
Two Motors and One Spring: Hypothetic Roles of Non-Muscle Myosin II and Submembrane Actin-Based Cytoskeleton in Cell Volume Sensing

 , , , , and
, , , , and 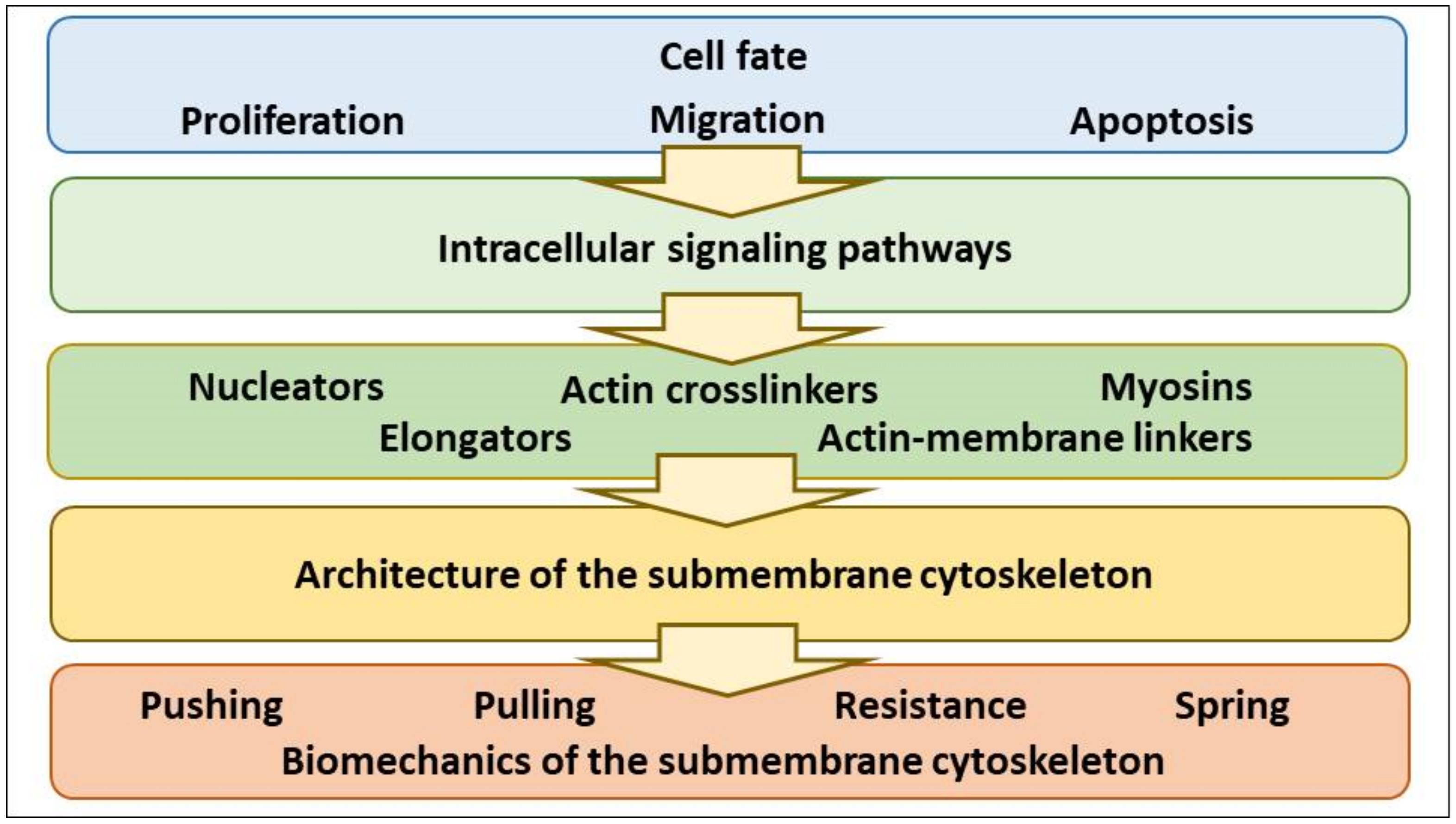{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tuning of Cell Volume Set Point by Cell Program (Proliferation, Apoptosis, Migration) via Control of NMMII, F-Actin Protrusive Force and PM-smACSK
