
Analysis of the Structural Mechanism of ATP Inhibition at the AAA1 Subunit of Cytoplasmic Dynein-1 Using a Chemical “Toolkit”

Abstract
:1. Introduction
2. Materials and Methods
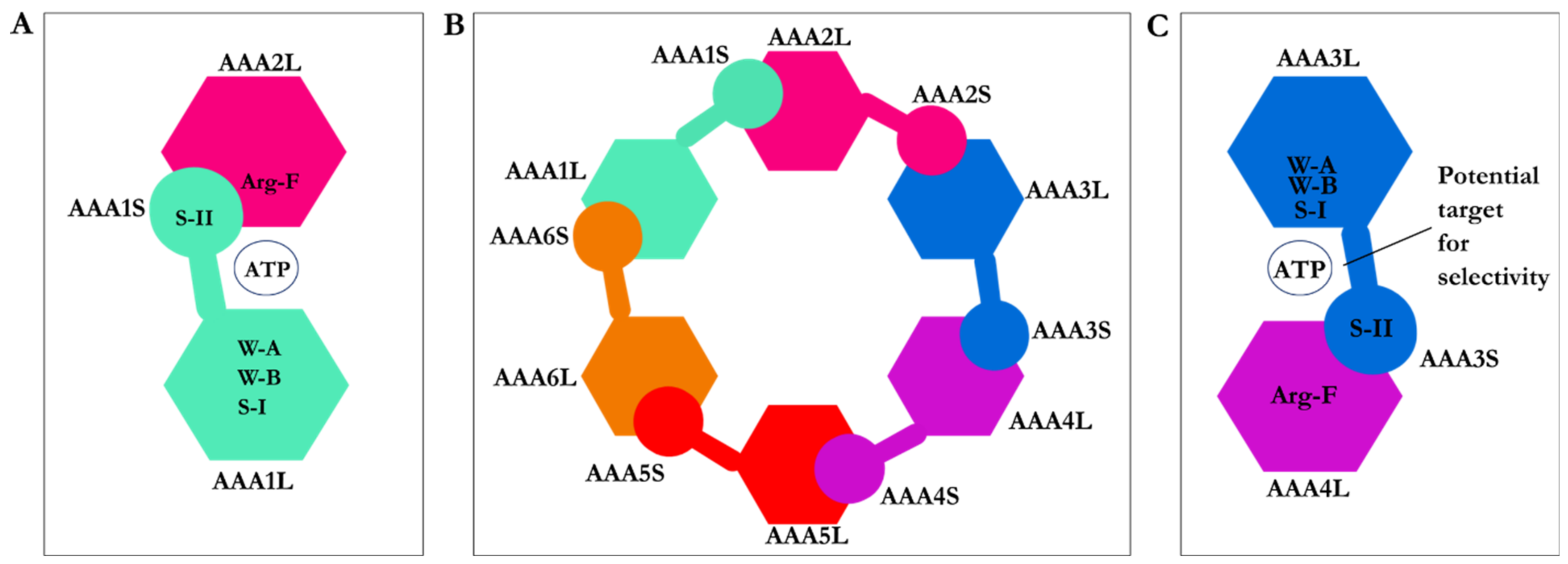
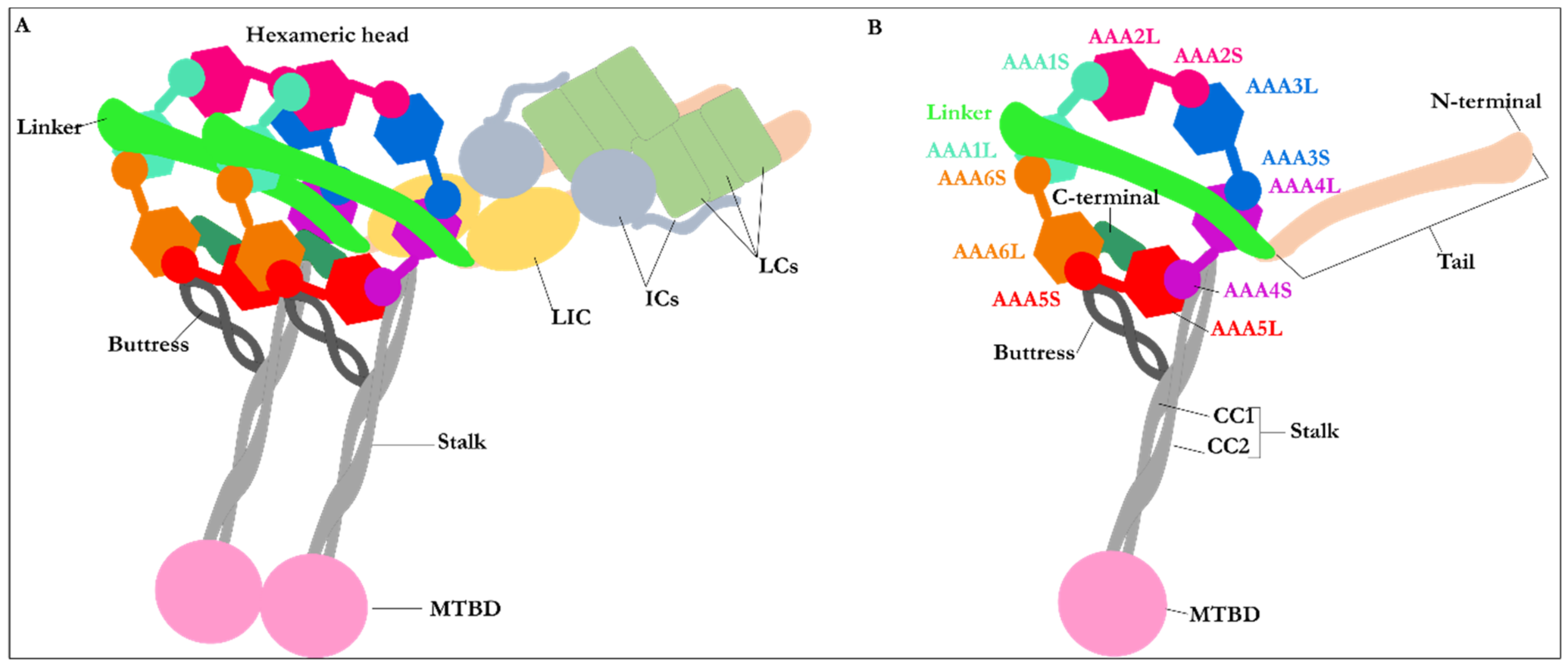
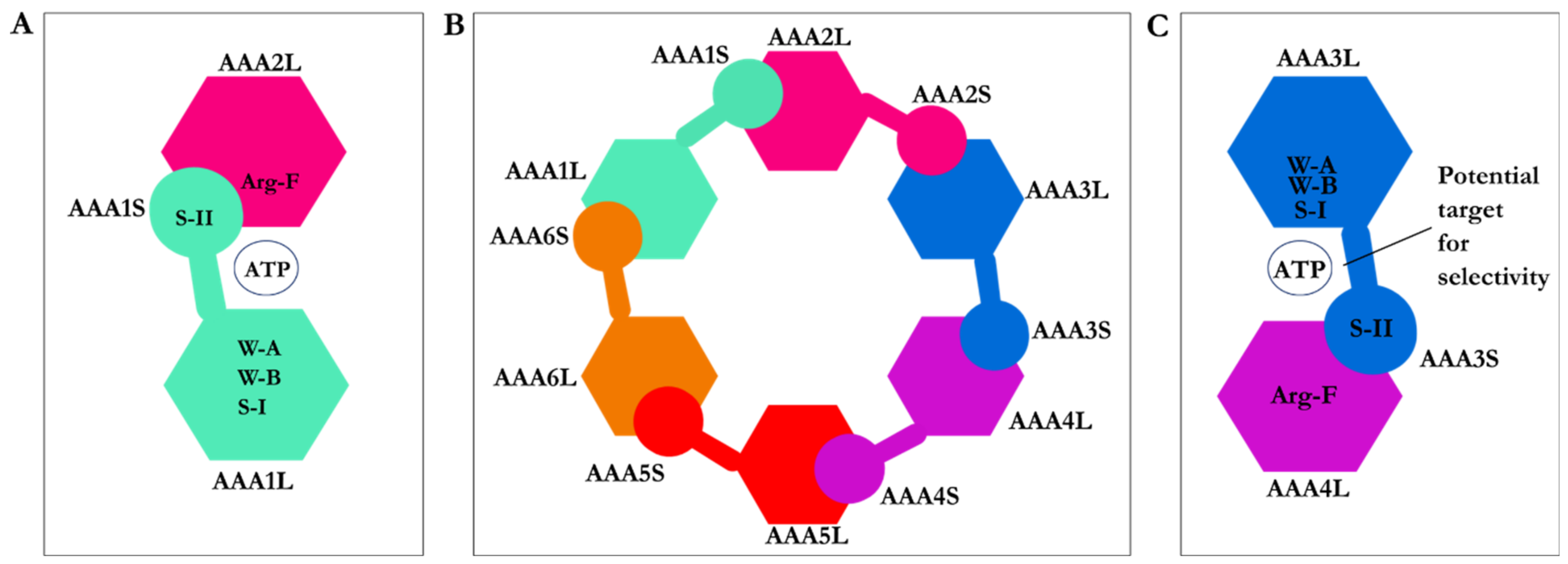
2.1. Structure of Dynein Motor Subdomains
2.2. Protein Structure Preparation
2.3. Ligands 3D Structure Preparation
3. Results and Discussion
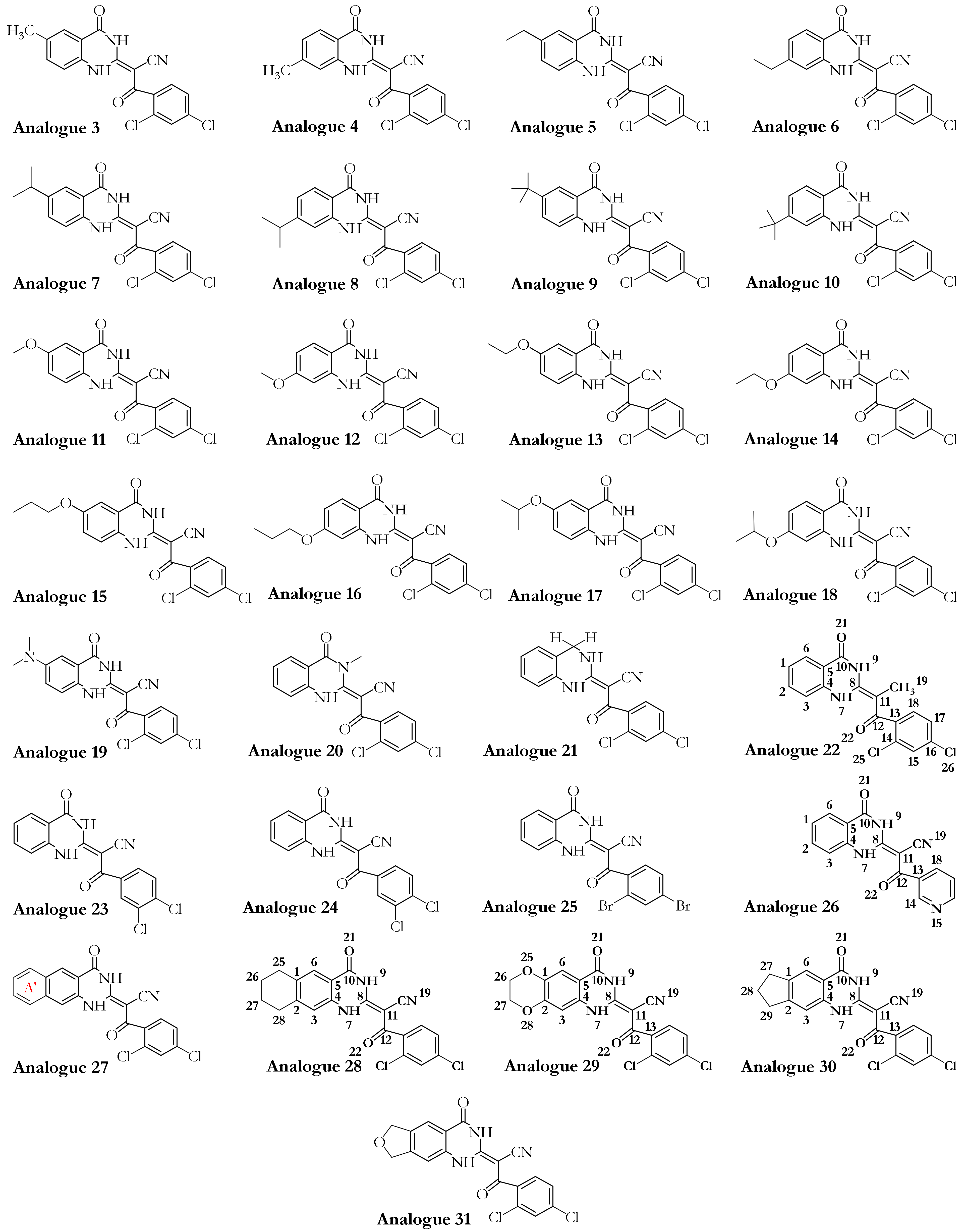
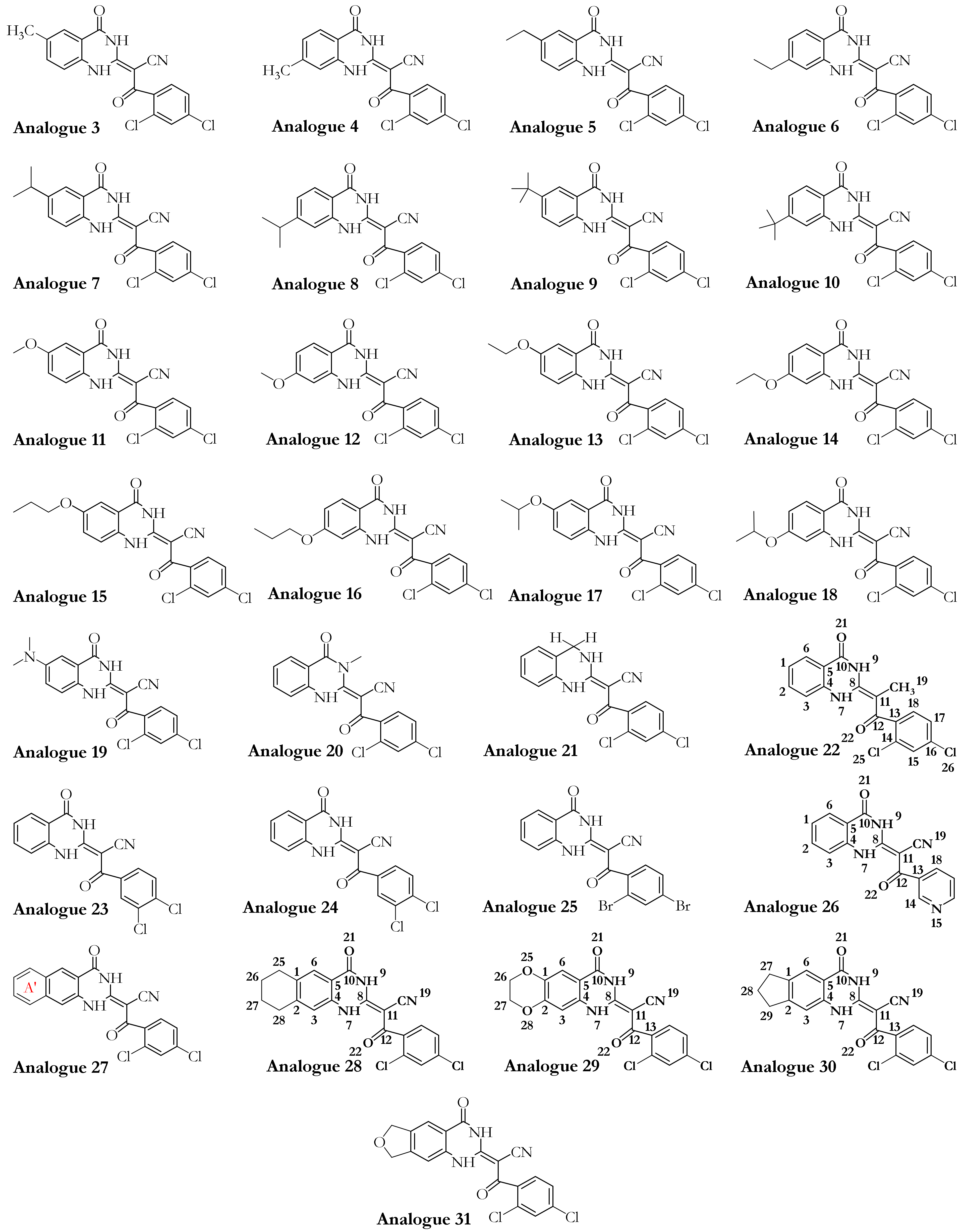
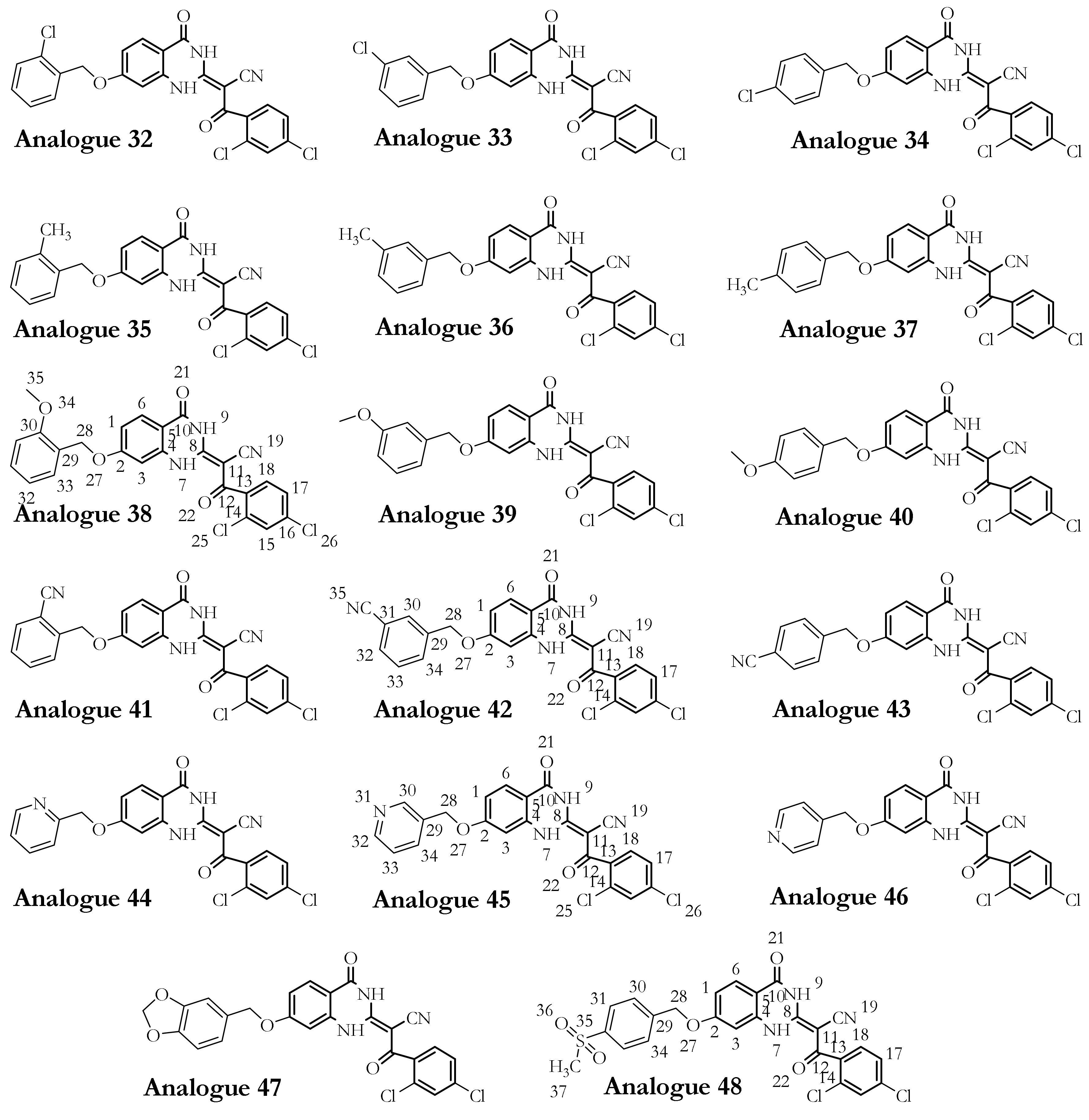
3.1. Dynapyrazole, Ciliobrevin, and Their Analogues
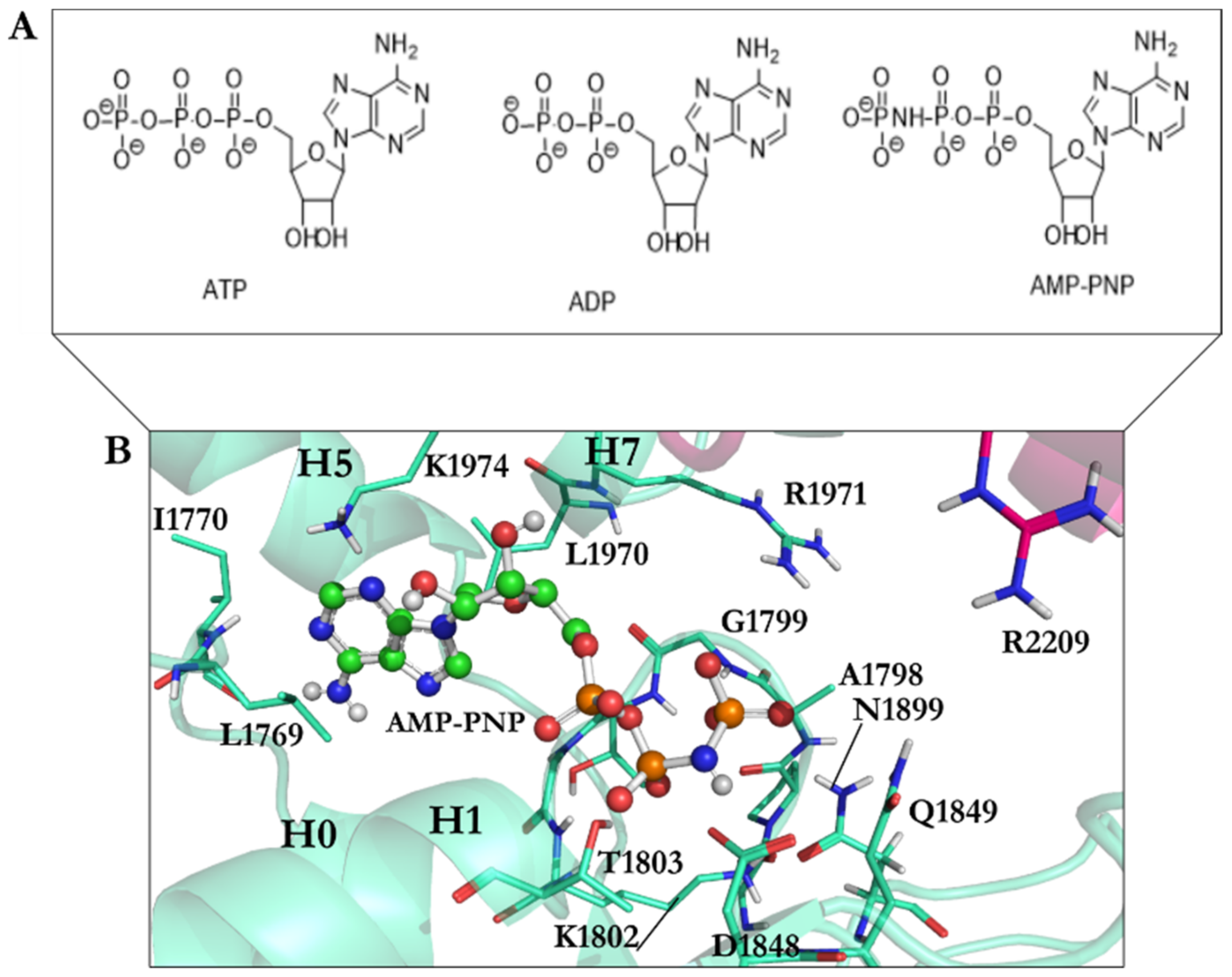
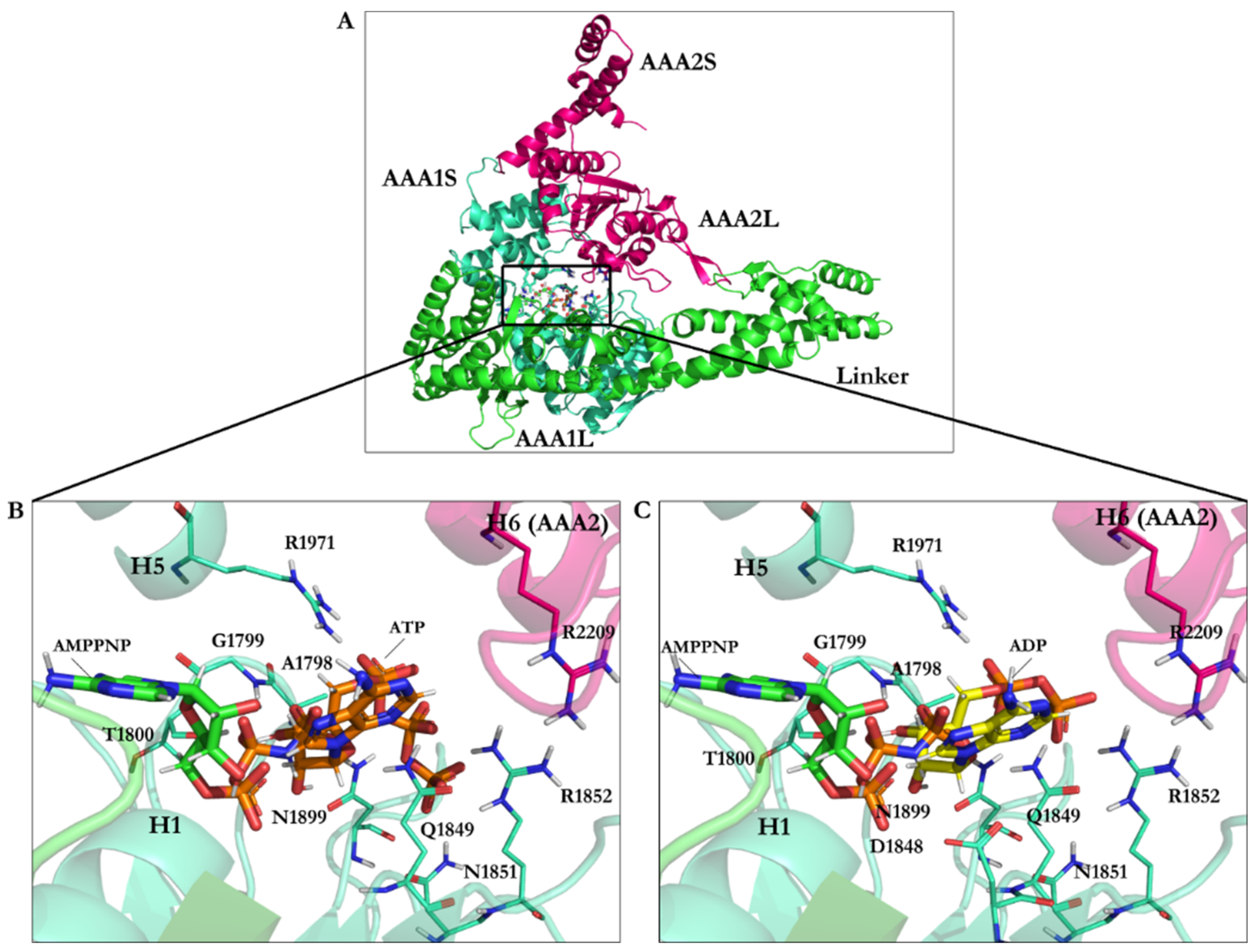
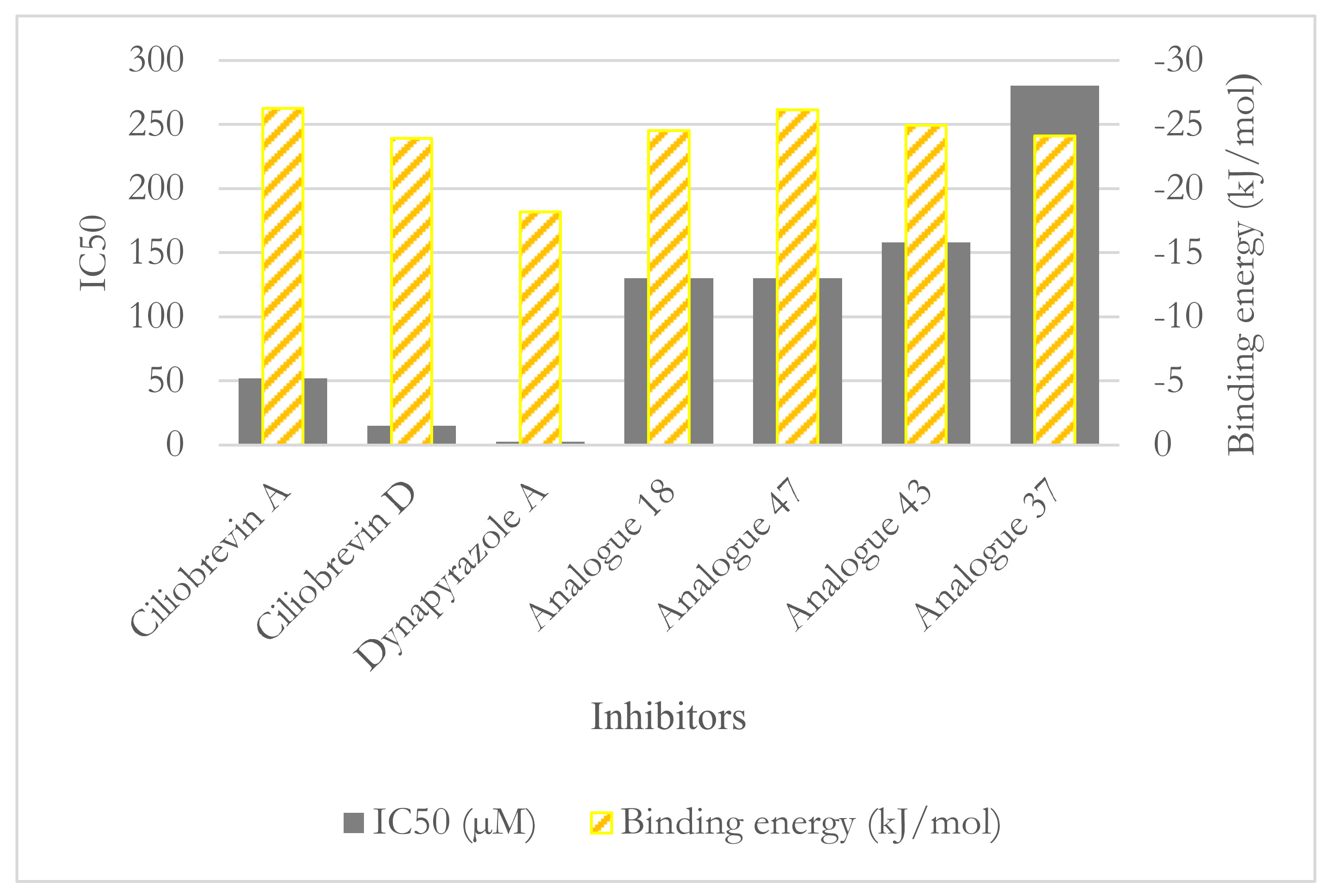
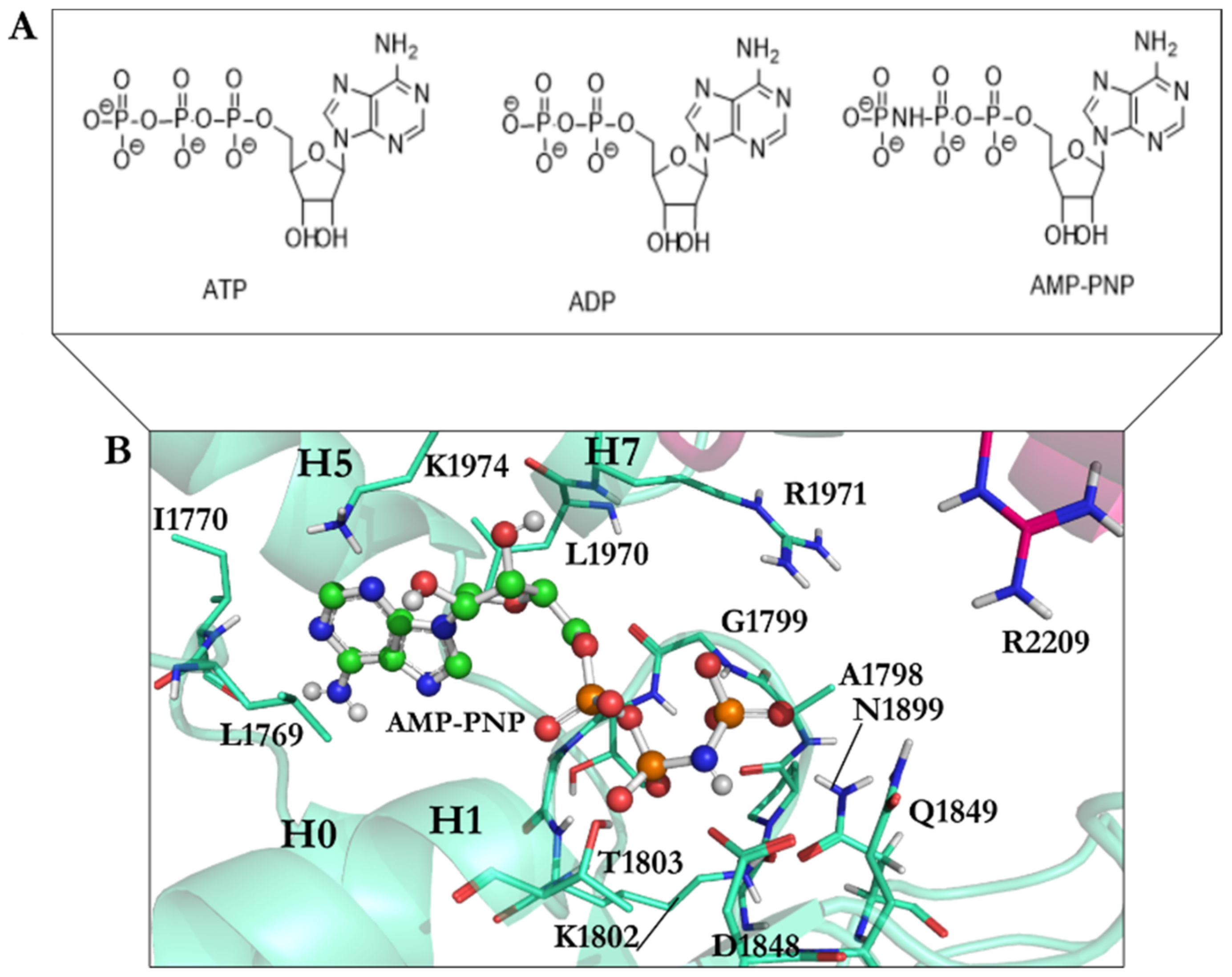
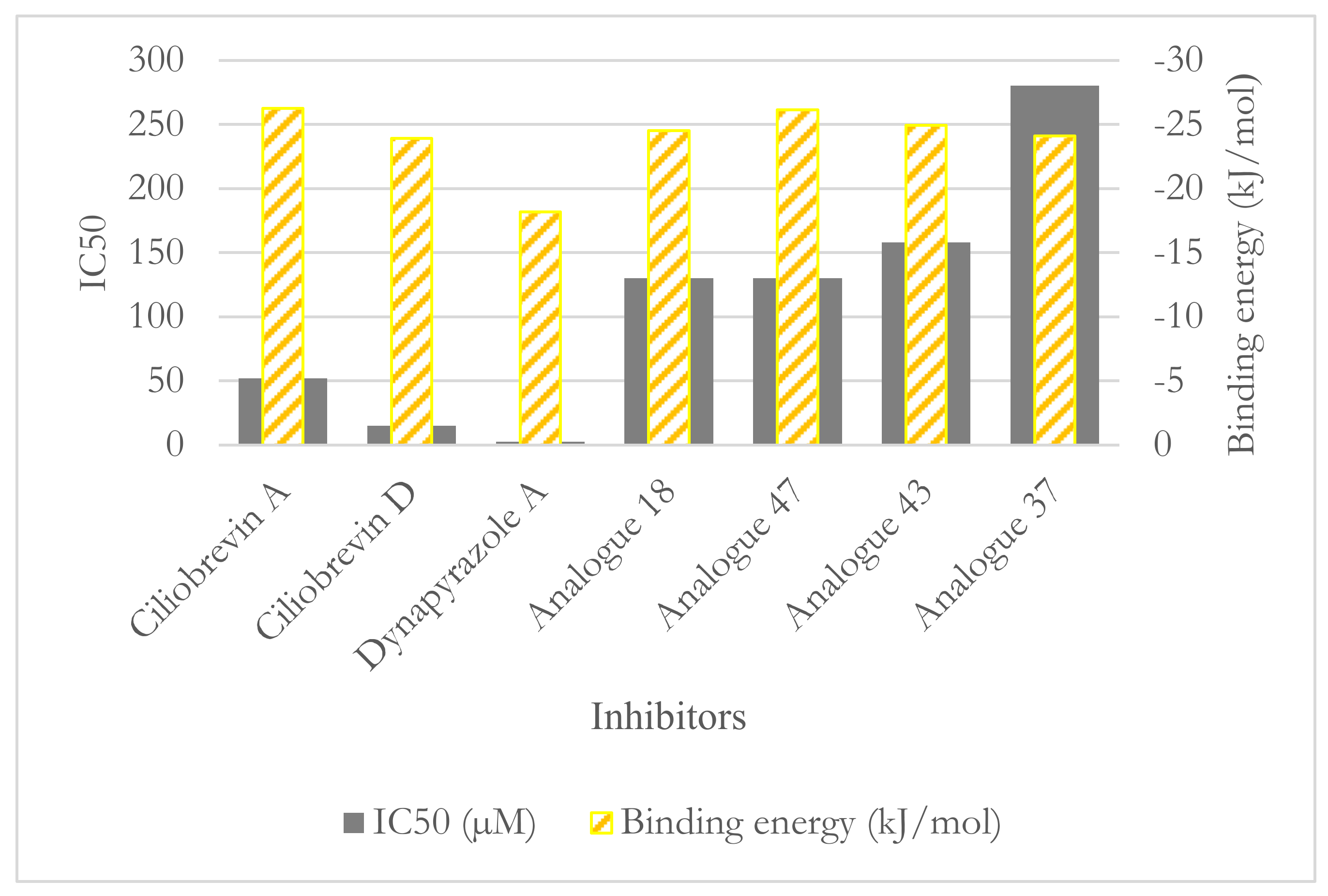
3.2. Binding Studies of Dynapyrazole, Ciliobrevin, and Their Analogues
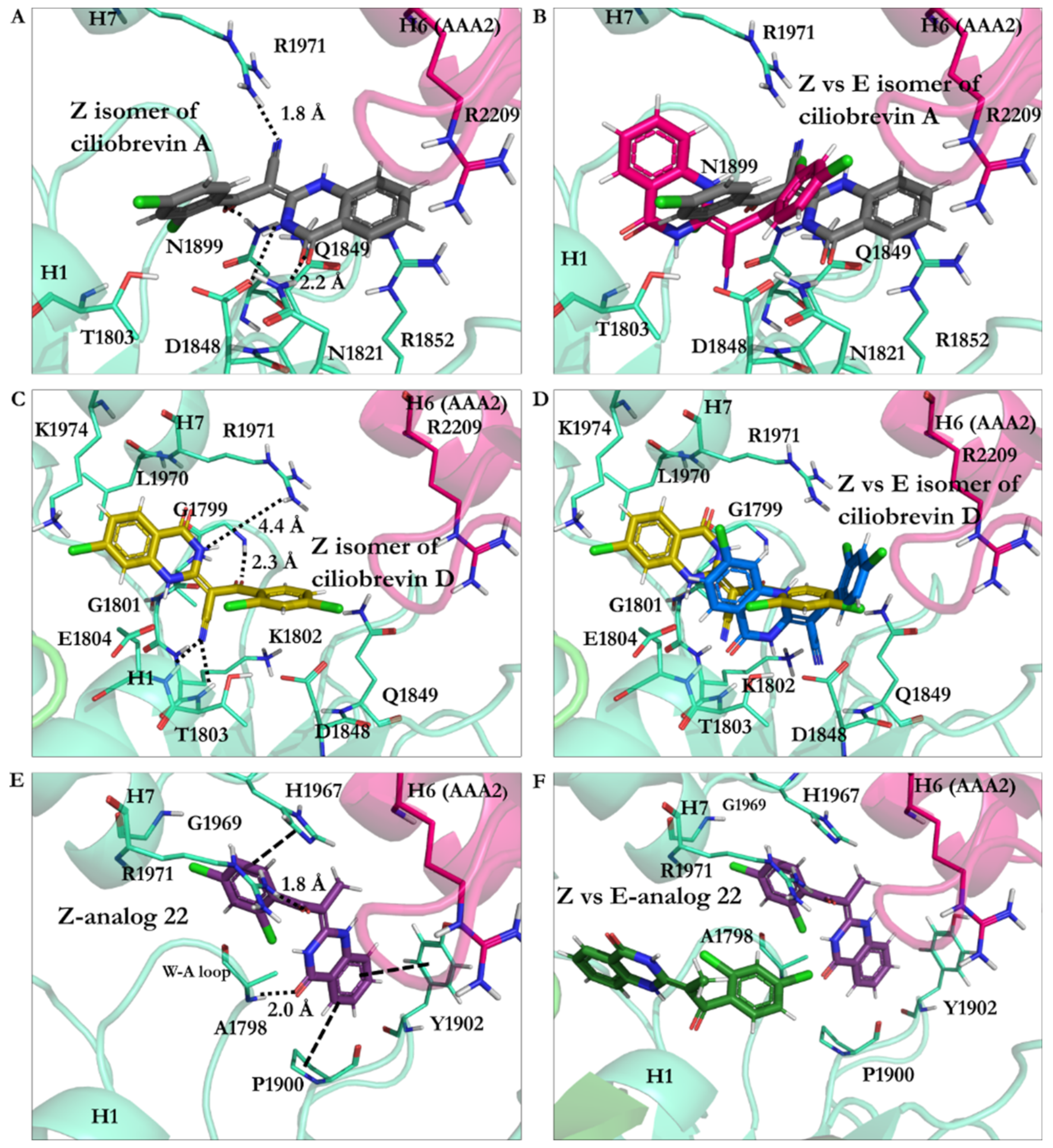
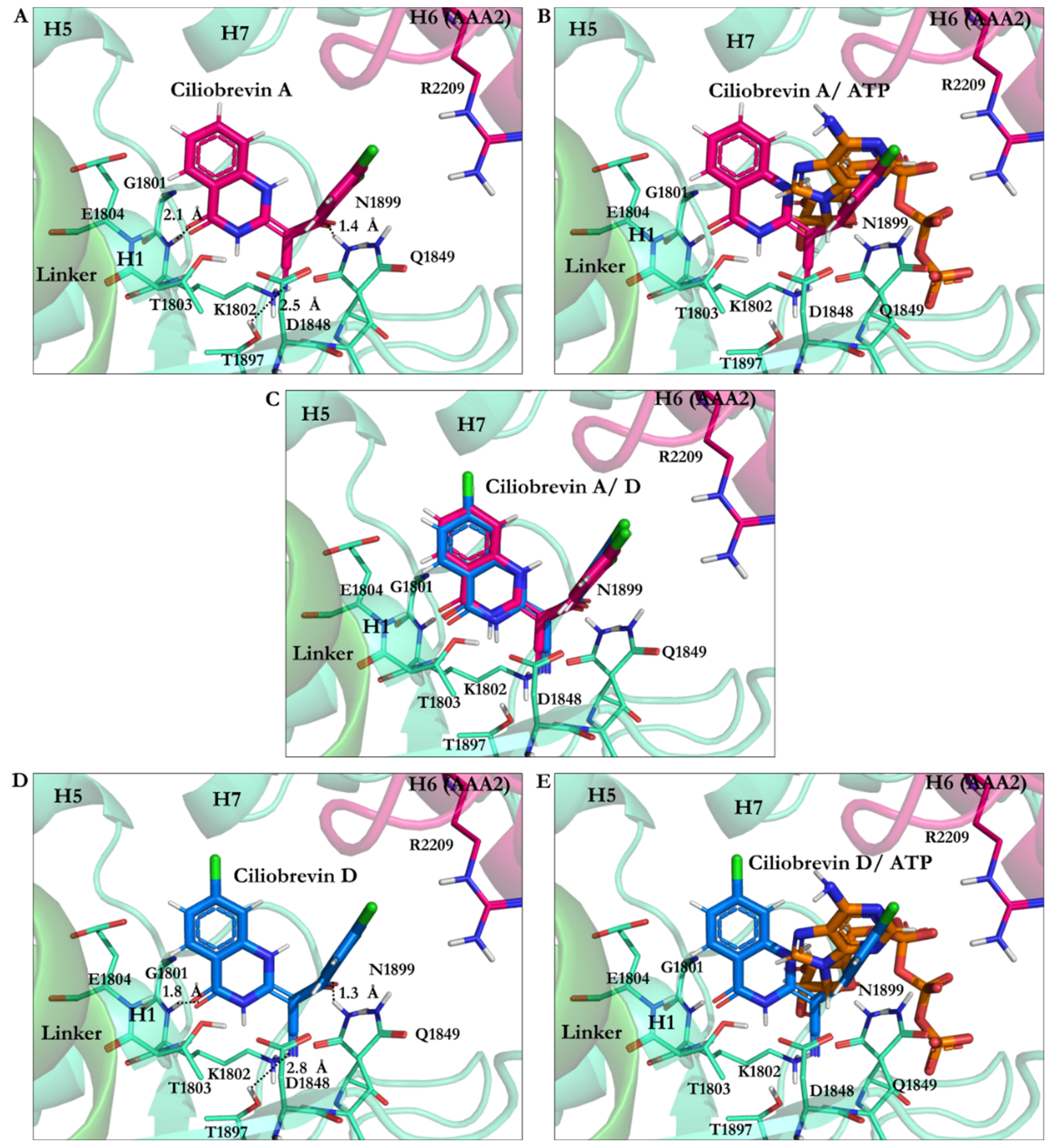
3.2.1. Ciliobrevin A and D
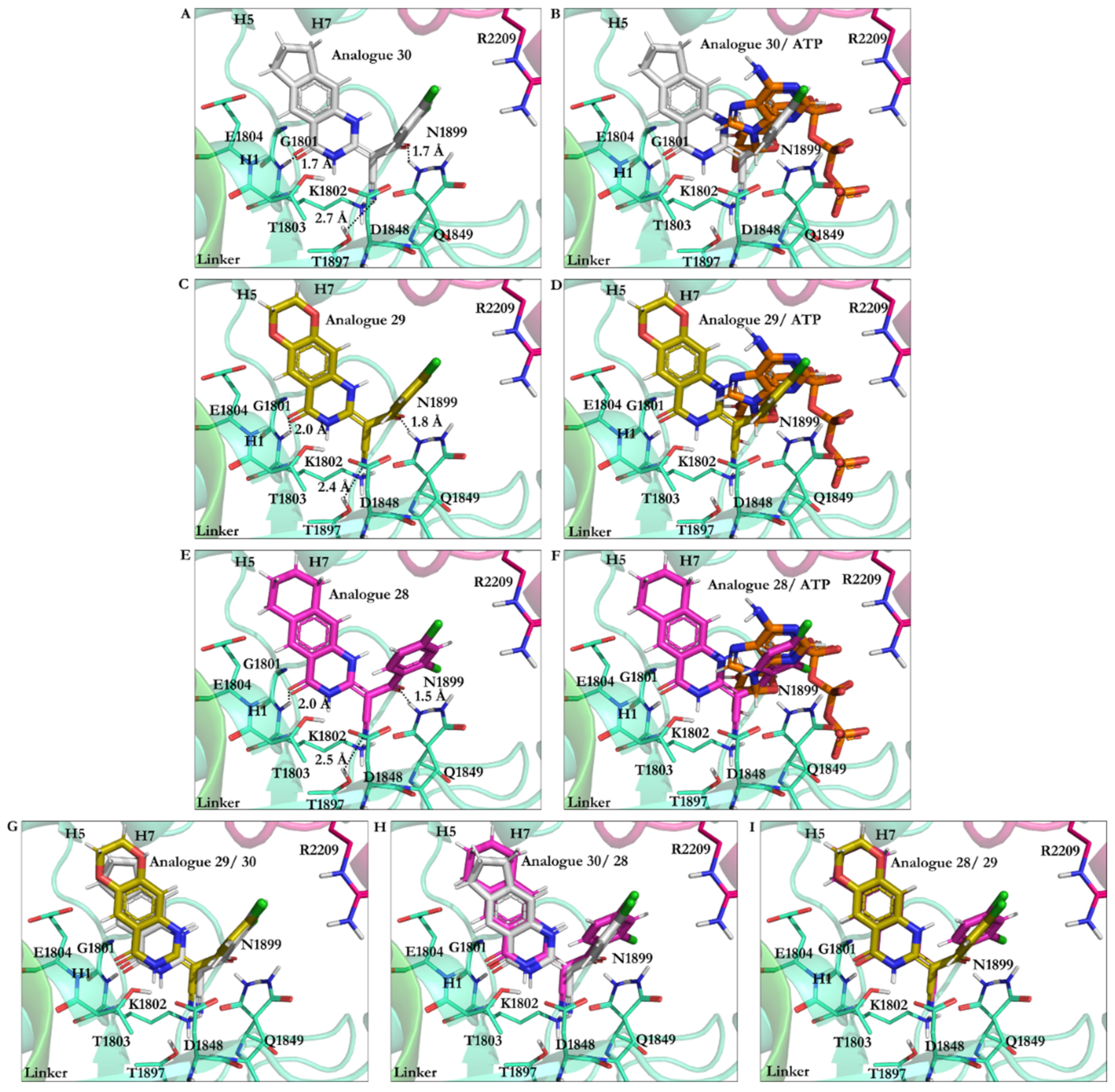
3.2.2. The Analogues Binding Profile
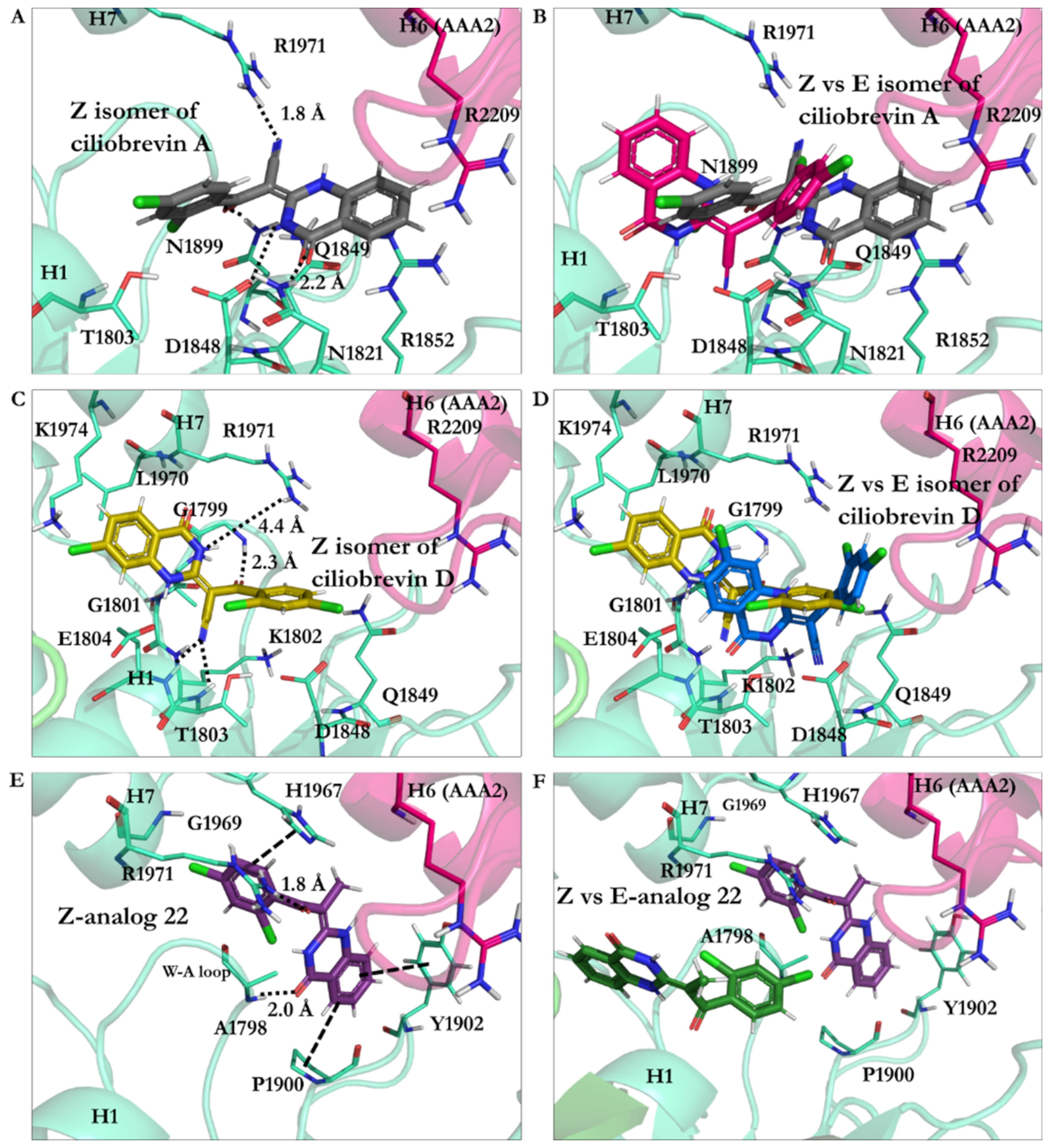
3.2.3. Geometrical Isomerization Effect on Ciliobrevin Binding to the AAA1
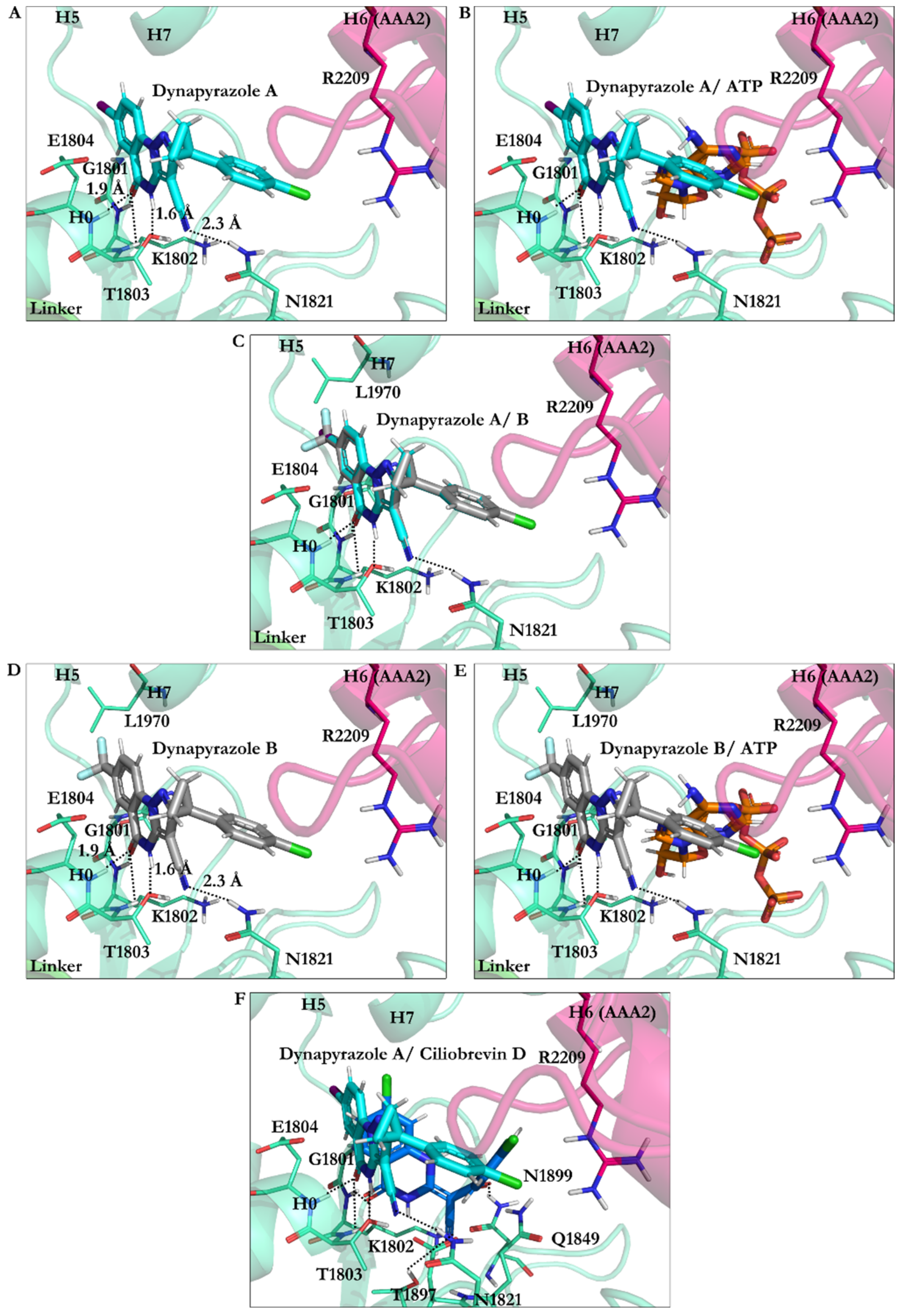
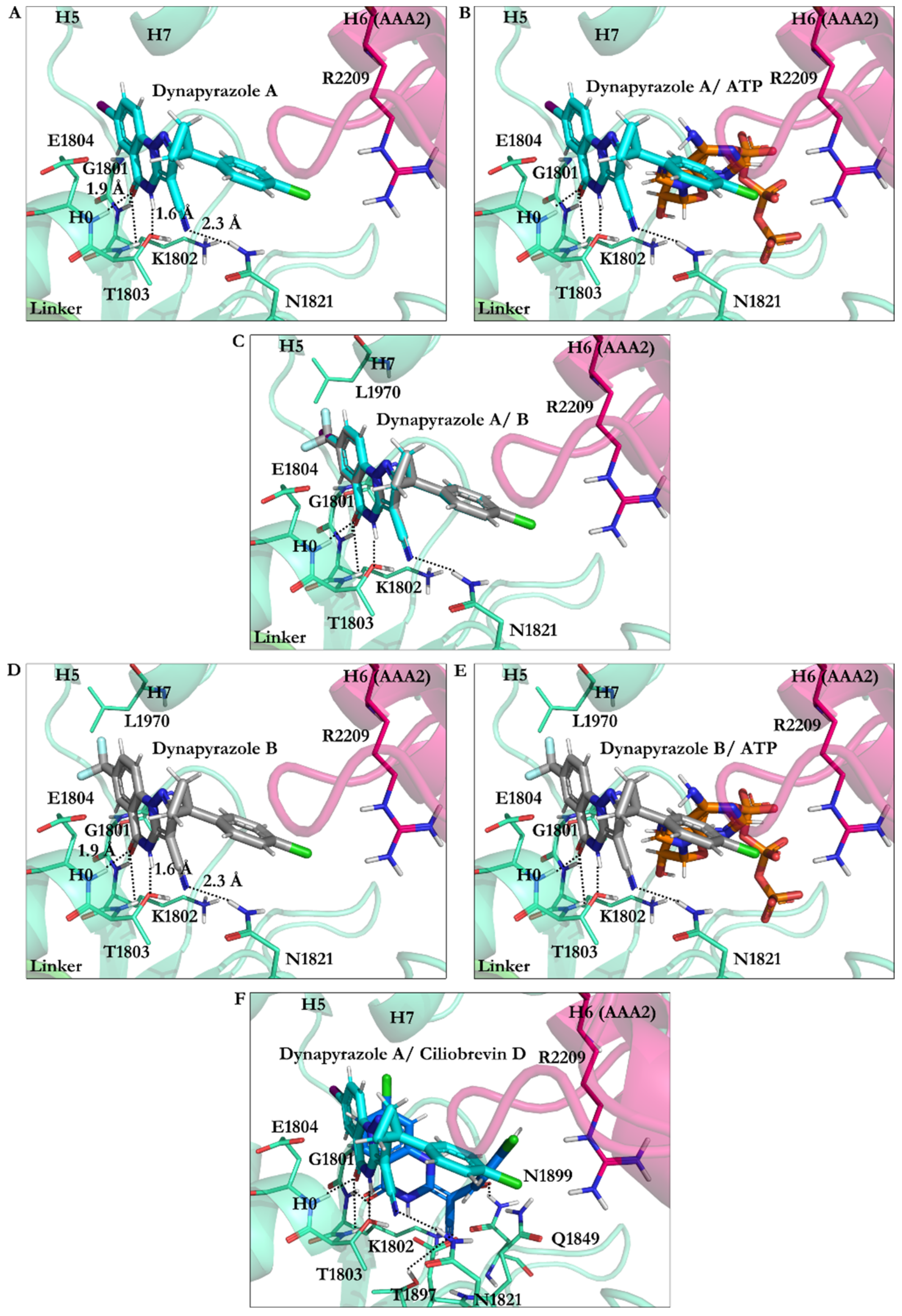
3.2.4. Dynapyrazole A and B
3.2.5. Impact of Elimination of Carbon Double Bond on the Affinity of Dynapyrazole and Analogues
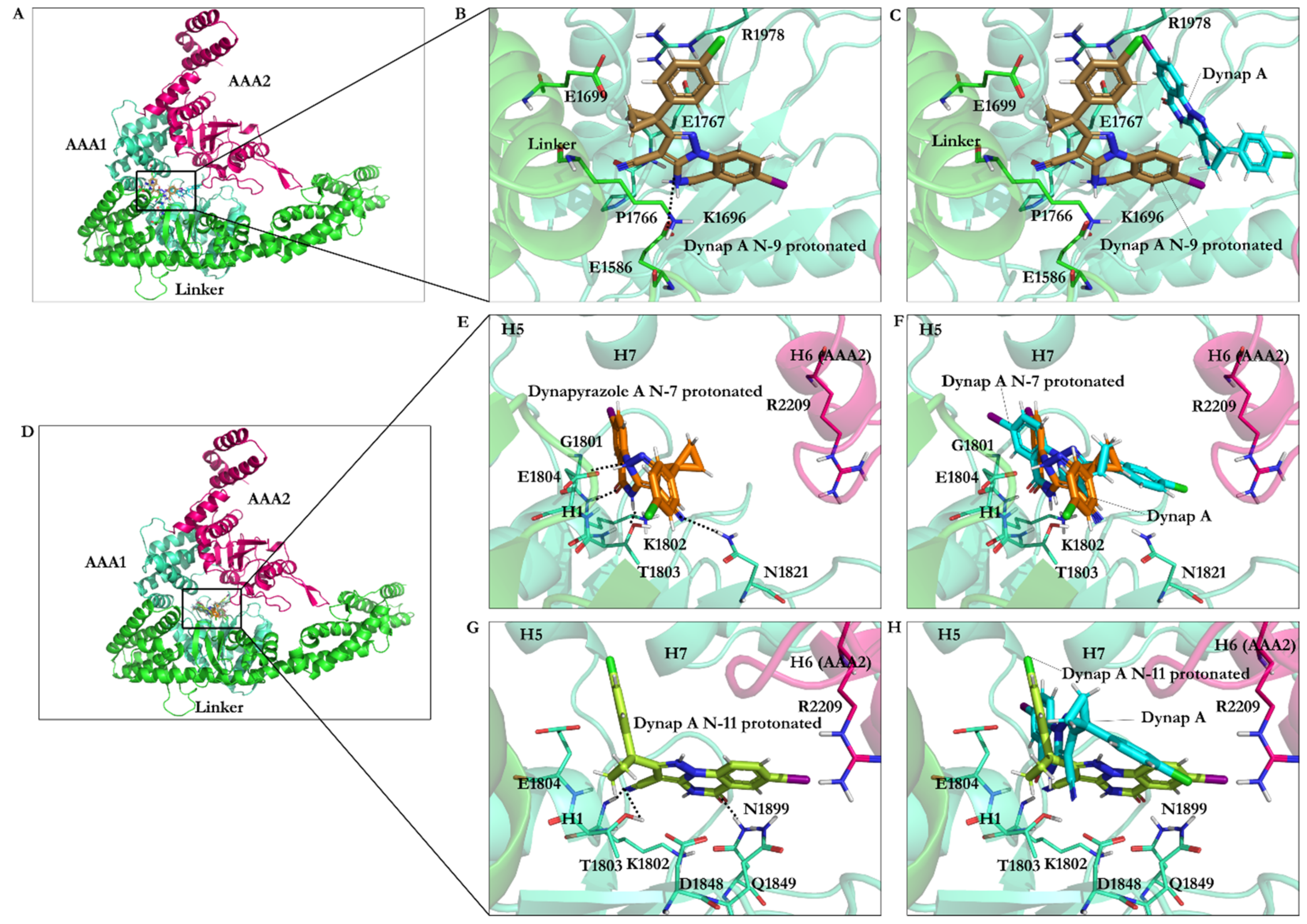
3.2.6. Protonation Effect on Ciliobrevin A and D Binding
