
Comprehensive Evaluation of Biological Effects of Pentathiepins on Various Human Cancer Cell Lines and Insights into Their Mode of Action

 , , and
, , and
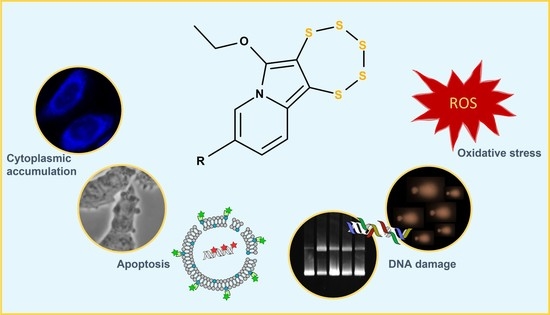
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.1.1. Synthesis of 11-methoxy-2-(4-methoxyphenyl)-3H-[1,2,3,4,5] pentathiepino[6′,7′:3,4]pyrrolo[1,2-a]pyrrolo[2,3-e]pyrazine (1)
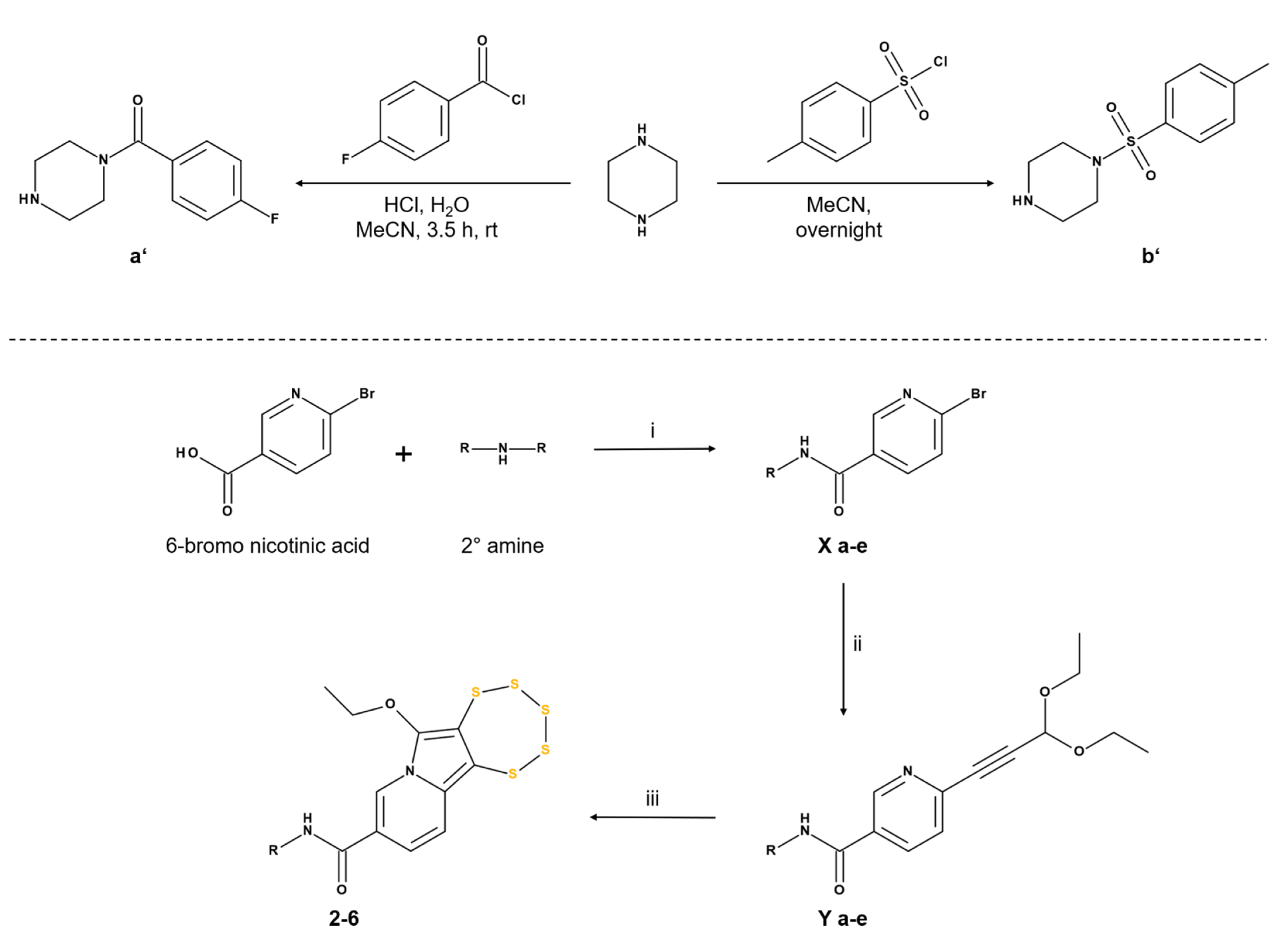
2.1.2. Synthesis of Nicotinamide-Fused Pentathiepins (2–6)
2.2. Biology
2.2.1. Inhibition of Glutathione Peroxidase 1
2.2.2. Cytotoxic and Antiproliferative Activity
2.2.3. GPx1 and Catalase Expression in Selected Cancer Cell Lines
2.2.4. Generation of Intracellular ROS
2.2.5. Induction of DNA Strand Breaks
2.2.6. Damage of Genomic DNA
2.2.7. Intracellular Distribution of Pentathiepin 1
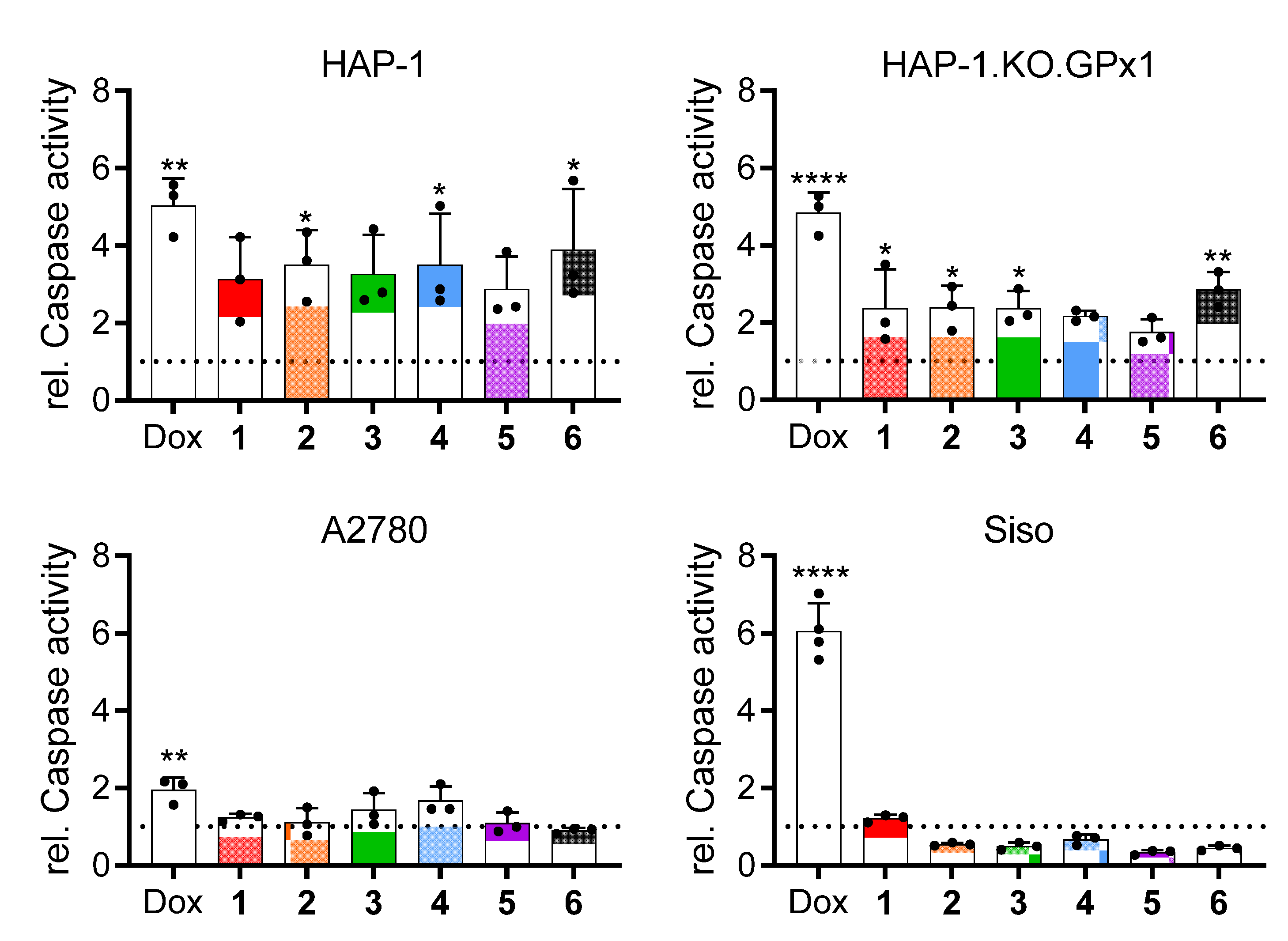
2.2.8. Induction of Apoptosis
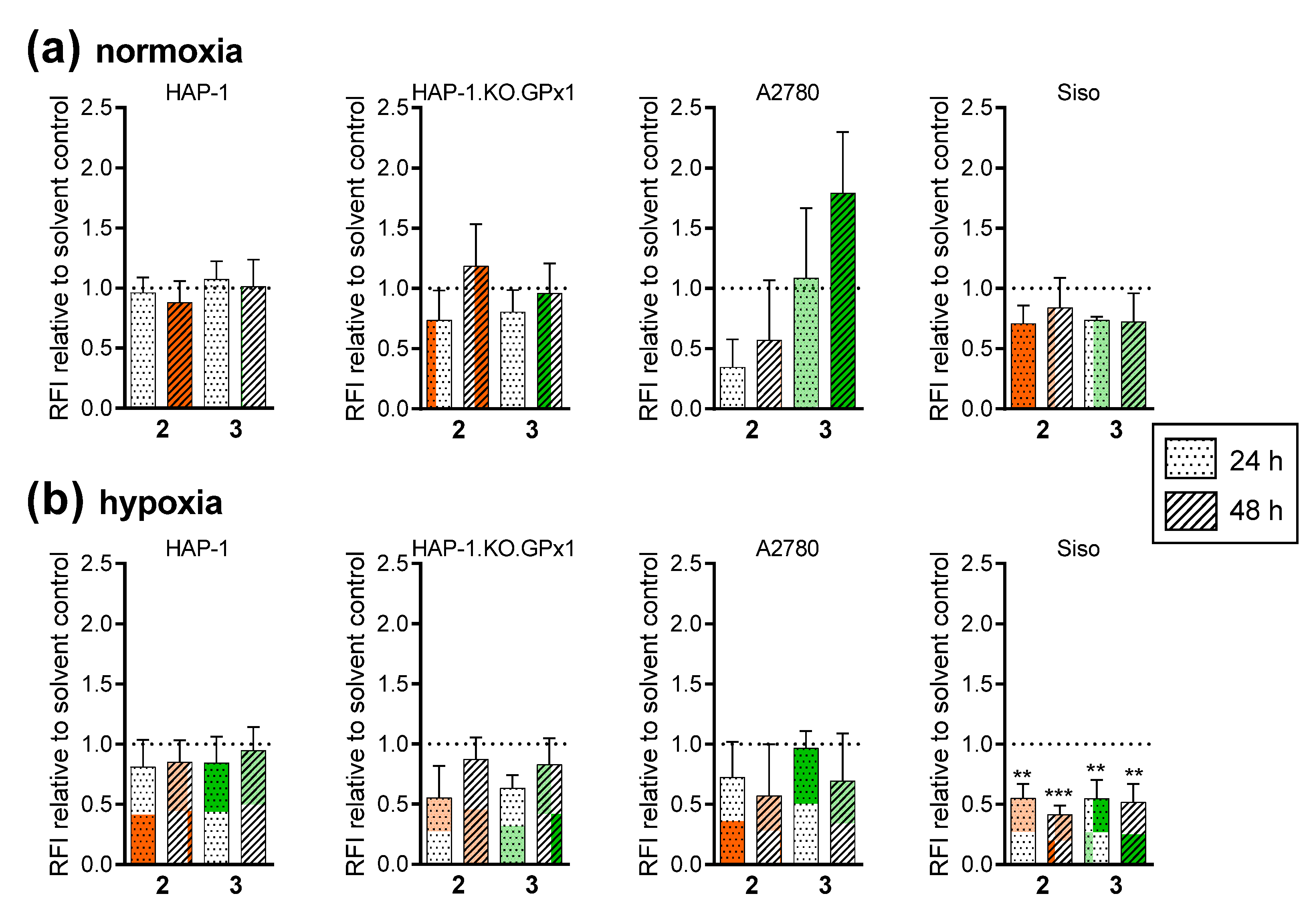
2.2.9. No Evidence for Ferroptosis
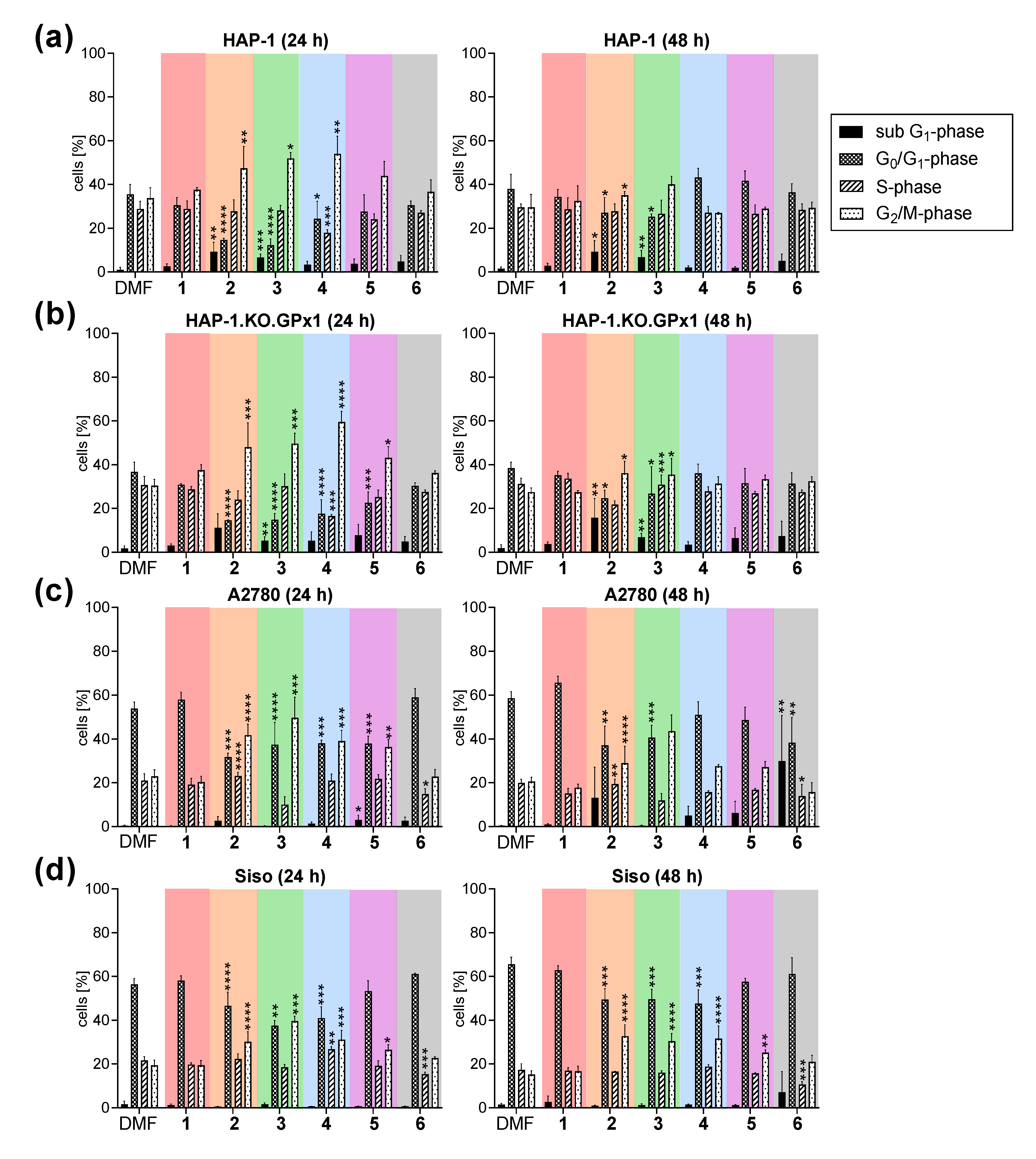
2.2.10. Influence on Cell Cycle Progression
2.2.11. Structure–Activity Relationships
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. General Procedure for Sonogashira Cross-Coupling Reaction
1: (D) 2-(3,3-Diethoxypropynyl)-6-(4-methoxyphenyl)-5H-pyrrolo[2,3-b]pyrazine
2: (Y a) (6-(3,3-Diethoxypropynyl)pyridin-3-yl)(piperidinyl)methanone
3: (Y b) (6-(3,3-Diethoxypropynyl)pyridin-3-yl)(morpholino)methanone
4: (Y c) 6-(3,3-Diethoxypropynyl)-N,N’-diethylnicotinamide
5: (Y d) (4-(6-(3,3-Diethoxypropynyl)nicotinoyl)piperazinyl)(4-fluorophenyl)methanone
6: (Y e) (6-(3,3-Diethoxypropynyl)pyridin-3-yl)(4-tosylpiperazinyl)methanone
3.1.3. Synthesis of Pentathiepins
1: 11-Ethoxy-2-(4-methoxyphenyl)-3H-[1,2,3,4,5]pentathiepino[6′,7′:3,4]pyrrolo[1,2-a]pyrrolo[2,3-e]pyrazine
2: (6-Ethoxy-[1,2,3,4,5]pentathiepino[6,7-a]indolizin-9-yl)(piperidinyl)methanone
3: (6-Ethoxy-[1,2,3,4,5]pentathiepino[6,7-a]indolizin-9-yl)(morpholino)methanone
4: 6-Ethoxy-N,N’-diethyl-[1,2,3,4,5]pentathiepino[6,7-a]indolizine-9-carboxamide
5: (6-Ethoxy-[1,2,3,4,5]pentathiepino[6,7-a]indolizin-9-yl)(4-(4-fluorobenzoyl)piperazinyl)methanone