2.1. NMMII, a Pulling Motor
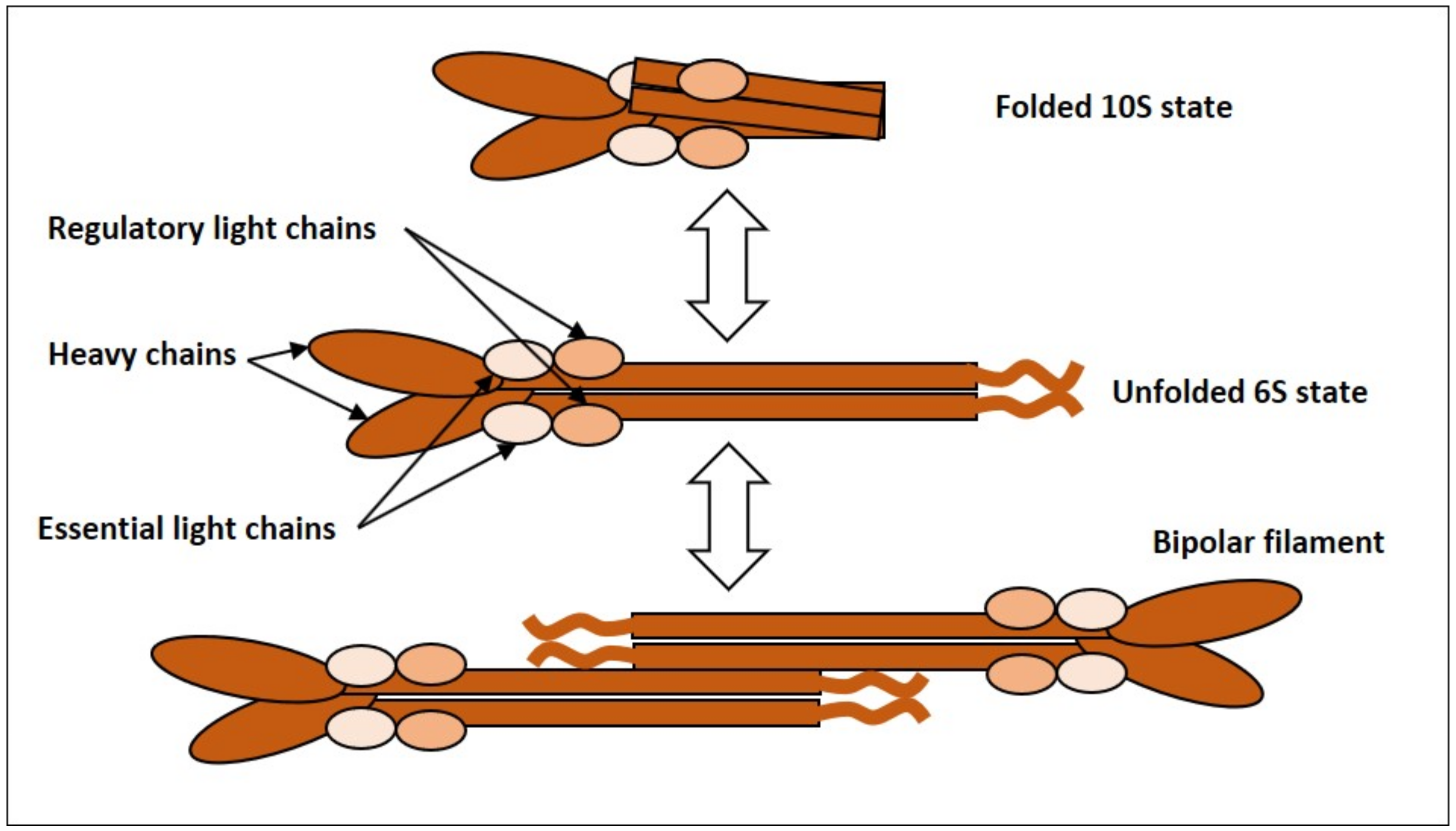
2.1.1. NMMII, Common Features and Regulation
2.1.2. Evidence for Oscillatory Activity of NMMII
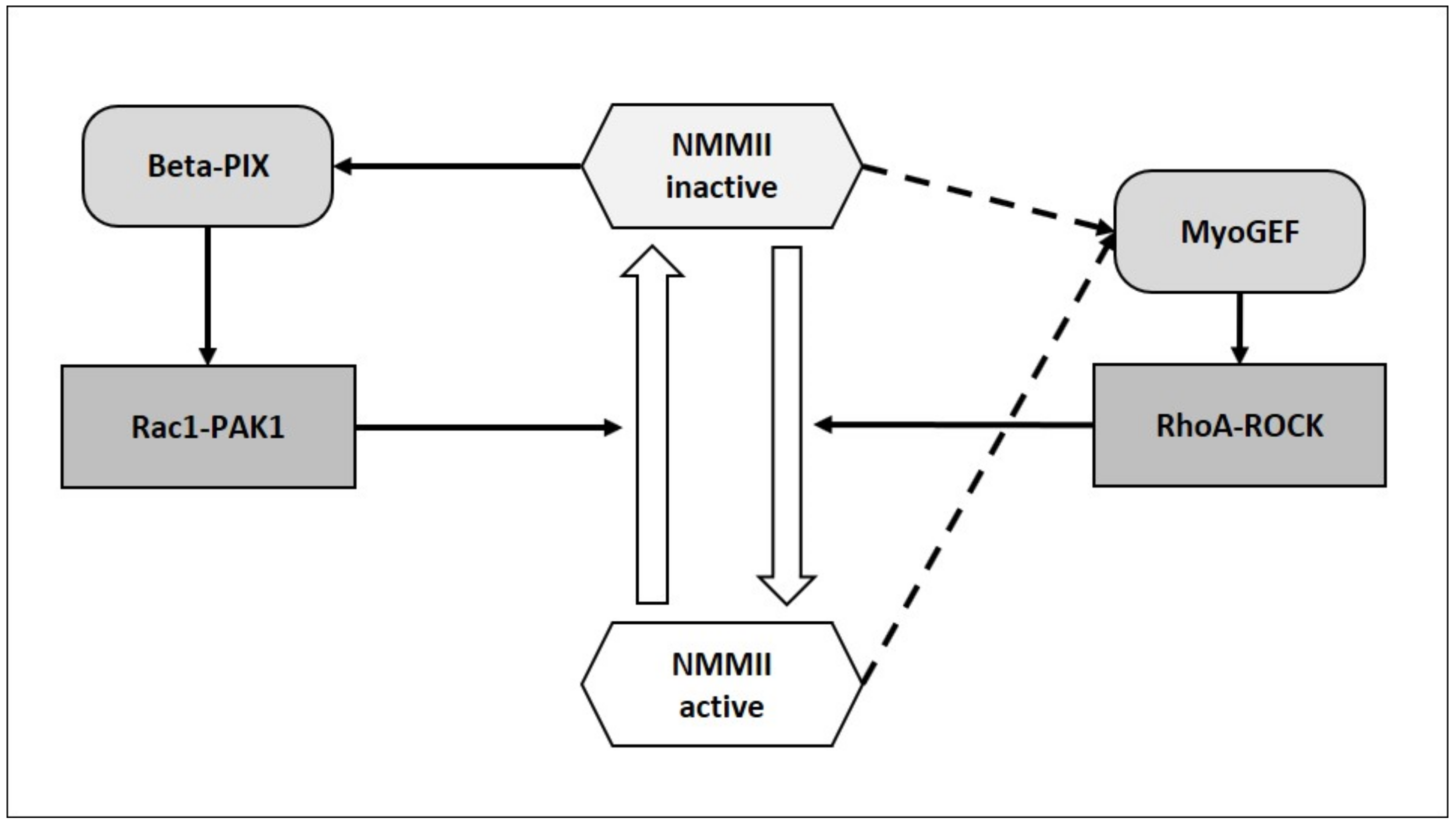
2.1.3. Can NMMII Reset Its Own Oscillatory Activity?
2.2. A Viscoelactic Solid Composed of the PM and the smACSK, a Pushing Motor, a Resistive Force and a Spring
2.2.1. The PM-smACSK Complex, Common Features
2.2.2. Protrusive F-Actin Polymerization, a Pushing Motor
2.2.3. A Resistive Force Generated by the PM-smACSK
2.2.4. Spring-Like Behavior of the PM—smACSK
2.3. Cell Fates Requiring Volume Change
2.3.1. Proliferation
2.3.2. Migration
2.3.3. Apoptosis
3. “Two Motors and One Spring” Model
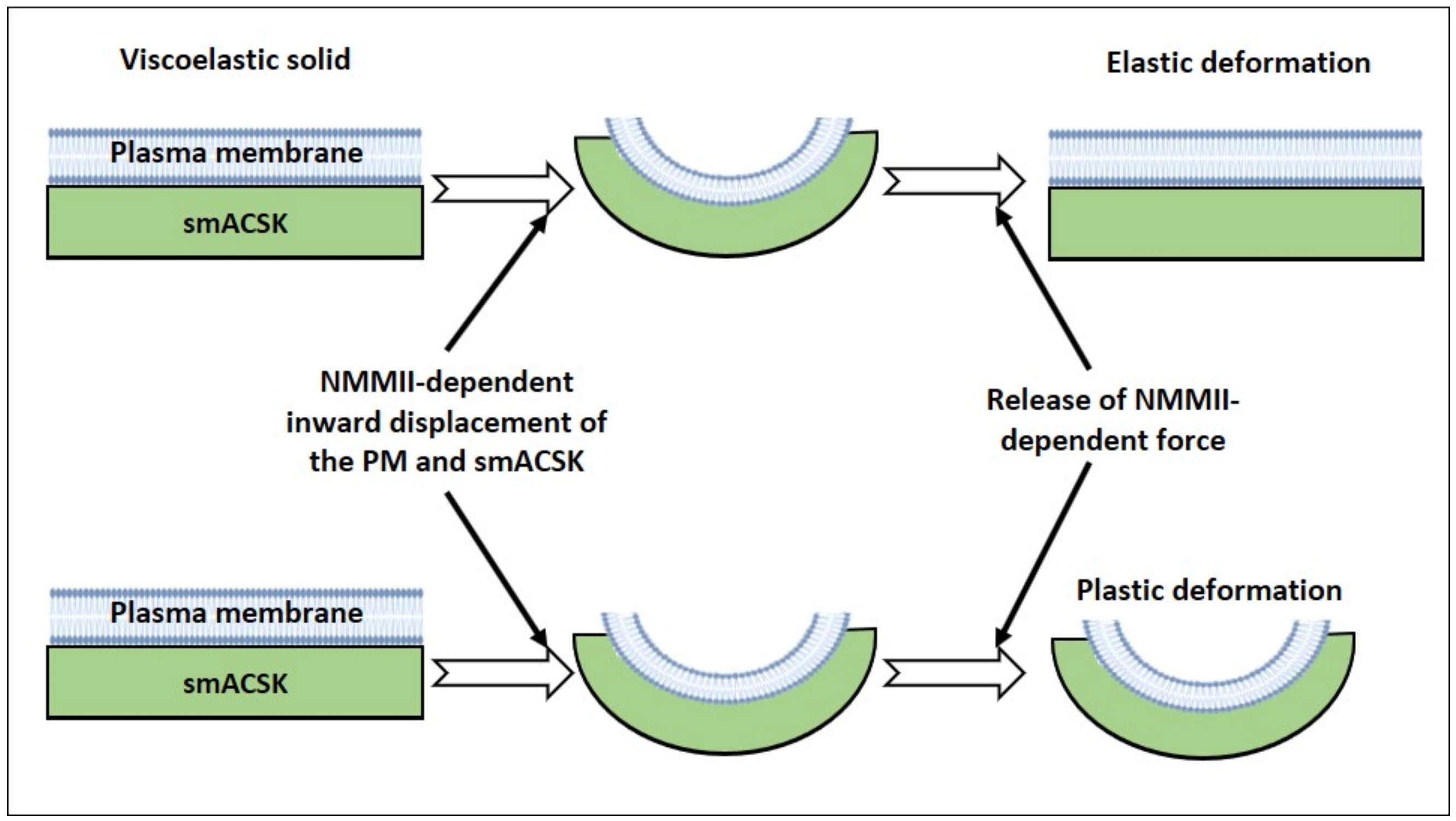
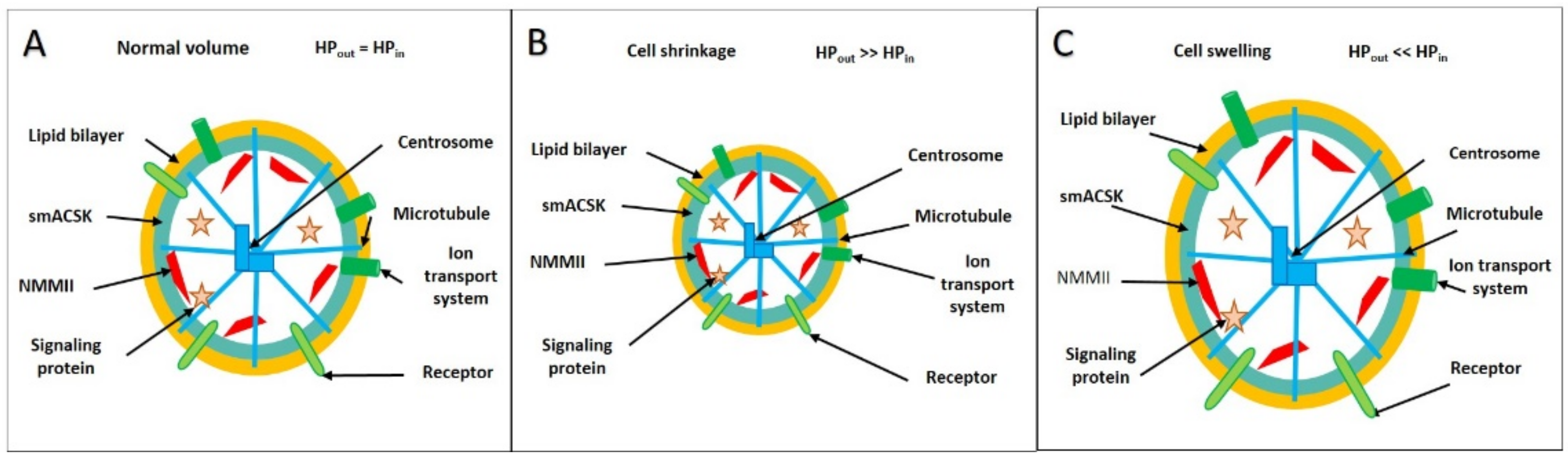
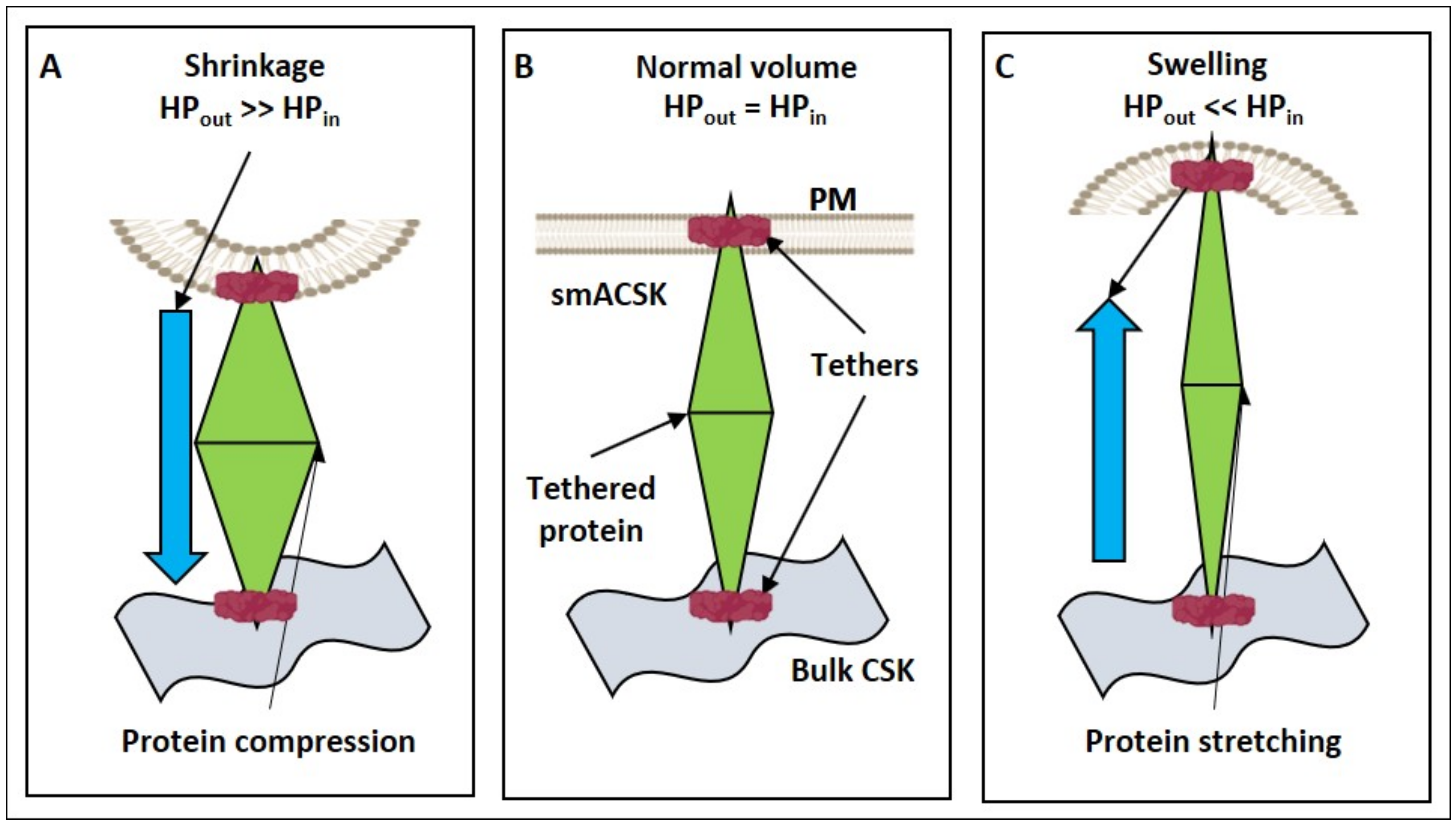
3.1. Hyperosmolarity-Driven Inward Movement of the PM—smACSK versus F-Actin-Driven Outward Movement of the PM—smACSK: Sensing of Cell Shrinkage
3.2. Relaxation of Both F-Actin-Driven Force and the PM—smACSK Spring
3.3. Hyposmolarity-Driven Outward Movement of the PM—smACSK versus NMMII-Driven Inward Movement of the PM—smACSK: Sensing of Cell Swelling
3.4. Relaxation of the Both NMMII and the PM—smACSK Spring
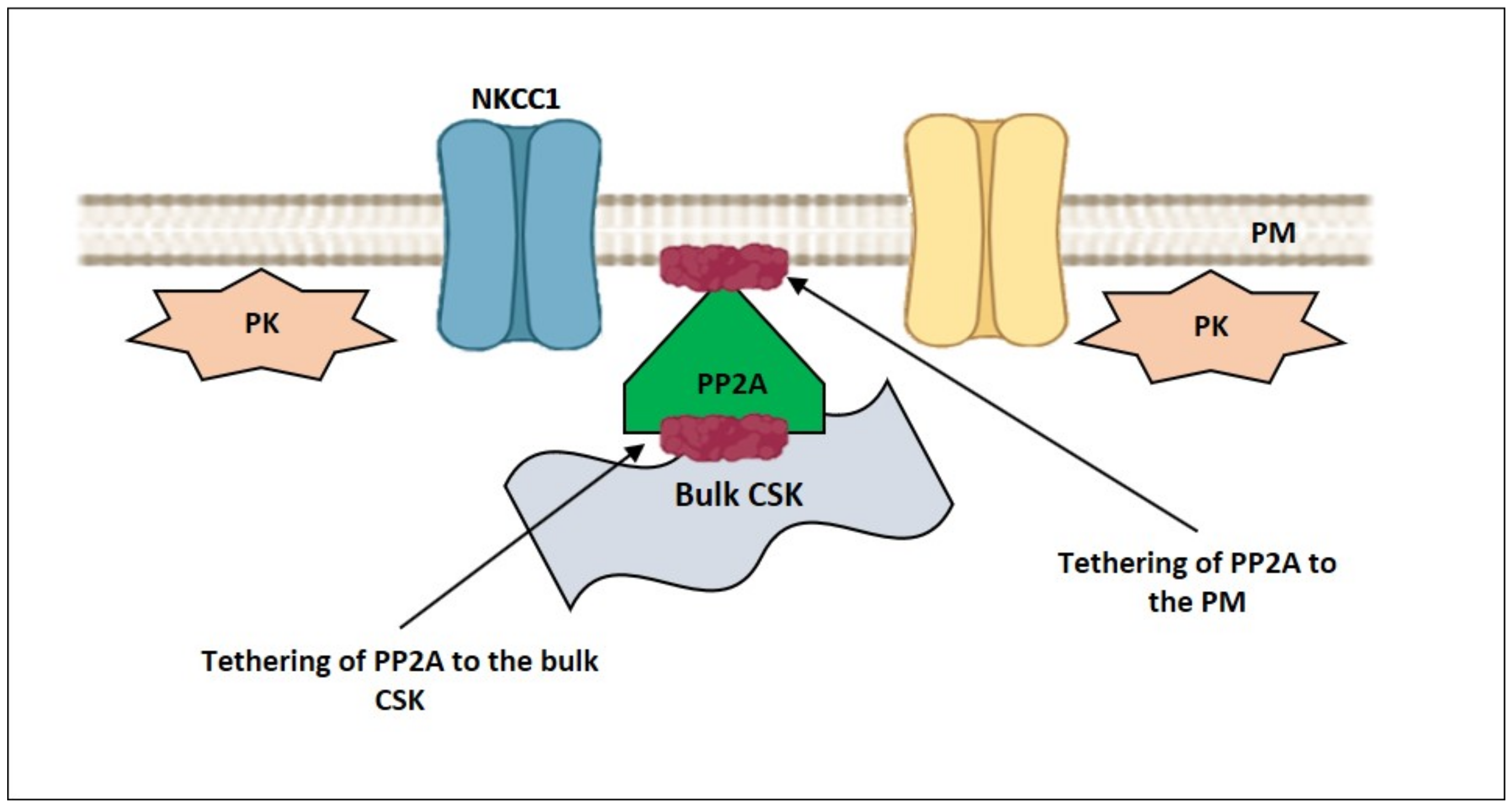
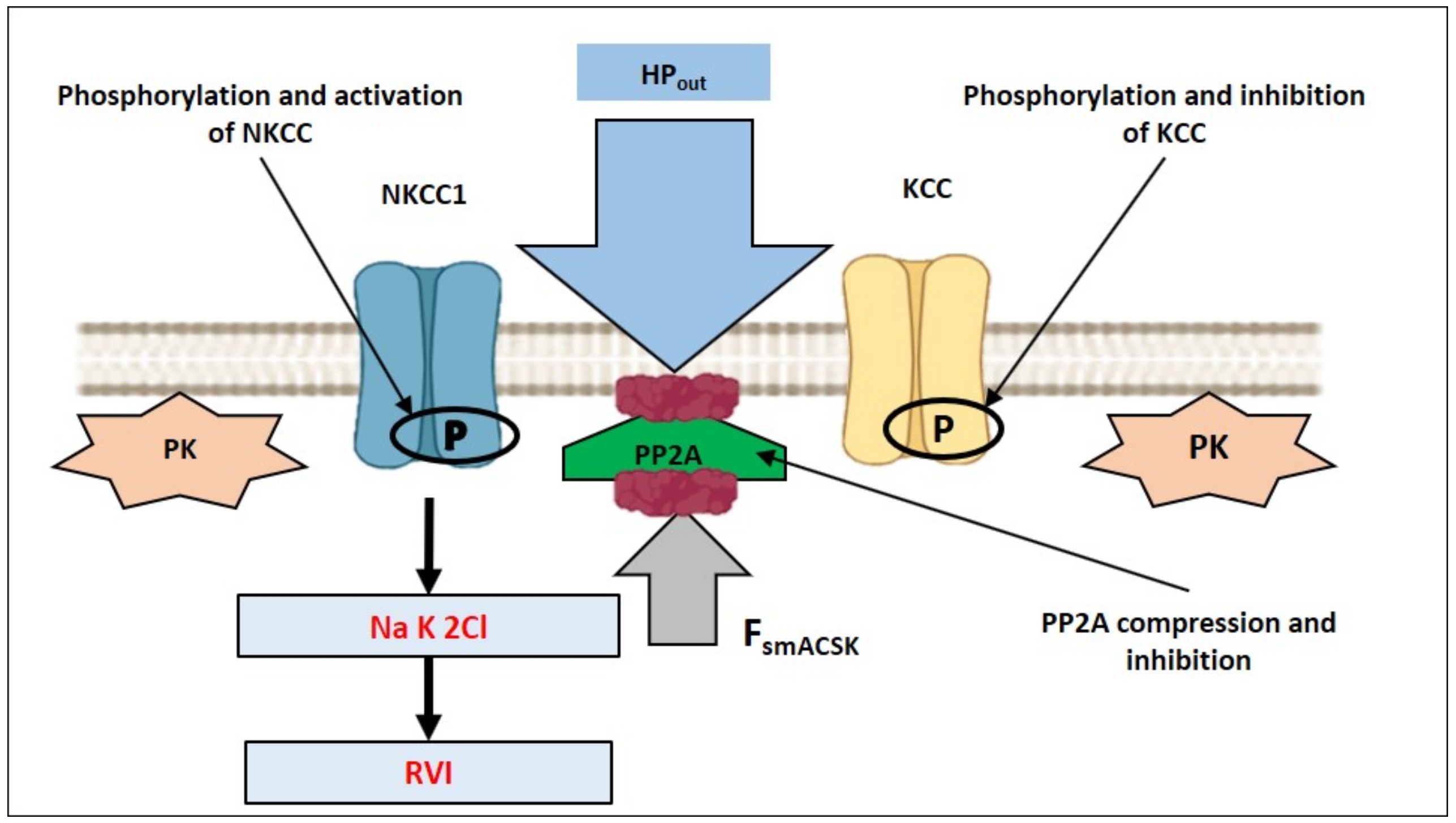
4. How Can NMMII, and the PM—smACSK Complex Sense Intracellular Ionic Strength?
5. NMMII Can Directly Interact with and Modulate Activities of Transmembrane Mechanosensors of the PM
5.1. Anionic Lipids
5.2. Ion Channels
5.3. Integrins
5.4. G protein Coupled Receptors (GPCRs)
5.5. Growth Factor Receptors or Receptor Tyrosine Kinases (RTKs)
5.6. Angiotensin-Converting Enzyme (ACE)
6. Towards the Experimental Testing of the Hypothesis in Live Cell in Real Time
- Visualization of activation of cell surface receptors—that is observation of signaling that fine-tune the NMMII and smACSK;
- Visualization of signaling proteins that transmit signal from cell surface receptors onto NMMII and smACSK;
- Observation of activation of NMMII and re-arrangements of the smACSK;
- Recording of pulling force and pushing force exerted by NMMII and actin protrusions, respectively;
- Visualization of activation of transmembrane signaling proteins by NMMII-driven pulling force and actin protrusion-driven pushing force;
- Observation of activation of signaling proteins that transduce signal from the transmembrane signaling proteins onto ion transport systems;
- Observation of activation of ion transport systems responsible for RVI and RVD.
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lang, F.; Ritter, M.; Gamper, N.; Huber, S.; Fillon, S.; Tanneur, V.; Lepple-Wienhues, A.; Szabo, I.; Bulbins, E. Cell Volume in the Regulation of Cell Proliferation and Apoptotic Cell Death. Cell. Physiol. Biochem. 2000, 10, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of Cell Volume Regulation in Vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef]
- Delpire, E.; Gagnon, K.B. Water Homeostasis and Cell Volume Maintenance and Regulation. Curr. Top. Membr. 2018, 81, 3–52. [Google Scholar]
- Föller, M.; Lang, F. Ion Transport in Eryptosis, the Suicidal Death of Erythrocytes. Front. Cell Dev. Biol. 2020, 8, 597. [Google Scholar] [CrossRef]