3.2.7. Effect of Protonation on Binding Mode of Dynapyrazole A and B
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carter, A.P. Crystal clear insights into how the dynein motor moves. J. Cell Sci. 2013, 126, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Bechstedt, S.; Brouhard, G.J. Fluorescence-based assays for microtubule architecture. Methods Cell Biol. 2013, 115, 343–354. [Google Scholar] [CrossRef]
- Li, Z.; Alisaraie, L. Microtubules dual chemo and thermo-responsive depolymerization. Proteins Struct. Funct. Bioinform. 2015, 83, 970–981. [Google Scholar] [CrossRef]
- Steinman, J.B.; Kapoor, T.M. 8-Chemical probes for dynein. In Dyneins: Structure, Biology and Disease, 2nd ed.; King, S.M., Ed.; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Eschbach, J.; Dupuis, L. Cytoplasmic dynein in neurodegeneration. Pharmacol. Ther. 2011, 130, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Roossien, D.; Miller, K.; Gallo, G. Ciliobrevins as tools for studying dynein motor function. Front. Cell. Neurosci. 2015, 9, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- See, S.K.; Hoogendoorn, S.; Chung, A.H.; Ye, F.; Steinman, J.B.; Sakata-Kato, T.; Miller, R.M.; Cupido, T.; Zalyte, R.; Carter, A.P.; et al. Cytoplasmic Dynein Antagonists with Improved Potency and Isoform Selectivity. ACS Chem. Biol. 2016, 11, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Höing, S.; Yeh, T.