 75–81. [Google Scholar] [CrossRef]
- Koch, C. Early infection and progression of cystic fibrosis lung disease. Pediatr. Pulmonol. 2002, 34, 232–236. [Google Scholar] [CrossRef]
- Goodman, A.L.; Kulasekara, B.; Rietsch, A.; Boyd, D.; Smith, R.S.; Lory, S. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 2004, 7, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; Bye, P.T. Mucociliary clearance in cystic fibrosis. Pediatr. Pulmonol. 2002, 33, 293–306. [Google Scholar] [CrossRef]
- Elborn, S.J. Cystic Fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 351–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Salinas, D.; Nielson, D.W.; Verkman, A.S. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am. J. Physiol. Cell Physiol. 2006, 290, C741–C749. [Google Scholar] [CrossRef] [PubMed]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A. Cystic fibrosis lung inflammation: Early, sustained, and severe. Am. J. Respir. Crit. Care Med. 1995, 151, 939–941. [Google Scholar] [CrossRef]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef] [Green Version]
- Jacquot, J.; Tabary, O.; Le Rouzic, P.; Clement, A. Airway epithelial cell inflammatory signalling in cystic fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 1703–1715. [Google Scholar] [CrossRef]
- Ratner, D.; Mueller, C. Immune responses in cystic fibrosis: Are they intrinsically defective? Am. J. Respir. Cell Mol. Biol. 2012, 46, 715–722. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, X.H.; Chen, H.; Guo, J.H.; Tsang, L.L.; Yu, M.K.; Xu, W.M.; Chan, H.C. CFTR negatively regulates cyclooxygenase-2-PGE(2) positive feedback loop in inflammation. J. Cell Physiol. 2012, 227, 2759–2766. [Google Scholar] [CrossRef]
- Gao, L.; Kim, K.J.; Yankaskas, J.R.; Forman, H.J. Abnormal glutathione transport in cystic fibrosis airway epithelia. Am. J. Physiol. 1999, 277, L113–L118. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.; Stecenko, A.A.; King, G.; Blackwell, T.R.; Brigham, K.L.; Christman, J.W.; Blackwell, T.S. Exaggerated activation of nuclear factor-kappaB and altered IkappaB-beta processing in cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2000, 23, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Roum, J.H.; Buhl, R.; McElvaney, N.G.; Borok, Z.; Crystal, R.G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Ren, C.; Aw, T.Y.; Ookhtens, M.; Kaplowitz, N. Effect of membrane potential and cellular ATP on glutathione efflux from isolated rat hepatocytes. Am. J. Physiol. 1988, 255 Pt 1, G403–G408. [Google Scholar] [CrossRef]
- Lash, L.H.; Jones, D.P. Renal glutathione transport. Characteristics of the sodium-dependent system in the basal-lateral membrane. J. Biol. Chem. 1984, 259, 14508–14514. [Google Scholar] [CrossRef]
- Lindén, A.; Laan, M.; Anderson, G.P. Neutrophils, interleukin-17A and lung disease. Eur. Respir. J. 2005, 25, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016, 28, 137–145. [Google Scholar] [CrossRef]
- Law, S.M.; Gray, R.D. Neutrophil extracellular traps and the dysfunctional innate immune response of cystic fibrosis lung disease: A review. J. Inflamm. 2017, 14, 29. [Google Scholar] [CrossRef]
- Dittrich, A.S.; Kühbandner, I.; Gehrig, S.; Rickert-Zacharias, V.; Twigg, M.; Wege, S.; Taggart, C.; Herth, F.; Schultz, C.; Mall, M.A. Elastase activity on sputum neutrophils correlates with severity of lung disease in cystic fibrosis. Eur. Respir. J. 2018, 51, 1701910. [Google Scholar] [CrossRef] [Green Version]
- Owen, C.A.; Campbell, M.A.; Sannes, P.L.; Boukedes, S.S.; Campbell, E.J. Cell surface-bound elastase and cathepsin G on human neutrophils: A novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J. Cell Biol. 1995, 131, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; Hardisty, G.; Regan, K.H.; Smith, M.; Robb, C.T.; Duffin, R.; Mackellar, A.; Felton, J.M.; Paemka, L.; McCullagh, B.N.; et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax 2018, 73, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storisteanu, D.M.; Pocock, J.M.; Cowburn, A.S.; Juss, J.K.; Nadesalingam, A.; Nizet, V.; Chilvers, E.R. Evasion of Neutrophil Extracellular Traps by Respiratory Pathogens. Am. J. Respir. Cell Mol. Biol. 2017, 56, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Pier, G.B. Pseudomonas aeruginosa lipopolysaccharide: A major virulence factor, initiator of inflammation and target for effective immunity. Int. J. Med. Microbiol. 2007, 297, 277–295. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Lévêque, M.; Le Trionnaire, S.; Del Porto, P.; Martin-Chouly, C. The impact of impaired macrophage functions in cystic fibrosis disease progression. J. Cyst. Fibros. 2017, 16, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Bruscia, E.M.; Zhang, P.X.; Ferreira, E.; Caputo, C.; Emerson, J.W.; Tuck, D.; Krause, D.S.; Egan, M.E. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator−/− mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Saini, Y.; Dang, H.; Livraghi-Butrico, A.; Kelly, E.J.; Jones, L.C.; O’Neal, W.K.; Boucher, R.C. Gene expression in whole lung and pulmonary macrophages reflects the dynamic pathology associated with airway surface dehydration. BMC Genom. 2014, 15, 726. [Google Scholar] [CrossRef] [Green Version]