- Wilson, C.S.; Mongin, A.A. Chapter Eleven—Cell Volume Control in Healthy Brain and Neuropathologies. Curr. Top. Membr. 2018, 81, 385–455. [Google Scholar]
- Bortner, C.D.; Cidlowski, J.A. Ions, the Movement of Water and the Apoptotic Volume Decrease. Front. Cell Dev. Biol. 2020, 8, 1415. [Google Scholar] [CrossRef]
- Okada, Y.; Okada, T.; Sato-Numata, K.; Islam, M.R.; Ando-Akatsuka, Y.; Numata, T.; Kubo, M.; Shimizu, T.; Kurbannazarova, R.S.; Marunaka, Y.; et al. Cell Volume-Activated and Volume-Correlated Anion Channels in Mammalian Cells: Their Biophysical, Molecular, and Pharmacological Properties. Pharmacol. Rev. 2019, 71, 49–88. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Sabirov, R.Z.; Sato-Numata, K.; Numata, T. Cell Death Induction and Protection by Activation of Ubiquitously Expressed Anion/Cation Channels. Part 1: Roles of VSOR/VRAC in Cell Volume Regulation, Release of Double-Edged Signals and Apoptotic/Necrotic Cell Death. Front. Cell Dev. Biol. 2021, 8, 1776. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Counillon, L. The SLC9A-C Mammalian Na+/H+ Exchanger Family: Molecules, Mechanisms, and Physiology. Physiol. Rev. 2019, 99, 2015–2113. [Google Scholar] [CrossRef] [PubMed]
- Adragna, N.C.; Ferrell, C.M.; Zhang, J.; Di Fulvio, M.; Temprana, C.F.; Sharma, A.; Fyffe, R.E.W.; Cool, D.R.; Lauf, P.K. Signal transduction mechanisms of K+-Cl− cotransport regulation and relationship to disease. Acta Physiol. 2006, 187, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.K.; Pedersen, S.F. Shrinkage insensitivity of NKCC1 in myosin II-depleted cytoplasts from Ehrlich ascites tumor cells. Am. J. Physiol. Cell Physiol. 2007, 292, C1854–C1866. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.D.; O’Neill, W.C. Volume-sensitive myosin phosphorylation in vascular endothelial cells: Correlation with Na-K-2Cl cotransport. Am. J. Physiol. Cell Physiol. 1995, 269, C1524–C1531. [Google Scholar] [CrossRef] [PubMed]
- Di Ciano-Oliveira, C.D.; Sirokmány, G.; Szászi, K.; Arthur, W.T.; Masszi, A.; Peterson, M.; Rotstein, O.D.; Kapus, A. Hyperosmotic stress activates Rho: Differential involvement in Rho kinase-dependent MLC phosphorylation and NKCC activation. Am. J. Physiol. Cell Physiol. 2003, 285, C555–C566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ciano-Oliveira, C.D.; Lodyga, M.; Fan, L.; Szászi, K.; Hosoya, H.; Rotstein, O.D.; Kapus, A. Is myosin light-chain phosphorylation a regulatory signal for the osmotic activation of the Na+-K+-2Cl− cotransporter? Am. J. Physiol. Cell Physiol. 2005, 289, C68–C81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishima, S.; Shimizu, T.; Kida, H.; Okada, Y. Volume Expansion Sensitivity of Swelling-Activated Cl− Channel in Human Epithelial Cells. Jpn. J. Physiol. 2000, 50, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Billington, N.; Wang, A.; Mao, J.; Adelstein, R.S.; Sellers, J.R. Characterization of Three Full-length Human Nonmuscle Myosin II Paralogs. J. Biol. Chem. 2013, 288, 33398–33410. [Google Scholar] [CrossRef] [Green Version]
