
Single Nucleotide Polymorphism Induces Divergent Dynamic Patterns in CYP3A5: A Microsecond Scale Biomolecular Simulation of Variants Identified in Sub-Saharan African Populations


Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Genome Analysis
2.2. System Preparation
2.3. Force Field Parameters for Heme Group, Ritonavir, and Artemether
2.4. Artemether Docking to CYP3A5
2.5. Generating the Structures for the Variants
2.6. Molecular Dynamics
2.7. Analysis of Molecular Dynamics Simulation
2.7.1. Essential Dynamics Analysis
2.7.2. Free Energy Landscape
2.7.3. Binding Free Energy Calculation
2.7.4. Cavity Volume and Tunnels Geometry Calculations
3. Results
3.1. Genetic Characterization of CYP3A5 Variants from WGS
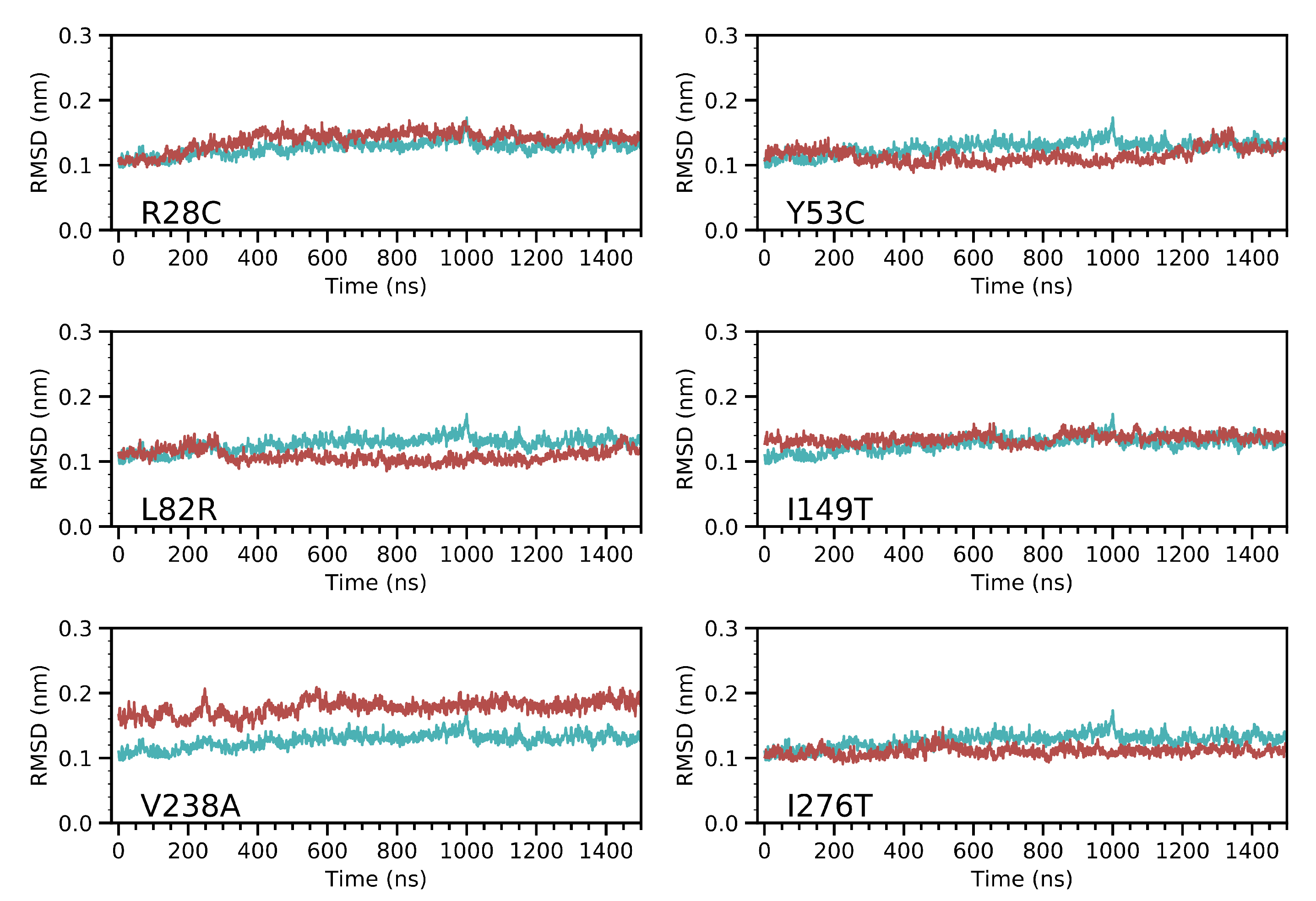
3.2. Molecular Dynamics Simulation of CYP3A5 Variants
Overall Effect of the Variants on CYP3A5 Structure
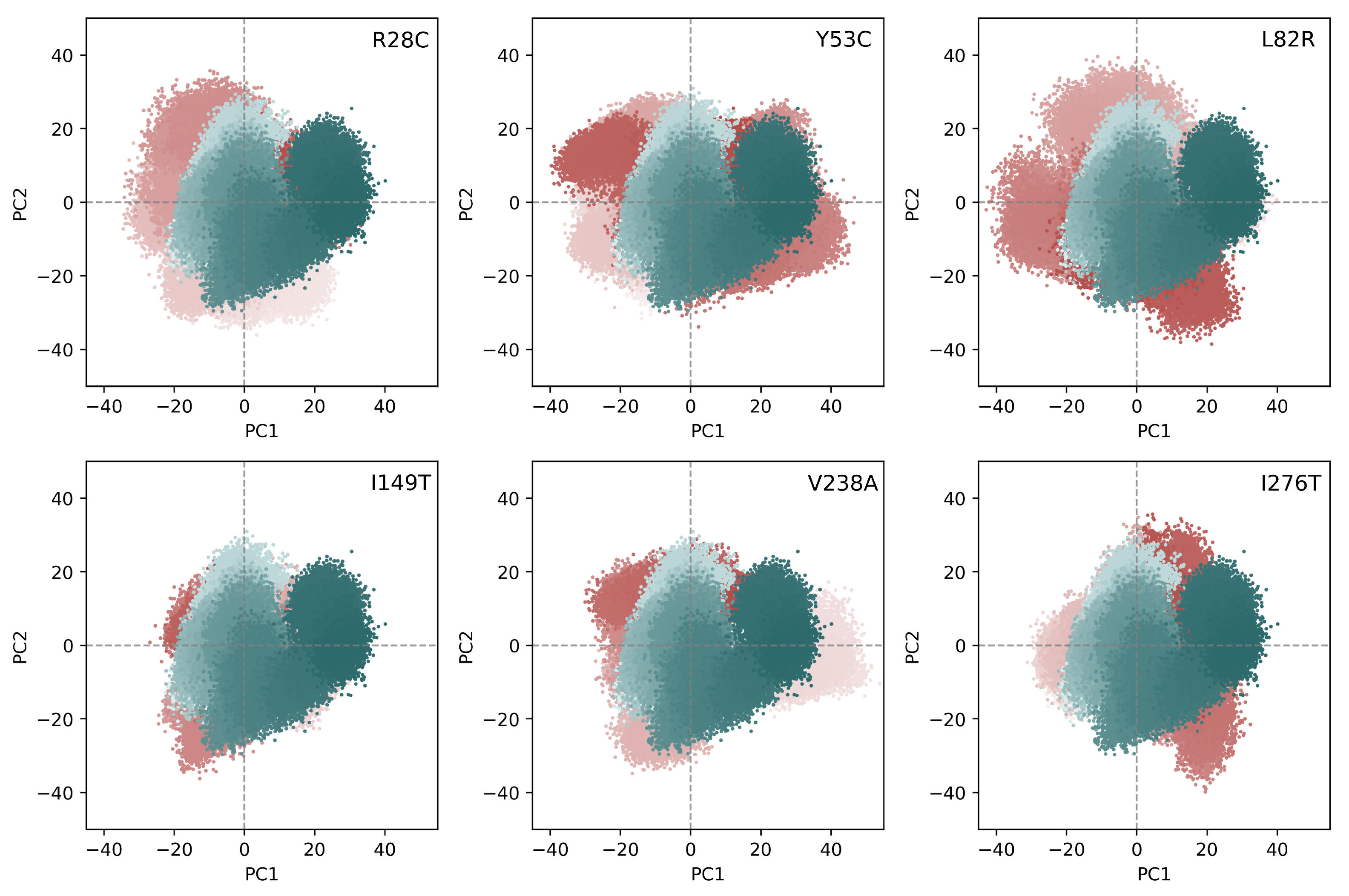
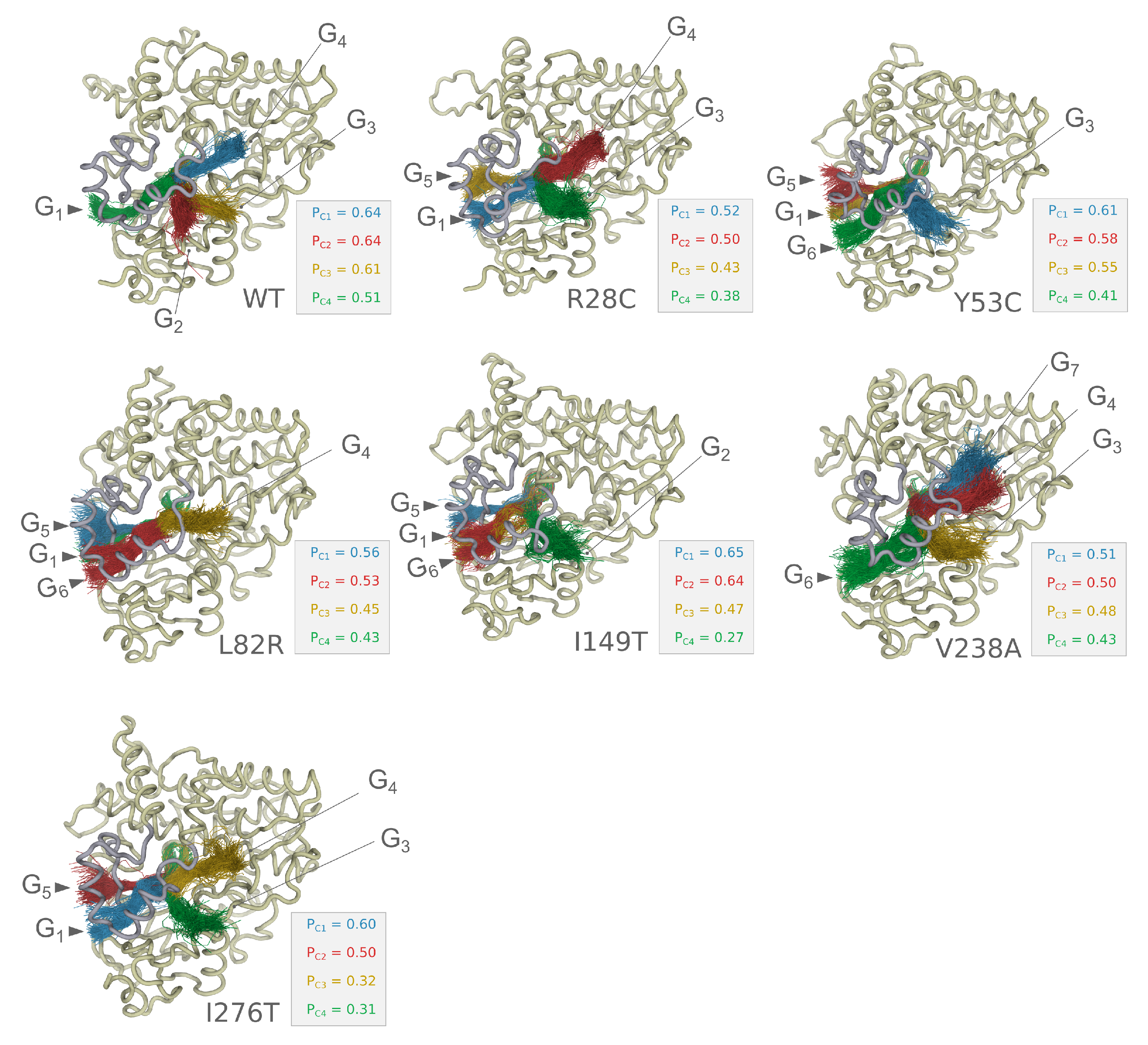
3.3. Analysis of the Collective Motions within CYP3A5
3.4. Essential Dynamics of CYP3A5
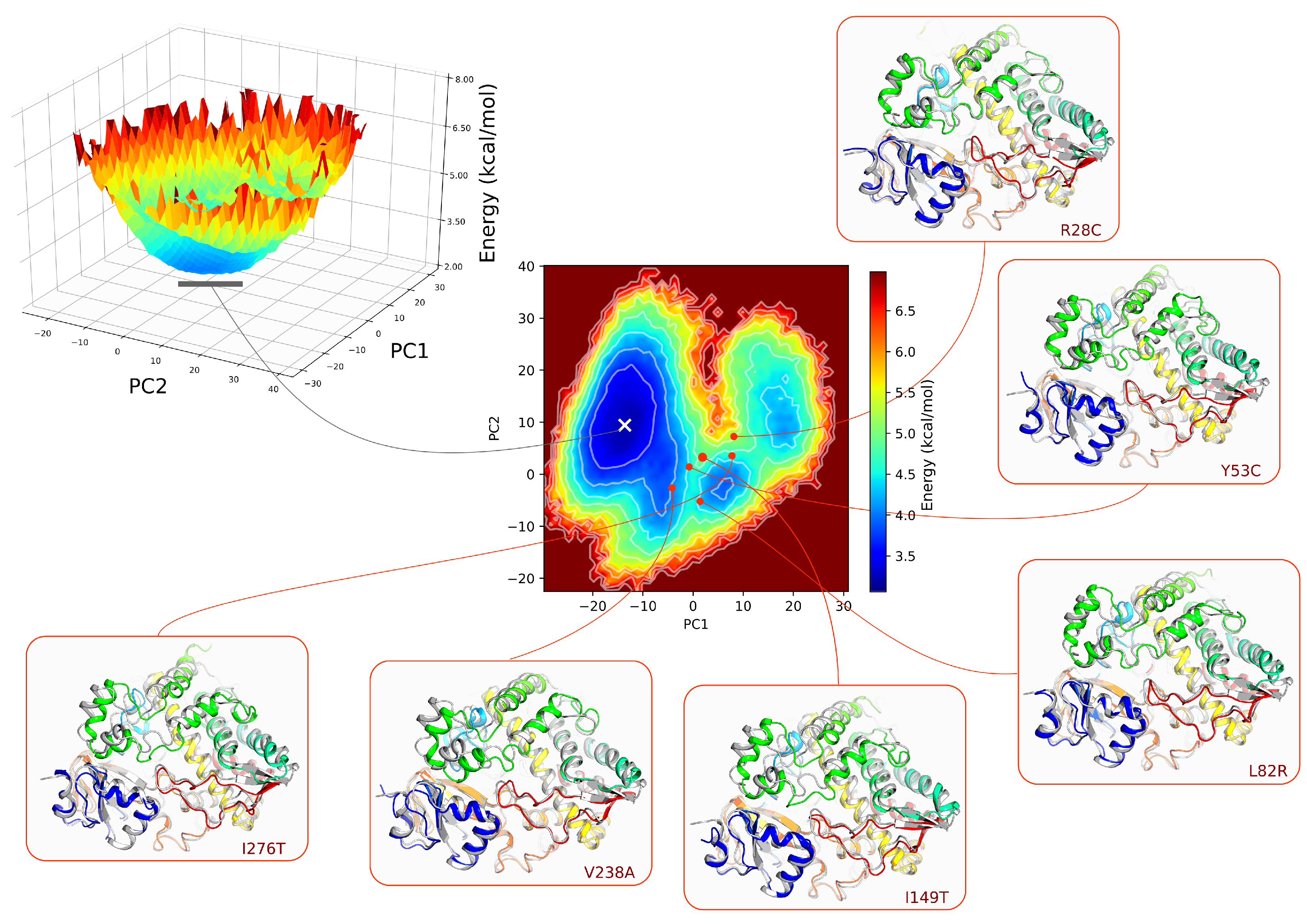
3.5. Analysis of the Free Energy Landscape
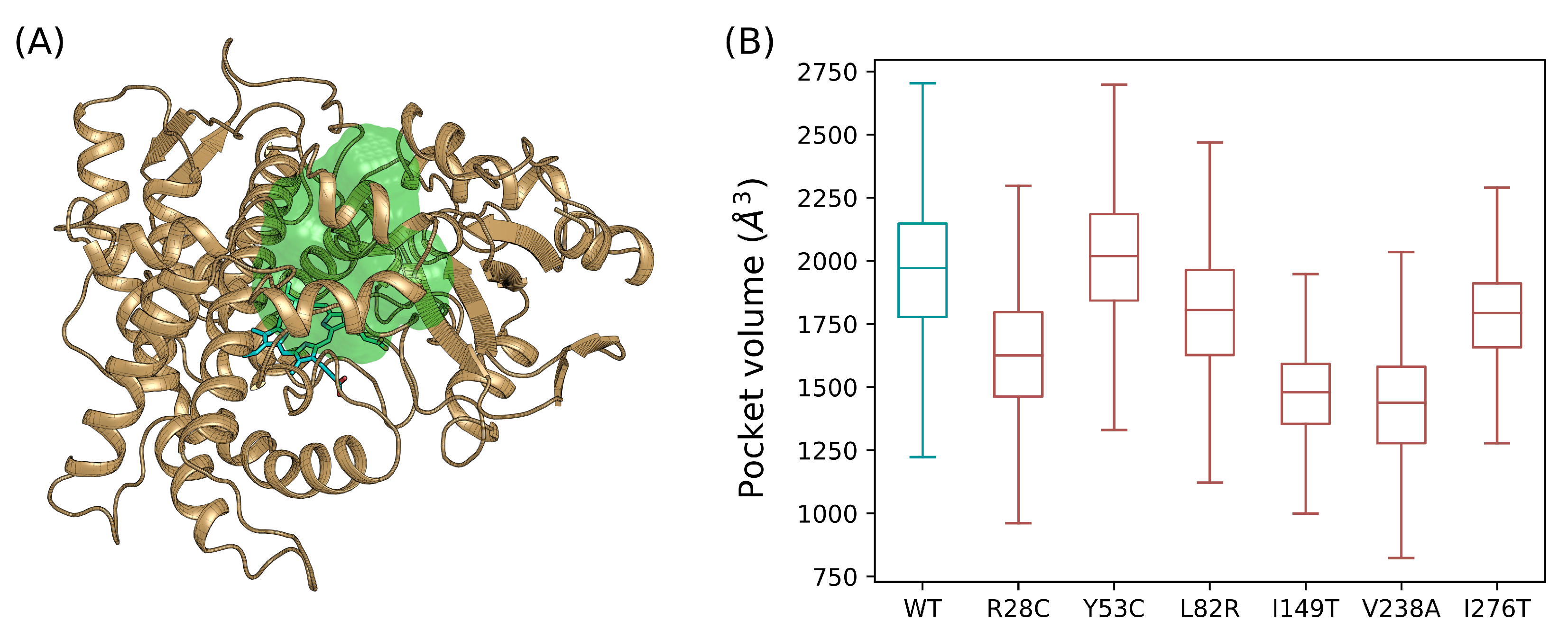
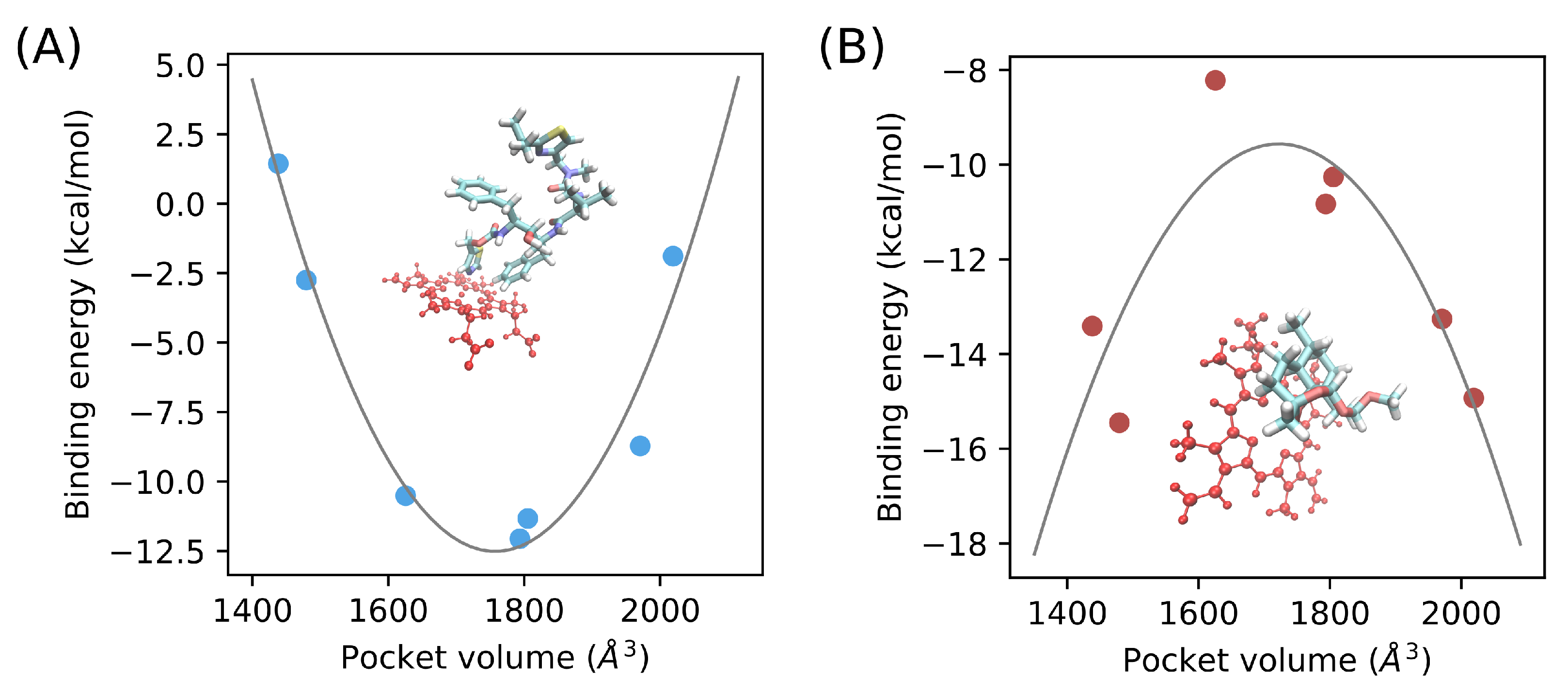
3.6. Catalytic Pocket Volume Calculation
3.7. Binding Free Energy Estimation of CYP3A5/Ritonavir and CYP3A5/Artemether Complexes
3.8. Analysis of the Tunnels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADME | Absorption, Distribution, Metabolism, and Excretion |