6: (6-Ethoxy-[1,2,3,4,5]pentathiepino[6,7-a]indolizin-9-yl)(4-tosylpiperazinyl)methanone
3.2. Biology
3.2.1. Materials
3.2.2. Cell Lines and Culturing
3.2.3. GPx1 Enzyme Activity Assay
3.2.4. Enzymatic Assays for the Activity of Glutathione Reductase and Catalase
3.2.5. MTT Assay
3.2.6. Adapted MTT Assay to Assess the Influence of Ferroptosis
3.2.7. Crystal Violet Proliferation Assay
3.2.8. Determination of Growth Rates and Doubling Times
3.2.9. DCFDA-Based Flow Cytometric Assay for Detection of Reactive Oxygen Species
3.2.10. Apoptosis Assay Based on Annexin V-FITC and PI
3.2.11. Cell Cycle Analysis Based on PI Staining
3.2.12. Western Blot Analysis
3.2.13. Plasmid Cleavage Assay
3.2.14. Alkaline Comet Assay
3.2.15. Fluorescence Microscopy
3.2.16. Luminescent Caspase Activity Assay
3.2.17. Statistic Evaluation and Correlation Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Konstantinova, L.S.; Rakitin, O.A.; Rees, C.W. Pentathiepins. Chem. Rev. 2004, 104, 2617–2630. [Google Scholar] [CrossRef]
- Feher, F.; Langer, M. Contribution to chemistry of sulfur. 104: Synthesis of pentathiepin and benzopentathiepin. Tetrahedron Lett. 1971, 12, 2125–2156. [Google Scholar] [CrossRef]
- Davidson, B.S.; Molinski, T.F.; Barrows, L.R.; Ireland, C.M. Varacin—A novel benzopentathiepin from Lissoclinum-Vareau that is cytotoxic toward a human colon-tumor. J. Am. Chem. Soc. 1991, 113, 4709–4710. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Laitinen, T.; Konstantinova, L.S.; Tizzard, G.; Poso, A.; Rakitin, O.A.; Hofmann-Lehmann, R.; Hilton, S.T. Investigation of the pentathiepin functionality as an inhibitor of feline immunodeficiency virus (FIV) via a potential zinc wjection mechanism, as a model for HIV infection. ChemMedChem 2019, 14, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Litaudon, M.; Trigalo, F.; Martin, M.T.; Frappier, F.; Guyot, M. Lissoclinotoxins—Antibiotic polysulfur derivatives from the Tunicate Lissoclinum-Perforatum—Revised structure of Lissoclinotoxin-A. Tetrahedron 1994, 50, 5323–5334. [Google Scholar] [CrossRef]