- Dulyaninova, N.G.; Bresnick, A.R. The heavy chain has its day. BioArchitecture 2013, 3, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Shutova, M.S.; Svitkina, T.M. Common and Specific Functions of Nonmuscle Myosin II Paralogs in Cells. Biochem. Mosc. 2018, 83, 1459–1468. [Google Scholar] [CrossRef]
- Sellers, J.R.; Heissler, S.M. Nonmuscle myosin-2 isoforms. Curr. Biol. 2019, 29, R275–R278. [Google Scholar] [CrossRef]
- Kamm, K.E.; Stull, J.T. Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 2001, 276, 4527–4530. [Google Scholar] [CrossRef] [Green Version]
- Hartshorne, D.J.; Ito, M.; Erdodi, F. Myosin light chain phosphatase: Subunit composition, interactions and regulation. J. Muscle Res. Cell Motil. 1998, 19, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Hartshorne, D. Myosin phosphatase: Subunits and interactions. Acta Physiol. Scand. 1998, 164, 483–493. [Google Scholar] [CrossRef]
- Kiss, A.; Erdődi, F.; Lontay, B. Myosin phosphatase: Unexpected functions of a long-known enzyme. Biochim. Biophys. Acta BBA Mol. Cell Res. 2019, 1866, 2–15. [Google Scholar] [CrossRef]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef]
- Sanders, L.C.; Matsumura, F.; Bokoch, G.M.; De Lanerolle, P. Inhibition of myosin light chain kinase by p21-activated kinase. Science 1999, 283, 2083–2085. [Google Scholar] [CrossRef]
- Van Leeuwen, F.N.; Van Delft, S.; Kain, H.E.; Van Der Kammen, R.A.; Collard, J.G. Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nat. Cell Biol. 1999, 1, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Giannone, G.; Dubin-Thaler, B.J.; Rossier, O.; Cai, Y.; Chaga, O.; Jiang, G.; Beaver, W.; Döbereiner, H.-G.; Freund, Y.; Borisy, G. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 2007, 128, 561–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, M.A.; Billington, N.; Wang, A.; Adelstein, R.S.; Sellers, J.R.; Fischer, R.S.; Waterman, C.M. Local pulsatile contractions are an intrinsic property of the myosin 2A motor in the cortical cytoskeleton of adherent cells. Mol. Biol. Cell 2016, 28, 240–251. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Hall, A. Guanine nucleotide exchange factors for Rho GTPases: Turning on the switch. Genes Dev. 2002, 16, 1587–1609. [Google Scholar] [CrossRef] [Green Version]
- Toma-Fukai, S.; Shimizu, T. Structural Insights into the Regulation Mechanism of Small GTPases by GEFs. Molecules 2019, 24, 3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-S.; Choi, C.-K.; Shin, E.-Y.; Schwartz, M.A.; Kim, E.-G. Myosin II directly binds and inhibits Dbl family guanine nucleotide exchange factors: A possible link to Rho family GTPases. J. Cell Biol. 2010, 190, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokoch, G.M. Biology of the p21-activated kinases. Annu. Rev. Biochem. 2003, 72, 743–781. [Google Scholar] [CrossRef]
- Sander, E.E.; ten Klooster, J.P.; van Delft, S.; van der Kammen, R.A.; Collard, J.G. Rac downregulates Rho activity: Reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999, 147, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Nimnual, A.S.; Taylor, L.J.; Bar-Sagi, D. Redox-dependent downregulation of Rho by Rac. Nat. Cell Biol. 2003, 5, 236–241. [Google Scholar] [CrossRef]