-Y.; Baumann, M.; Martinez, N.E.; Habenberger, P.; Kremer, L.; Drexler, H.C.A.; Küchler, P.; Reinhardt, P.; Choidas, A.; et al. Dynarrestin, a Novel Inhibitor of Cytoplasmic Dynein. Cell Chem. Biol. 2018, 25, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguether, T.; Cordelieres, F.P.; Pazour, G.J. Intraflagellar transport is deeply integrated in hedgehog signaling. Mol. Biol. Cell 2018, 29, 1178–1189. [Google Scholar] [CrossRef]
- Firestone, A.J.; Weinger, J.S.; Maldonado, M.; Barlan, K.; Langston, L.D.; O’Donnell, M.; Gelfand, V.I.; Kapoor, T.M.; Chen, J.K. Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 2012, 484, 125–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.-J.; Xu, H.; Cooper, H.M.; Liu, Y. Cytoplasmic dynein: A key player in neurodegenerative and neurodevelopmental diseases. Sci. China Life Sci. 2014, 57, 372–377. [Google Scholar] [CrossRef]
- Kikkawa, M. Big steps toward understanding dynein. J. Cell Biol. 2013, 202, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Redwine, W.B.; Hernandez-Lopez, R.; Zou, S.; Huang, J.; Reck-Peterson, S.L.; Leschziner, A.E. Structural basis for microtubule binding and release by dynein. Science 2012, 337, 1532–1536. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.P.; Cho, C.; Jin, L.; Vale, R.D. Crystal structure of the dynein motor domain. Science 2011, 331, 1159–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heale, K.A.; Alisaraie, L. C-terminal Tail of β-Tubulin and its Role in the Alterations of Dynein Binding Mode. Cell Biochem. Biophys. 