- Zaman, M.M.; Gelrud, A.; Junaidi, O.; Regan, M.M.; Warny, M.; Shea, J.C.; Kelly, C.; O’Sullivan, B.P.; Freedman, S.D. Interleukin 8 secretion from monocytes of subjects heterozygous for the deltaF508 cystic fibrosis transmembrane conductance regulator gene mutation is altered. Clin. Diagn. Lab. Immunol. 2004, 11, 819–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruscia, E.M.; Zhang, P.X.; Satoh, A.; Caputo, C.; Medzhitov, R.; Shenoy, A.; Egan, M.E.; Krause, D.S. Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis. J. Immunol. 2011, 186, 6990–6998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Shrestha, C.L.; Wisniewski, B.L.; Pham, H.; Hou, X.; Li, W.; Dong, Y.; Kopp, B.T. Consequences of CRISPR-Cas9-Mediated CFTR Knockout in Human Macrophages. Front. Immunol. 2020, 11, 1871. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Brown, M.E.; Deriy, L.V.; Li, C.; Szeto, F.L.; Chen, Y.; Huang, P.; Tong, J.; Naren, A.P.; Bindokas, V.; et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol. 2006, 8, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Barriere, H.; Bagdany, M.; Bossard, F.; Okiyoneda, T.; Wojewodka, G.; Gruenert, D.; Radzioch, D.; Lukacs, G.L. Revisiting the role of cystic fibrosis transmembrane conductance regulator and counterion permeability in the pH regulation of endocytic organelles. Mol. Biol. Cell 2009, 20, 3125–3141. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Hazlett, H.F.; Hampton, T.H.; Aridgides, D.S.; Armstrong, D.A.; Dessaint, J.A.; Mellinger, D.L.; Nymon, A.B.; Ashare, A. Altered iron metabolism in cystic fibrosis macrophages: The impact of CFTR modulators and implications for Pseudomonas aeruginosa survival. Sci. Rep. 2020, 10, 10935. [Google Scholar] [CrossRef]
- Bruscia, E.M.; Bonfield, T.L. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. J. Innate. Immun. 2016, 8, 550–563. [Google Scholar] [CrossRef]
- Moreau-Marquis, S.; Bomberger, J.M.; Anderson, G.G.; Swiatecka-Urban, A.; Ye, S.; O’Toole, G.A.; Stanton, B.A. The DeltaF508-CFTR mutation results in increased biofilm formation by Pseudomonas aeruginosa by increasing iron availability. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L25–L37. [Google Scholar] [CrossRef] [Green Version]
- Tiringer, K.; Treis, A.; Fucik, P.; Gona, M.; Gruber, S.; Renner, S.; Dehlink, E.; Nachbaur, E.; Horak, F.; Jaksch, P.; et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 2013, 187, 621–629. [Google Scholar] [CrossRef]
- Dubin, P.J.; Kolls, J.K. IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L519–L528. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.L.; Regamey, N.; Brown, S.; Bush, A.; Lloyd, C.M.; Davies, J.C. The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2011, 184, 252–258. [Google Scholar] [CrossRef]
- Casaulta, C.; Schöni, M.H.; Weichel, M.; Crameri, R.; Jutel, M.; Daigle, I.; Akdis, M.; Blaser, K.; Akdis, C.A. IL-10 controls Aspergillus fumigatus- and Pseudomonas aeruginosa-specific T-cell response in cystic fibrosis. Pediatr. Res. 2003, 53, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Moss, R.B.; Hsu, Y.P.; Olds, L. Cytokine dysregulation in activated cystic fibrosis (CF) peripheral lymphocytes. Clin. Exp. Immunol. 2000, 120, 518–525. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Cahalan, M.D.; Chandy, K.G. The functional network of ion channels in T lymphocytes. Immunol. Rev. 2009, 231, 59–87. [Google Scholar] [CrossRef] [Green Version]
- Kayani, K.; Mohammed, R.; Mohiaddin, H. Cystic Fibrosis-Related Diabetes. Front. Endocrinol. 2018, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Schwarzenberg, S.J. Pancreatic insufficiency in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16 (Suppl. 2), S70–S78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, A.; Komaki, Y.; Komaki, F.; Micic, D.; Zullow, S.; Sakuraba, A. Risk of gastrointestinal cancers in patients with cystic fibrosis: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 758–767. [Google Scholar] [CrossRef]
- Zolin, A.; McKone, E.; Van Rens, J.; Fox, A.; Iansa, P.; Preftitsi, A. ECFSPR annual report 2013. Eur. Cyst. Fibros. Soc. 2016. Available online: https://www.ecfs.eu/projects/ecfs-patient-registry/annual-reports (accessed on 15 July 2021).