- Wu, D.; Asiedu, M.; Adelstein, R.; Wei, Q. A Novel Guanine Nucleotide Exchange Factor, MyoGEF, is Required for Cytokinesis. Cell Cycle 2006, 5, 1234–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamill, O.P.; Martinac, B. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 2001, 81, 685–740. [Google Scholar] [CrossRef]
- Jacobs, M.L.; Faizi, H.A.; Peruzzi, J.A.; Vlahovska, P.M.; Kamat, N.P. EPA and DHA differentially modulate membrane elasticity in the presence of cholesterol. Biophys. J. 2021, 120, 2317–2329. [Google Scholar] [CrossRef]
- Barvitenko, N.; Aslam, M.; Lawen, A.; Pantaleo, A.; Saldanha, C.; Matteucci, E. Effects of oxygen depletion on transmembrane protein activities. Curr. Org. Chem. 2015, 19, 2002–2010. [Google Scholar] [CrossRef]
- Bennett, V.; Baines, A.J. Spectrin and ankyrin-based pathways: Metazoan inventions for integrating cells into tissues. Physiol. Rev. 2001, 81, 1353–1392. [Google Scholar] [CrossRef] [Green Version]
- Kapus, A.; Janmey, P. Plasma membrane—Cortical cytoskeleton interactions: A cell biology approach with biophysical considerations. Compr. Physiol. 2013, 3, 1231–1281. [Google Scholar] [PubMed]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, S.; Saldanha, C. An overview about erythrocyte membrane. Clin. Hemorheol. Microcirc. 2010, 44, 63–74. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, S.; Silva-Herdade, A.S.; Saldanha, C. Modulation of erythrocyte deformability by PKC activity. Clin. Hemorheol. Microcirc. 2008, 39, 363–373. [Google Scholar] [CrossRef]
- Saldanha, C.; Silva, A.S.; Gonçalves, S.; Martins-Silva, J. Modulation of erythrocyte hemorheological properties by band 3 phosphorylation and dephosphorylation. Clin. Hemorheol. Microcirc. 2007, 36, 183–194. [Google Scholar]
- de Almeida, J.P.L.; Freitas-Santos, T.; Saldanha, C. Erythrocyte deformability dependence on band 3 protein in an in-vitro model of hyperfibrinogenemia. Clin. Hemorheol. Microcirc. 2012, 50, 213–219. [Google Scholar] [CrossRef]
- Pantaleo, A.; Ferru, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Mattè, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 Erythrocyte Membrane Protein Acts as Redox Stress Sensor Leading to Its Phosphorylation by p72 Syk. Oxid. Med. Cell. Longev. 2016, 2016, 6051093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferru, E.; Giger, K.; Pantaleo, A.; Campanella, E.; Grey, J.; Ritchie, K.; Vono, R.; Turrini, F.; Low, P.S. Regulation of membrane-cytoskeletal interactions by tyrosine phosphorylation of erythrocyte band 3. Blood 2011, 117, 5998–6006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Huang, Z.; Wu, Z.; Ali, A.; Qian, A. Mammalian plakins, giant cytolinkers: Versatile biological functions and roles in cancer. Int. J. Mol. Sci. 2018, 19, 974. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Su, P.; Li, R.; Yin, C.; Zhang, Y.; Shang, P.; Yang, T.; Qian, A. Isoforms, structures, and functions of versatile spectraplakin MACF1. BMB Rep. 2016, 49, 37. [Google Scholar] [CrossRef] [Green Version]
- Barvitenko, N.; Lawen, A.; Aslam, M.; Pantaleo, A.; Saldanha, C.; Skverchinskaya, E.; Regolini, M.; Tuszynski, J.A. Integration of intracellular signaling: Biological analogues of wires, processors and memories organized by a centrosome 3D reference system. Comput. Theor. Exp. Approaches Morphog. 2018, 173, 191–206. [Google Scholar] [CrossRef]