2020, 78, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Kon, T.; Nishiura, M.; Ohkura, R.; Toyoshima, Y.Y.; Sutoh, K. Distinct functions of nucleotide-binding/hydrolysis sites in the four AAA modules of cytoplasmic dynein. Biochemistry 2004, 43, 11266–11274. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, T.; Oyama, T.; Shimo-Kon, R.; Imamula, K.; Shima, T.; Sutoh, K.; Kurisu, G. The 2.8 Å crystal structure of the dynein motor domain. Nature 2012, 484, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Gleave, E.S.; Carter, A.P. Insights into dynein motor domain function from a 3.3-Å crystal structure. Nat. Struct. Mol. Biol. 2012, 19, 492–497. [Google Scholar] [CrossRef] [Green Version]
- Bhabha, G.; Cheng, H.C.; Zhang, N.; Moeller, A.; Liao, M.; Speir, J.A.; Cheng, Y.; Vale, R.D. Allosteric communication in the dynein motor domain. Cell 2014, 159, 857–868. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Raghava, G.P.S.; Searle, S.M.J.; Audley, P.C.; Barber, J.D.; Barton, G.J. OXBench: A benchmark for evaluation of protein multiple sequence alignment accuracy. BMC Bioinform. 2003, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843. [Google Scholar] [CrossRef] [PubMed]
- Steinman, J.B.; Santarossa, C.C.; Miller, R.M.; Yu, L.S.; Serpinskaya, A.S.; Furukawa, H.; Morimoto, S.; Tanaka, Y.; Nishitani, M.; Asano, M.; et al. Chemical structure-guided design of dynapyrazoles, cell-permeable dynein inhibitors with a unique mode of action. eLife 2017, 6, e25174. [Google Scholar] [CrossRef] [PubMed]