- Chatterji, T.; Gates, K.S. DNA cleavage by 7-methylbenzopentathiepin: A simple analog of the antitumor antibiotic varacin. Bioorg. Med. Chem. Lett. 1998, 8, 535–538. [Google Scholar] [CrossRef]
- Mahendran, A.; Ghogare, A.A.; Bittman, R.; Arthur, G.; Greer, A. Synthesis and antiproliferative properties of a new ceramide analog of varacin. Chem. Phys. Lipids 2016, 194, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagnone, R.S.; Faulkner, D.J.; Carte, B.K.; Chan, G.; Freyer, A.; Hemling, M.E.; Hofmann, G.A.; Mattern, M.R. Pentathiepins and trithianes from 2 Lissoclinum Species and a Eudistoma Sp—Inhibitors of protein-kinase-C. Tetrahedron 1994, 50, 12785–12792. [Google Scholar] [CrossRef]
- Baguley, T.D.; Nairn, A.C.; Lombroso, P.J.; Ellman, J.A. Synthesis of benzopentathiepin analogs and their evaluation as inhibitors of the phosphatase STEP. Bioorg. Med. Chem. Lett. 2015, 25, 1044–1046. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Chatterjee, M.; Baguley, T.D.; Brouillette, J.; Kurup, P.; Ghosh, D.; Kanyo, J.; Zhang, Y.; Seyb, K.; Ononenyi, C.; et al. Inhibitor of the tyrosine phosphatase STEP reverses cognitive deficits in a mouse model of Alzheimer’s disease. PLoS Biol. 2014, 12, e1001923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V.; et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Behnisch-Cornwell, S.; Bandaru, S.S.M.; Napierkowski, M.; Wolff, L.; Zubair, M.; Urbainsky, C.; Lillig, C.; Schulzke, C.; Bednarski, P.J. Pentathiepins: A novel class of glutathione peroxidase 1 inhibitors that induce oxidative stress, loss of mitochondrial membrane potential and apoptosis in human cancer cells. ChemMedChem 2020, 15. [Google Scholar] [CrossRef]
- Flohe, L.; Gunzler, W.A. Gsh Peroxidase. H-S Z Physiol. Chem. 1973, 354, 831–832. [Google Scholar]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.R.; Roh, J.-L.; Lee, S.M.; Park, Y.; Cho, K.-J.; Choi, S.-H.; Nam, S.Y.; Kim, S.Y. Overexpression of glutathione peroxidase 1 predicts poor prognosis in oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol 2017, 143, 2257–2265. [Google Scholar] [CrossRef]