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Svitkina, T.M. Ultrastructure of protrusive actin filament arrays. Curr. Opin. Cell Biol. 2013, 25, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Svitkina, T.M. Ultrastructure of the actin cytoskeleton. Curr. Opin. Cell Biol. 2018, 54, 1–8. [Google Scholar] [CrossRef]
- Lieber, A.D.; Yehudai-Resheff, S.; Barnhart, E.L.; Theriot, J.A.; Keren, K. Membrane Tension in Rapidly Moving Cells Is Determined by Cytoskeletal Forces. Curr. Biol. 2013, 23, 1409–1417. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Li, L.; Ballermann, B.; Wang, Z. Phosphorylation of Rac1 T108 by extracellular signal-regulated kinase in response to epidermal growth factor: A novel mechanism to regulate Rac1 function. Mol. Cell. Biol. 2013, 33, 4538–4551. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Li, L.; Ballermann, B.; Wang, Z. Phosphorylation and activation of RhoA by ERK in response to epidermal growth factor stimulation. PLoS ONE 2016, 11, e0147103. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Somlyo, A.P.; Somlyo, A.V. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J. Physiol. 2000, 522, 177–185. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase signalling in cell migration. Cell Adhes. Migr. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, L.; Qu, R.; Zhang, L.; Huang, W. Rho A Regulates Epidermal Growth Factor-Induced Human Osteosarcoma MG63 Cell Migration. Int. J. Mol. Sci. 2018, 19, 1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakiashvili, E.; Dan, Q.; Vandermeer, M.; Zhang, Y.; Waheed, F.; Pham, M.; Szászi, K. The Epidermal Growth Factor Receptor Mediates Tumor Necrosis Factor-α-induced Activation of the ERK/GEF-H1/RhoA Pathway in Tubular Epithelium. J. Biol. Chem. 2011, 286, 9268–9279. [Google Scholar] [CrossRef] [Green Version]
- Rao, T.C.; Ma, V.P.-Y.; Blanchard, A.; Urner, T.M.; Grandhi, S.; Salaita, K.; Mattheyses, A.L. EGFR activation attenuates the mechanical threshold for integrin tension and focal adhesion formation. J. Cell Sci. 2020, 133, jcs238840. [Google Scholar] [CrossRef]
- Bagnato, A.; Rosanò, L. New Routes in GPCR/β-Arrestin-Driven Signaling in Cancer Progression and Metastasis. Front. Pharmacol. 2019, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Juárez, E.; Ramos-Mandujano, G.; Hernández-Benítez, R.; Pasantes-Morales, H. On the Role of G-Protein Coupled Receptors in Cell Volume Regulation. Cell. Physiol. Biochem. 2008, 21, 001–014. [Google Scholar] [CrossRef]
- Rubtsova, S.N.; Zhitnyak, I.Y.; Gloushankova, N.A. Phenotypic Plasticity of Cancer Cells Based on Remodeling of the Actin Cytoskeleton and Adhesive Structures. Int. J. Mol. Sci. 2021, 22, 1821. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of Ion Channels and Transporters in Cell Migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Biol. 2004, 265, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Straussman, R.; Even, L.; Ravid, S. Myosin II heavy chain isoforms are phosphorylated in an EGF-dependent manner: Involvement of protein kinase C. J. Cell Sci. 2001, 114, 3047–3057. [Google Scholar] [CrossRef]