- Tribolet, R.; Sigel, H. Influence of the protonation degree on the self-association properties of adenosine 5′-triphosphate (ATP). Eur. J. Biochem. 1988, 170, 617–626. [Google Scholar] [CrossRef] [PubMed]
- David, A.W. Foye’s Principles of Medicinal Chemistry, 7th ed.; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2012. [Google Scholar]
- Baumgarten, H.E. Advances in heterocyclic chemistry. J. Chem. Educ. 1963, 40, 559. [Google Scholar] [CrossRef] [Green Version]
- Schellhammer, I.; Rarey, M. FlexX-Scan: Fast, structure-based virtual screening. Proteins Struct. Funct. Bioinform. 2004, 57, 504–517. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T. Docking of hydrophobic ligands with interaction-based matching algorithms. Bioinformatics 1999, 15, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Rarey, M.; Kramer, B.; Lengauer, T. Multiple automatic base selection: Protein–ligand docking based on incremental construction without manual intervention. J. Comput.-Aided Mol. Des. 1997, 11, 369–384. [Google Scholar] [CrossRef]
- Böhm, H.J. The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J. Comput.-Aided Mol. Des. 1994, 8, 243–256. [Google Scholar] [CrossRef]
- Böhm, H.J. Prediction of binding constants of protein ligands: A fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J. Comput.-Aided Mol. Des. 1998, 12, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Ecklund, K.H.; Morisaki, T.; Lammers, L.G.; Marzo, M.G.; Stasevich, T.J.; Markus, S.M. She1 affects dynein through direct interactions with the microtubule and the dynein microtubule-binding domain. Nat. Commun. 