| VEP | Variant Effect Predictor |
| GAFF | General Amber Force Field |
| RMSD | Root Mean Square Deviation |
| WT | Wild Type |
| RMSIP | Root Mean Square Inner Product |
| PCA | Principal Component Analysis |
| PC | Principal Component |
| DCCM | Dynamic Cross-Correlation Matrix |
| FEL | Free Energy Landscape |
| HAAD | High coverage African ADME Dataset |
| KGP | 1000 Genomes Project |
| RMSF | Root Mean Square Fluctuation |
References
- Rendic, S.; Guengerich, F.P. Survey of Human Oxidoreductases and Cytochrome P450 Enzymes Involved in the Metabolism of Xenobiotic and Natural Chemicals. Chem. Res. Toxicol. 2015, 28, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Robarge, J.D.; Li, L.; Desta, Z.; Nguyen, A.; Flockhart, D.A. The star-allele nomenclature: Retooling for translational genomics. Clin. Pharmacol. Ther. 2007, 82, 244–248. [Google Scholar] [CrossRef]
- Lamba, J.; Hebert, J.M.; Schuetz, E.G.; Klein, T.E.; Altman, R.B. PharmGKB summary: Very important pharmacogene information for CYP3A5. Pharmacogenet. Genom. 2012, 22, 555–558. [Google Scholar] [CrossRef] [Green Version]
- Mpeta, B.; Kampira, E.; Castel, S.; Mpye, K.L.; Soko, N.D.; Wiesner, L.; Smith, P.; Skelton, M.; Lacerda, M.; Dandara, C. Differences in genetic variants in lopinavir disposition among HIV-infected Bantu Africans. Pharmacogenomics 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Kiaco, K.; Rodrigues, A.S.; do Rosário, V.; Gil, J.P.; Lopes, D. The drug transporter ABCB1 c.3435C>T SNP influences artemether-lumefantrine treatment outcome. Malar J. 2017, 16, 383. [Google Scholar] [CrossRef] [PubMed]