- Wieczorek, E.; Jablonowski, Z.; Tomasik, B.; Gromadzinska, J.; Jablonska, E.; Konecki, T.; Fendler, W.; Sosnowski, M.; Wasowicz, W.; Reszka, E. Different gene expression and activity pattern of antioxidant enzymes in bladder cancer. Anticancer Res. 2017, 37, 841–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, J.J.; Mitros, F.A.; Oberley, L.W. Expression of antioxidant enzymes in diseases of the human pancreas: Another link between chronic pancreatitis and pancreatic cancer. Pancreas 2003, 26, 23–27. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Factor, V.M.; Housseau, F.; Hatfield, D.L. Contrasting patterns of regulation of the antioxidant selenoproteins, thioredoxin reductase, and glutathione peroxidase, in cancer cells. Biochem. Biophys. Res. Commun. 1998, 251, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Emmrich, T.; Lemmerhirt, H.; Leffler, U.; Sydow, K.; Hirt, C.; Kiefer, T.; Link, A.; Bednarski, P.J. Identification of a glutathione peroxidase inhibitor that reverses resistance to anticancer drugs in human B-cell lymphoma cell lines. Bioorganic Med. Chem. Lett. 2012, 22, 6712–6715. [Google Scholar] [CrossRef]
- Behnisch-Cornwell, S.; Wolff, L.; Bednarski, P.J. The effect of Gglutathione peroxidase-1 knockout on anticancer drug sensitivities and reactive oxygen species in haploid HAP-1 cells. Antioxidant 2020, 9, 1300. [Google Scholar] [CrossRef] [PubMed]
- Chaudiere, J.; Wilhelmsen, E.C.; Tappel, A.L. Mechanism of selenium-glutathione peroxidase and its inhibition by mercaptocarboxylic acids and other mercaptans. J. Biol. Chem. 1984, 259, 1043–1050. [Google Scholar] [CrossRef]
- Chatterji, T.; Gates, K.S. Reaction of thiols with 7-methylbenzopentathiepin. Bioorganic Med. Chem. Lett. 2003, 13, 1349–1352. [Google Scholar] [CrossRef]
- Tatu, M.-L.; Georgescu, E.; Boscornea, C.; Popa, M.-M.; Ungureanu, E.-M. Synthesis and fluorescence of new 3-biphenylpyrrolo[1,2-c]pyrimidines. Arab. J. Chem. 2017, 10, 643–652. [Google Scholar] [CrossRef] [Green Version]