- Kolega, J. Asymmetric distribution of myosin IIB in migrating endothelial cells is regulated by a rho-dependent kinase and contributes to tail retraction. Mol. Biol. Cell 2003, 14, 4745–4757. [Google Scholar] [CrossRef] [Green Version]
- Kolega, J. The role of myosin II motor activity in distributing myosin asymmetrically and coupling protrusive activity to cell translocation. Mol. Biol. Cell 2006, 17, 4435–4445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandquist, J.C.; Means, A.R. The C-Terminal Tail Region of Nonmuscle Myosin II Directs Isoform-specific Distribution in Migrating Cells. Mol. Biol. Cell 2008, 19, 5156–5167. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Manzanares, M.; Zareno, J.; Whitmore, L.; Choi, C.K.; Horwitz, A.F. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol. 2007, 176, 573–580. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Liu, Q.; Zhang, Y.; Gao, Z.; Yin, M.; Jiang, N.; Cao, G.; Yu, B.; Cao, Z.; et al. Myosin IIA-related Actomyosin Contractility Mediates Oxidative Stress-induced Neuronal Apoptosis. Front. Mol. Neurosci. 2017, 10, 75. [Google Scholar] [CrossRef] [Green Version]
- Lang, E.; Qadri, S.M.; Zelenak, C.; Gu, S.; Rotte, A.; Draeger, A.; Lang, F. Inhibition of suicidal erythrocyte death by blebbistatin. Am. J. Physiol. Cell Physiol. 2011, 301, C490–C498. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Cheng, C.; Wang, M.; Jiang, P.; Zhang, L.; Wang, Y.; Wu, H.; Zeng, X.; Wang, H.; Gao, X.; et al. Blebbistatin Inhibits Neomycin-Induced Apoptosis in Hair Cell-Like HEI-OC-1 Cells and in Cochlear Hair Cells. Front. Cell. Neurosci. 2020, 13, 590. [Google Scholar] [CrossRef]
- Schein, S.J.; Colombini, M.; Finkelstein, A. Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J. Membr. Biol. 1976, 30, 99–120. [Google Scholar] [CrossRef]
- De Pinto, V.; Messina, A.; Lane, D.J.; Lawen, A. Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett. 2010, 584, 1793–1799. [Google Scholar] [CrossRef] [Green Version]
- Lisanti, M.P.; Scherer, P.E.; Vidugiriene, J.; Tang, Z.; Hermanowski-Vosatka, A.; Tu, Y.-H.; Cook, R.F.; Sargiacomo, M. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: Implications for human disease. J. Cell Biol. 1994, 126, 111–126. [Google Scholar] [CrossRef]
- Fernandez-Echevarria, C.; Díaz, M.; Ferrer, I.; Canerina-Amaro, A.; Marin, R. Aβ promotes VDAC1 channel dephosphorylation in neuronal lipid rafts. Relevance to the mechanisms of neurotoxicity in Alzheimer’s disease. Neuroscience 2014, 278, 354–366. [Google Scholar] [CrossRef]
- Okada, S.F.; O’Neal, W.K.; Huang, P.; Nicholas, R.A.; Ostrowski, L.E.; Craigen, W.J.; Lazarowski, E.R.; Boucher, R.C. Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J. Gen. Physiol. 2004, 124, 513–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Forbes, J.; Colombini, M. Actin modulates the gating of Neurospora crassa VDAC. J. Membr. Biol. 2001, 180, 73–81. [Google Scholar] [CrossRef]