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Hisert; Birkland, T.P.; Schoenfelt, K.Q.; Long, M.E.; Grogan, B.; Carter, S.; Liles, W.C.; McKone, E.F.; Becker, L.; Manicone, A.M. Ivacaftor decreases monocyte sensitivity to interferon-γ in people with cystic fibrosis. ERJ Open Res. 2020, 6, 00318–2019. [Google Scholar] [CrossRef]
- Barnaby, R.; Koeppen, K.; Nymon, A.; Hampton, T.H.; Berwin, B.; Ashare, A.; Stanton, B.A. Lumacaftor (VX-809) restores the ability of CF macrophages to phagocytose and kill Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L432–L438. [Google Scholar] [CrossRef] [Green Version]
- Bratcher, P.E.; Rowe, S.M.; Reeves, G.; Roberts, T.; Szul, T.; Harris, W.T.; Tirouvanziam, R.; Gaggar, A. Alterations in blood leukocytes of G551D-bearing cystic fibrosis patients undergoing treatment with ivacaftor. J. Cyst. Fibros. 2016, 15, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, S.M.; McColley, S.A.; Rietschel, E.; Li, X.; Bell, S.C.; Konstan, M.W.; Marigowda, G.; Waltz, D.; Boyle, M.P.; Group, V.X.S. Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del-CFTR. Ann. Am. Thorac. Soc. 2017, 14, 213–219. [Google Scholar] [CrossRef]
- Zhang, S.; Shrestha, C.L.; Kopp, B.T. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Schneider, E.K.; Reyes-Ortega, F.; Li, J.; Velkov, T. Can Cystic Fibrosis Patients Finally Catch a Breath With Lumacaftor/Ivacaftor? Clin. Pharm. 2017, 101, 130–141. [Google Scholar] [CrossRef] [Green Version]
- Schneider, E.K. Cytochrome P450 3A4 Induction: Lumacaftor versus Ivacaftor Potentially Resulting in Significantly Reduced Plasma Concentration of Ivacaftor. Drug Metab. Lett. 2018, 12, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Hanafin, P.; Sermet-Gaudelus, I.; Griese, M.; Kappler, M.; Ellemunter, H.; Tan, M.; Schwarz, C.; Wilson, J.; Velkov, T.; Rao, G.G.; et al. Insights into patient variability. during ivacaftor-lumacaftor therapy in cystic fibrosis. Front. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Kopp, B.T.; Fitch, J.; Jaramillo, L.; Shrestha, C.L.; Robledo-Avila, F.; Zhang, S.; Palacios, S.; Woodley, F.; Hayes, D., Jr.; Partida-Sanchez, S.; et al. Whole-blood transcriptomic responses to lumacaftor/ivacaftor therapy in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Gentzsch, M.; Cholon, D.M.; Quinney, N.L.; Boyles, S.E.; Martino, M.E.B.; Ribeiro, C.M.P. The cystic fibrosis airway milieu enhances rescue of F508del in a pre-clinical model. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Keown, K.; Brown, R.; Doherty, D.F.; Houston, C.; McKelvey, M.C.; Creane, S.; Linden, D.; McAuley, D.F.; Kidney, J.C.; Weldon, S.; et al. Airway Inflammation and Host Responses in the Era of CFTR Modulators. Int. J. Mol. Sci. 2020, 21, 6379. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harwood, K.H.; McQuade, R.M.; Jarnicki, A.; Schneider-Futschik, E.K. Anti-Inflammatory Influences of Cystic Fibrosis Transmembrane Conductance Regulator Drugs on Lung Inflammation in Cystic Fibrosis. Int. J. Mol. Sci. 2021, 22, 7606. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147606
Harwood KH, McQuade RM, Jarnicki A, Schneider-Futschik EK. Anti-Inflammatory Influences of Cystic Fibrosis Transmembrane Conductance Regulator Drugs on Lung Inflammation in Cystic Fibrosis. International Journal of Molecular Sciences. 2021; 22(14):7606. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147606
Chicago/Turabian StyleHarwood, Kiera H., Rachel M. McQuade, Andrew Jarnicki, and Elena K. Schneider-Futschik. 2021. "Anti-Inflammatory Influences of Cystic Fibrosis Transmembrane Conductance Regulator Drugs on Lung Inflammation in Cystic Fibrosis" International Journal of Molecular Sciences 22, no. 14: 7606. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147606