- DiPalma, J.R.; Thayer, W.S. Use of niacin as a drug. Annu. Rev. Nutr. 1991, 11, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Rathi, A.K.; Syed, R.; Shin, H.-S.; Patel, R.V. Piperazine derivatives for therapeutic use: A patent review (2010-present). Expert Opin. Ther. Pat. 2016, 26, 777–797. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef]
- Ojima, I. Use of Fluorine in the medicinal chemistry and chemical biology of bioactive compounds—A case study on fluorinated taxane anticancer agents. ChemBioChem 2004, 5, 628–635. [Google Scholar] [CrossRef]
- Kirk, K.L.; Filler, R. Recent advances in the biomedicinal chemistry of fluorine-containing compounds. In Biomedical Frontiers of Fluorine Chemistry; ACS Symposium Series; American Chemical Society: Washington DC, USA, 1996; Volume 639, pp. 1–24. [Google Scholar]
- Lange, P.P.; Bogdan, A.R.; James, K. A New flow methodology for the expedient synthesis of drug-like 3-aminoindolizines. Adv. Synth. Catal. 2012, 354, 2373–2379. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Slater, T.F.; Sawyer, B.; Straeuli, U. Studies on succinate-tetrazolium reductase systems. III. Points of coupling of four different tetrazolium salts. Biochim. Biophys. Acta 1963, 77, 383–393. [Google Scholar] [CrossRef]
- Gillies, R.J.; Didier, N.; Denton, M. Determination of cell number in monolayer cultures. Anal. Biochem. 1986, 159, 109–113. [Google Scholar] [CrossRef]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B 2014, 369, 20130099. [Google Scholar] [CrossRef] [Green Version]
- Schober, P.; Boer, C.; Schwarte, L.A. Correlation coefficients: Appropriate use and interpretation. Anesth. Analg. 2018, 126, 1763–1768. [Google Scholar] [CrossRef]
- Bracht, K.; Boubakari; Grünert, R.; Bednarski, P.J. Correlations between the activities of 19 anti-tumor agents and the intracellular glutathione concentrations in a panel of 14 human cancer cell lines: Comparisons with the National Cancer Institute data. Anticancer Drugs 2006, 17, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Gutierrez, B.; Anzola, A.; Martinez-Augustin, O.; de Medina, F.S. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal. Biochem. 2014, 467, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colella, A.D.; Chegenii, N.; Tea, M.N.; Gibbins, I.L.; Williams, K.A.; Chataway, T.K. Comparison of Stain-Free gels with traditional immunoblot loading control methodology. Anal. Biochem. 2012, 430, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Gurtler, A.; Kunz, N.; Gomolka, M.; Hornhardt, S.; Friedl, A.A.; McDonald, K.; Kohn, J.E.; Posch, A. Stain-Free technology as a normalization tool in Western blot analysis. Anal. Biochem. 2013, 433, 105–111. [Google Scholar] [CrossRef]
- Lee, A.H.F.; Chan, A.S.C.; Li, T.H. Acid-accelerated DNA-cleaving activities of antitumor antibiotic varacin. Chem. Commun. 2002, 2112–2113. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Chen, J.; Liu, D.; Leung, T.Y.; Chan, A.S.; Li, T. Acid-promoted DNA-cleaving activities and total synthesis of varacin C. J. Am. Chem. Soc. 2002, 124, 13972–13973. [Google Scholar] [CrossRef]
- Martin, S.J.; Reutelingsperger, C.P.; McGahon, A.J.; Rader, J.A.; van Schie, R.C.; LaFace, D.M.; Green, D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995, 182, 1545–1556. [Google Scholar] [CrossRef] [Green Version]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Sagasser, J.; Ma, B.N.; Baecker, D.; Salcher, S.; Hermann, M.; Lamprecht, J.; Angerer, S.; Obexer, P.; Kircher, B.; Gust, R. A new approach in cancer treatment: Discovery of chlorido[N,N′-disalicylidene-1,2-phenylenediamine]iron(III) complexes as ferroptosis inducers. J. Med. Chem. 2019, 62, 8053–8061. [Google Scholar] [CrossRef] [PubMed]
- Chabner, B. Bleomycin. In Cancer Chemotherapy: Principles and Practice; Chabner, B., Collins, J.M., Eds.; Lippincott: Philadelphia, PA, USA, 1990; pp. 341–355. [Google Scholar]