- Bains, R.K. African variation at Cytochrome P450 genes: Evolutionary aspects and the implications for the treatment of infectious diseases. Evol. Med. Public Health 2013, 2013, 118–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.R.; Raza, A.; Firasat, S.; Abid, A. CYP3A5 gene polymorphisms and their impact on dosage and trough concentration of tacrolimus among kidney transplant patients: A systematic review and meta-analysis. Pharmacogenom. J. 2020, 20, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Esu, E.B.; Effa, E.E.; Opie, O.N.; Meremikwu, M.M. Artemether for severe malaria. Cochrane Database Syst. Rev. 2019, 6, CD010678. [Google Scholar] [CrossRef]
- Ernest, C.S.; Hall, S.D.; Jones, D.R. Mechanism-based inactivation of CYP3A by HIV protease inhibitors. J. Pharmacol. Exp. Ther. 2005, 312, 583–591. [Google Scholar] [CrossRef]
- Da Rocha, J.E.B.; Othman, H.; Botha, G.; Cottino, L.; Twesigomwe, D.; Ahmed, S.; Drögemöller, B.I.; Fadlelmola, F.M.; Machanick, P.; Mbiyavanga, M.; et al. The Extent and Impact of Variation in ADME Genes in Sub-Saharan African Populations. Front. Pharmacol. 2021, 12, 366. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Mkrtchian, S.; Kumondai, M.; Hiratsuka, M.; Lauschke, V.M. An optimized prediction framework to assess the functional impact of pharmacogenetic variants. Pharmacogenom. J. 2019, 19, 115–126. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.H.; Johnson, E.F. Active-site differences between substrate-free and ritonavir-bound cytochrome P450 (CYP) 3A5 reveal plasticity differences between CYP3A5 and CYP3A4. J. Biol. Chem. 2019, 294, 8015–8022. [Google Scholar] [CrossRef]
- Hsu, M.H.; Savas, U.; Johnson, E.F. The X-Ray Crystal Structure of the Human Mono-Oxygenase Cytochrome P450 3A5-Ritonavir Complex Reveals Active Site Differences between P450s 3A4 and 3A5. Mol. Pharmacol. 2018, 93, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Mustafa, G.; Nandekar, P.P.; Mukherjee, G.; Bruce, N.J.; Wade, R.C. The Effect of Force-Field Parameters on Cytochrome P450-Membrane Interactions: Structure and Dynamics. Sci. Rep. 2020, 10, 7284. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Shahrokh, K.; Orendt, A.; Yost, G.S.; Cheatham, T.E. Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012, 33, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Moody, P.C.E.; Raven, E.L. The Nature and Reactivity of Ferryl Heme in Compounds I and II. Acc. Chem. Res. 2018, 51, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; van der Heiden, I.P.; Goldstein, J.A.; van Schaik, R.H. A new CYP3A5 variant, CYP3A5*11, is shown to be defective in nifedipine metabolism in a recombinant cDNA expression system. Drug Metab. Dispos. 