2017, 8, 2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarossa, C.C.; Mickolajczyk, K.J.; Steinman, J.B.; Urnavicius, L.; Chen, N.; Hirata, Y.; Fukase, Y.; Coudray, N.; Ekiert, D.C.; Bhabha, G.; et al. Targeting Allostery in the Dynein Motor Domain with Small Molecule Inhibitors. Cell Chem. Biol. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Carter, A.P. Review: Structure and mechanism of the dynein motor ATPase. Biopolymers 2016, 105, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Mogni, M.E.; Costa, A.; Ioannou, C.; Bell, S.D. The glutamate switch is present in all seven clades of AAA+ protein. Biochemistry 2009, 48, 8774–8775. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Uniprot Code | Species | Resolution (Å) | Exp. pH | Nucleotide Binding Domain | Missing Residues | Released Date |
|---|---|---|---|---|---|---|---|
| 4AKG | P36022 | S. cerevisiae | 3.3 | 5.6 | AAA1 (apo), AAA2 (ATP), AAA3 (ADP) | 2944–2959 (AAA4) and 3658–3669 (AAA5–AAA6) | 14 March 2012 |
| 4W8F | P36022 | S. cerevisiae | 3.54 | 8.0 | AMPPNP in AAA1, AAA2, AAA3 and AAA4 | 2025–2029 (AAA1–AAA2), 2950–2953 (AAA4), 3659–3668 (AAA5–AAA6) | 12 December 2014 |
| 3VKG | P34036 | Dictyostelium discoideum | 2.81 | 7.0 | ADP in AAA1, AAA2, AAA3 and AAA4 | 2061–2063 (AAA1), 2454–2488 (AAA2), 3212–3215 (AAA4), 3699–3703 (AAA5), 3725–3758 (AAA5), 4114–4115 (AAA6) | 14 March 2012 |
| Dynein (S. cerevisiae and D. discoideum) | Sequence Identity (%) (No. of Residues) | Sequence Similarity (Residues) |
|---|---|---|