- Mettey, Y.; Gompel, M.; Thomas, V.; Garnier, M.; Leost, M.; Ceballos-Picot, I.; Noble, M.; Endicott, J.; Vierfond, J.-M.; Meijer, L. Aloisines, a new family of CDK/GSK-3 inhibitors. SAR study, crystal structure in complex with CDK2, enzyme selectivity, and cellular effects. J. Med. Chem. 2003, 46, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Ritzén, A.; Sindet, R.; Hentzer, M.; Svendsen, N.; Brodbeck, R.M.; Bundgaard, C. Discovery of a potent and brain penetrant mGluR5 positive allosteric modulator. Bioorganic Med. Chem. Lett. 2009, 19, 3275–3278. [Google Scholar] [CrossRef]
- Lemmerhirt, H.; Behnisch, S.; Bodtke, A.; Lillig, C.H.; Pazderova, L.; Kasparkova, J.; Brabec, V.; Bednarski, P.J. Effects of cytotoxic cis- and trans-diammine monochlorido platinum(II) complexes on selenium-dependent redox enzymes and DNA. J. Inorg. Biochem. 2018, 178, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Wilde, F.; Chamseddin, C.; Lemmerhirt, H.; Bednarski, P.J.; Jira, T.; Link, A. Evaluation of (S)- and (R)-misonidazole as GPx inhibitors: Synthesis, characterization including circular dichroism and in vitro testing on bovine GPx-1. Arch. Pharm. 2014, 347, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.K. Colorimetric assay of catalase. Anal. Biochem. 1972, 47, 389. [Google Scholar] [CrossRef]
- Wang, H.; Joseph, J.A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radical. Biol. Med. 1999, 27, 612–616. [Google Scholar] [CrossRef]
- Amer, J.; Goldfarb, A.; Fibach, E. Flow cytometric measurement of reactive oxygen species production by normal and thalassaemic red blood cells. Eur. J. Haematol. 2003, 70, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Behroozi, S.J.; Kim, W.; Dannaldson, J.; Gates, K.S. 1,2-Dithiolan-3-one 1-oxides: A class of thiol-activated DNA-cleaving agents that are structurally related to the natural product leinamycin. Biochemistry 1996, 35, 1768–1774. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banath, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
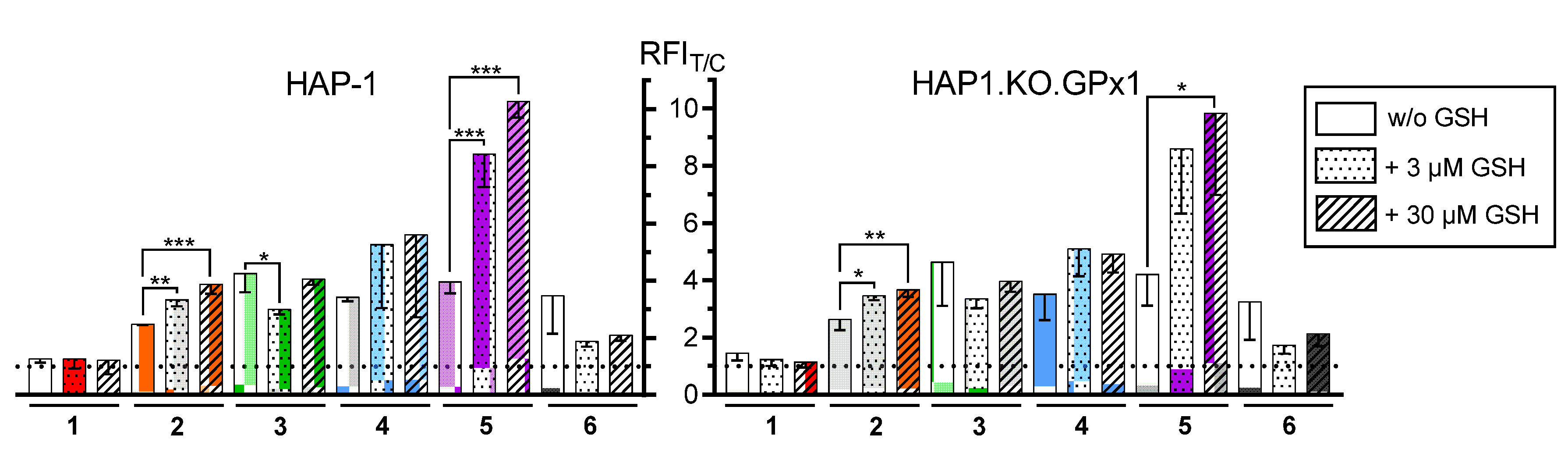{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Rel. IC50 (CI 95%) | Abs. IC50 (SD) | Residual Activity (%) |
|---|---|---|---|
| 1 | 0.92 (n.d.–2.10) | 1.49 (0.76) | 16.4 |
| 2 | 2.28 (1.52–13.74) | 3.25 (0.95) | 27.5 |