2007, 35, 67–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, I.T.E.; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z. Interaction entropy: A new paradigm for highly efficient and reliable computation of protein–ligand binding free energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate—DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Schmidtke, P.; Bidon-Chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar] [CrossRef] [Green Version]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.T.; Wilson, R.F.; Rupniewski, I.; Goodin, D.B. P450cam visits an open conformation in the absence of substrate. Biochemistry 2010, 49, 3412–3419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiani, Y.S.; Ranaghan, K.E.; Jabeen, I.; Mulholland, A.J. Molecular Dynamics Simulation Framework to Probe the Binding Hypothesis of CYP3A4 Inhibitors. Int. J. Mol. Sci. 2019, 20, 4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevrioukova, I.F.; Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 2010, 107, 18422–18427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestres, J. Structure conservation in cytochromes P450. Proteins 2005, 58, 596–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Jang, H.; Tsai, C.J.; Cheng, F. Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput. Biol. 2019, 15, e1006658. [Google Scholar]
- Soars, M.G.; Grime, K.; Riley, R.J. Comparative analysis of substrate and inhibitor interactions with CYP3A4 and CYP3A5. Xenobiotica 2006, 36, 287–299. [Google Scholar] [CrossRef]
- Ariza Márquez, Y.V.; Briceño, I.; Aristizábal, F.; Niño, L.F.; Yosa Reyes, J. Dynamic Effects of CYP2D6 Genetic Variants in a Set of Poor Metaboliser Patients with Infiltrating Ductal Cancer Under Treatment with Tamoxifen. Sci. Rep. 2019, 9, 2521. [Google Scholar] [CrossRef]
- Lee, S.J.; Usmani, K.A.; Chanas, B.; Ghanayem, B.; Xi, T.; Hodgson, E.; Mohrenweiser, H.W.; Goldstein, J.A. Genetic findings and functional studies of human CYP3A5 single nucleotide polymorphisms in different ethnic groups. Pharmacogenetics 2003, 13, 461–472. [Google Scholar] [CrossRef]
- De Marco, A.; Vigh, L.; Diamant, S.; Goloubinoff, P. Native folding of aggregation-prone recombinant proteins in Escherichia coli by osmolytes, plasmid- or benzyl alcohol-overexpressed molecular chaperones. Cell Stress Chaperones 2005, 10, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Kuehl, P.; Zhang, J.; Lin, Y.; Lamba, J.; Assem, M.; Schuetz, J.; Watkins, P.B.; Daly, A.; Wrighton, S.A.; Hall, S.D.; et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 2001, 27, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Masimirembwa, C.; Dandara, C.; Hasler, J. Population Diversity and Pharmacogenomics in Africa. In Handbook of Pharmacogenomics and Stratified Medicine; Padmanabhan, S., Ed.; Academic Press: San Diego, CA, USA, 2014; Chapter 43; pp. 971–998. [Google Scholar] [CrossRef]