| Entire amino-acid sequence of cytoplasmic dynein | 24.83% (1193 residues) | 1668 |
| AAA1 | 52.02% (116 residues) | 63 |
| AAA2 | 28.14% (83 residues) | 95 |
| AAA1 and AAA2 | 34.67% (207 residues) | 185 |
| ATP Motifs | S. Cerevisiae | D. Discoideum |
|---|---|---|
| Walker-A | Gly1796–Thr1803 | Gly1974–Thr1980 |
| Walker-B | Asp1848–Glu1849 | Asp2026–Glu2027 |
| Sensor I | Asn1899 | Asn2078 |
| Sensor II | Arg1971 | Arg2150 |
| Arg finger | Arg2209 | Arg2410 |
| N-loop | Leu1769–Ile1770 | Leu1947–Val1948 |
| Compounds | IC50 (µM) Dynein 1 | IC50 (µM) Dynein 2 |
|---|---|---|
| Ciliobrevin A | 52.0 | 55.0 |
| Ciliobrevin D | 15.0 | 15.5 |
| Dynapyrazole A | 2.3 | 2.6 |
| Dynapyrazole B * | _ | 2.9 |
| Analogue 18 | 130.0 | 21.0 |
| Analogue 37 | 280.0 | 11.0 |
| Analogue 43 | 158.0 | 16.0 |
| Analogue 47 | 130.0 | 11.0 |
| Ligand | RMSD vs. X-ray Structure | Binding Energy (kJ/mol) | Residues Interacting with the Compound |
|---|---|---|---|
| AMPPNP | 1.67 | −22.06 | Leu1769, Ile1770, Gly1799, Gly1801, Lys1802, Thr1803, Glu1804, Asn1899, Ile1929, Leu1970, Lys1974 |
| AMPPNP energy-minimized structure | 4.75 | −40.18 | Glu1767, Gly1799, Gly1801, Lys1802, Thr1803, Glu1804, Gln1849, Asn1899, Lys1974 |
| Minimized ATP | — | −42.33 | Ala1798, Gly1799, Thr1800, Gln1849, Asn1851, Arg1852, Asn1899, Arg1971 |
| Minimized ADP | — | −31.89 | Ala1798, Gly1799, Thr1800, Asp1848, Gln1849, Arg1852, Asn1899, Arg1971 |
| Compound | Binding Energy (kJ/mol) | Residues Interacting with the Compound |
|---|---|---|