| 3 | 0.56 (0.24–0.80) | 0.91 (0.33) | 29.0 |
| 4 | 0.60 (0.32–0.84) | 0.75 (0.27) | 27.9 |
| 5 | 0.63 (0.50–0.75) | 1.17 (0.20) | 28.7 |
| 6 | >12.5 | >12.5 | 53.3 |
| Investigation | 1 | 2 | 3 | 4 | 5 | 6 | (c) |
|---|---|---|---|---|---|---|---|
| GPx1 inhibition | ++ | + | ++ | ++ | ++ | 0 | + |
| GR/CAT inhibition | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cytotoxicity | ++ | + | ++ | ++ | ++ | + | + |
| Antiproliferative effect | ++ | + | ++ | ++ | ++ | + | + |
| ROS induction | 0 | ++ | ++ | ++ | ++ | + | + |
| DNA cleavage | + | ++ | ++ | ++ | ++ | + | + |
| Comet formation | + | + | ++ | ++ | ++ | + | + |
| Apoptosis | + | + | ++ | + | ++ | + | + |
| Ferroptosis | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| G0/G1 arrest | + | 0 | 0 | 0 | 0 | + | n.d. |
| G2/M arrest | 0 | + | + | + | + | 0 | n.d. |
| Morphological changes | 0 | + | ++ | ++ | ++ | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolff, L.; Bandaru, S.S.M.; Eger, E.; Lam, H.-N.; Napierkowski, M.; Baecker, D.; Schulzke, C.; Bednarski, P.J. Comprehensive Evaluation of Biological Effects of Pentathiepins on Various Human Cancer Cell Lines and Insights into Their Mode of Action. Int. J. Mol. Sci. 2021, 22, 7631. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147631
Wolff L, Bandaru SSM, Eger E, Lam H-N, Napierkowski M, Baecker D, Schulzke C, Bednarski PJ. Comprehensive Evaluation of Biological Effects of Pentathiepins on Various Human Cancer Cell Lines and Insights into Their Mode of Action. International Journal of Molecular Sciences. 2021; 22(14):7631. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147631
Chicago/Turabian StyleWolff, Lisa, Siva Sankar Murthy Bandaru, Elias Eger, Hoai-Nhi Lam, Martin Napierkowski, Daniel Baecker, Carola Schulzke, and Patrick J. Bednarski. 2021. "Comprehensive Evaluation of Biological Effects of Pentathiepins on Various Human Cancer Cell Lines and Insights into Their Mode of Action" International Journal of Molecular Sciences 22, no. 14: 7631. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147631