- Inoue, E.; Takahashi, Y.; Imai, Y.; Kamataki, T. Development of bacterial expression system with high yield of CYP3A7, a human fetus-specific form of cytochrome P450. Biochem. Biophys. Res. Commun. 2000, 269, 623–627. [Google Scholar] [CrossRef]
- Giantin, M.; Rahnasto-Rilla, M.; Tolosi, R.; Lucatello, L.; Pauletto, M.; Guerra, G.; Pezzato, F.; Lopparelli, R.M.; Merlanti, R.; Carnier, P.; et al. Functional impact of cytochrome P450 3A (CYP3A) missense variants in cattle. Sci. Rep. 2019, 9, 19672. [Google Scholar] [CrossRef] [Green Version]
- Emoto, C.; Iwasaki, K. Enzymatic characteristics of CYP3A5 and CYP3A4: A comparison of in vitro kinetic and drug-drug interaction patterns. Xenobiotica 2006, 36, 219–233. [Google Scholar] [CrossRef]
- Saiz-Rodríguez, M.; Almenara, S.; Navares-Gómez, M.; Ochoa, D.; Román, M.; Zubiaur, P.; Koller, D.; Santos, M.; Mejía, G.; Borobia, A.M.; et al. Effect of the Most Relevant CYP3A4 and CYP3A5 Polymorphisms on the Pharmacokinetic Parameters of 10 CYP3A Substrates. Biomedicines 2020, 8, 94. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RS ID | Nucleotide | Amino Acid Variant | Frequency in HAAD + KGP |
|---|---|---|---|
| rs55817950 | c.82C > T | R28C | 0.0021 |
| rs56244447 | c.245T > G | L82R | 0.0054 |
| rs142823108 | c.446T > C | I149T | 0.0043 |
| rs542523237 | c.713T > C | V238A | 0.001 |
| rs145774441 | c.827T > G | I276T | 0.0021 |
| rs72552791 | c.158A > G | Y53C | 0.000 |
| Drug | Variant | (kcal/mol) | (kcal/mol) | (kcal/mol) | (kcal/mol) |
|---|---|---|---|---|---|
| ritonavir | WT | 11.01 | 38.72 | ||
| R28C | 14.01 | 40.66 | |||