| Ciliobrevin A N9 protonated | −28.22 | Ala1798, Lys1802, Thr1803, Glu1804, Asp1848, Gln1849, Thr1897, Asn1899, Arg1971 |
| Ciliobrevin A | −26.23 | Gly1801, Lys1802, Thr1803, Glu1804, Asp1848, Gln1849, Thr1897, Asn1899 |
| Ciliobrevin D N9 protonated | −26.15 | Lys1802, Thr1803, Glu1804, Asp1848, Gln1849, Thr1897, Asn1899 |
| Ciliobrevin D | −23.92 | Gly1801, Lys1802, Thr1803, Glu1804, Asp1848, Gln1849, Thr1897, Asn1899 |
| Ciliobrevin A N7 protonated | −23.84 | Gly1799, Thr1800, Gly1801, Lys1802, Thr1803, Glu1804, Leu1970, Arg1971 |
| Ciliobrevin D N7 protonated | −23.55 | Gly1801, Lys1802, Thr1803, Glu1804, Asp1848, Gln1849, Thr1897, Asn1899 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tati, S.; Alisaraie, L. Analysis of the Structural Mechanism of ATP Inhibition at the AAA1 Subunit of Cytoplasmic Dynein-1 Using a Chemical “Toolkit”. Int. J. Mol. Sci. 2021, 22, 7704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147704
Tati S, Alisaraie L. Analysis of the Structural Mechanism of ATP Inhibition at the AAA1 Subunit of Cytoplasmic Dynein-1 Using a Chemical “Toolkit”. International Journal of Molecular Sciences. 2021; 22(14):7704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147704
Chicago/Turabian StyleTati, Sayi’Mone, and Laleh Alisaraie. 2021. "Analysis of the Structural Mechanism of ATP Inhibition at the AAA1 Subunit of Cytoplasmic Dynein-1 Using a Chemical “Toolkit”" International Journal of Molecular Sciences 22, no. 14: 7704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147704