| Y53C | 21.08 | 41.11 | |||
| L82R | 12.69 | 36.84 | |||
| I149T | 15.29 | 39.79 | |||
| V238A | +1.44 | 19.45 | 37.96 | ||
| I276T | 13.58 | 35.20 | |||
| artemether | WT | 7.75 | 8.55 | ||
| R28C | 12.08 | 12.85 | |||
| Y53C | 6.13 | 8.24 | |||
| L82R | 10.36 | 10.08 | |||
| I149T | 7.66 | 7.88 | |||
| V238A | 8.43 | 7.74 | |||
| I276T | 9.47 | 9.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Othman, H.; da Rocha, J.E.B.; Hazelhurst, S. Single Nucleotide Polymorphism Induces Divergent Dynamic Patterns in CYP3A5: A Microsecond Scale Biomolecular Simulation of Variants Identified in Sub-Saharan African Populations. Int. J. Mol. Sci. 2021, 22, 7786. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157786
Othman H, da Rocha JEB, Hazelhurst S. Single Nucleotide Polymorphism Induces Divergent Dynamic Patterns in CYP3A5: A Microsecond Scale Biomolecular Simulation of Variants Identified in Sub-Saharan African Populations. International Journal of Molecular Sciences. 2021; 22(15):7786. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157786
Chicago/Turabian StyleOthman, Houcemeddine, Jorge E. B. da Rocha, and Scott Hazelhurst. 2021. "Single Nucleotide Polymorphism Induces Divergent Dynamic Patterns in CYP3A5: A Microsecond Scale Biomolecular Simulation of Variants Identified in Sub-Saharan African Populations" International Journal of Molecular Sciences 22, no. 15: 7786. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157786