- Roman, I.; Figys, J.; Steurs, G.; Zizi, M. Direct measurement of VDAC–actin interaction by surface plasmon resonance. Biochim. Biophys. Acta BBA Biomembr. 2006, 1758, 479–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elinder, F.; Akanda, N.; Tofighi, R.; Shimizu, S.; Tsujimoto, Y.; Orrenius, S.; Ceccatelli, S. Opening of plasma membrane voltage-dependent anion channels (VDAC) precedes caspase activation in neuronal apoptosis induced by toxic stimuli. Cell Death Differ. 2005, 12, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Grinthal, A.; Adamovic, I.; Weiner, B.; Karplus, M.; Kleckner, N. PR65, the HEAT-repeat scaffold of phosphatase PP2A, is an elastic connector that links force and catalysis. Proc. Natl. Acad. Sci. USA 2010, 107, 2467–2472. [Google Scholar] [CrossRef] [Green Version]
- Liedtke, C.M.; Wang, X.; Smallwood, N.D. Role for Protein Phosphatase 2A in the Regulation of Calu-3 Epithelial Na+-K+-2Cl–, Type 1 Co-transport Function. J. Biol. Chem. 2005, 280, 25491–25498. [Google Scholar] [CrossRef] [Green Version]
- Bize, I.; Güvenç, B.; Buchbinder, G.; Brugnara, C. Stimulation of Human Erythrocyte K-Cl Cotransport and Protein Phosphatase Type 2A by n-Ethylmaleimide: Role of Intracellular Mg++. J. Membr. Biol. 2000, 177, 159–168. [Google Scholar] [CrossRef]
- Fowler, V.M.; Davis, J.Q.; Bennett, V. Human erythrocyte myosin: Identification and purification. J. Cell Biol. 1985, 100, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.S.; Nowak, R.B.; Zhou, S.; Giannetto, M.; Gokhin, D.S.; Papoin, J.; Ghiran, I.C.; Blanc, L.; Wan, J.; Fowler, V.M. Myosin IIA interacts with the spectrin-actin membrane skeleton to control red blood cell membrane curvature and deformability. Proc. Natl. Acad. Sci. USA 2018, 115, E4377–E4385. [Google Scholar] [CrossRef] [Green Version]
- Straussman, R.; Squire, J.M.; Ben-Ya’acov, A.; Ravid, S. Skip residues and charge interactions in myosin II coiled-coils: Implications for molecular packing. J. Mol. Biol. 2005, 353, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Straussman, R.; Ben-Ya’acov, A.; Ronen, D.; Ravid, S. MHC-IIB filament assembly and cellular localization are governed by the rod net charge. PLoS ONE 2008, 3, e1496. [Google Scholar] [CrossRef] [Green Version]
- Schroer, C.F.E.; Baldauf, L.; van Buren, L.; Wassenaar, T.A.; Melo, M.N.; Koenderink, G.H.; Marrink, S.J. Charge-dependent interactions of monomeric and filamentous actin with lipid bilayers. Proc. Natl. Acad. Sci. USA 2020, 117, 5861–5872. [Google Scholar] [CrossRef] [Green Version]
- Beck, M.R.; Dixon, R.D.S.; Goicoechea, S.M.; Murphy, G.S.; Brungardt, J.G.; Beam, M.T.; Srinath, P.; Patel, J.; Mohiuddin, J.; Otey, C.A.; et al. Structure and Function of Palladin’s Actin Binding Domain. J. Mol. Biol. 2013, 425, 3325–3337. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, E.; Domański, J.; Naughton, F.B.; Best, R.B.; Kalli, A.C.; Stansfeld, P.J.; Sansom, M.S. Multiple lipid binding sites determine the affinity of PH domains for phosphoinositide-containing membranes. Sci. Adv. 2020, 6, eaay5736. [Google Scholar] [CrossRef] [Green Version]
- Cox, C.D.; Bavi, N.; Martinac, B. Biophysical Principles of Ion-Channel-Mediated Mechanosensory Transduction. Cell Rep. 2019, 29, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Martinac, B.; Bavi, N.; Ridone, P.; Nikolaev, Y.A.; Martinac, A.D.; Nakayama, Y.; Rohde, P.R.; Bavi, O. Tuning ion channel mechanosensitivity by asymmetry of the transbilayer pressure profile. Biophys. Rev. 2018, 10, 1377–1384. [Google Scholar] [CrossRef]
- Senju, Y.; Lappalainen, P. Regulation of actin dynamics by PI (4, 5) P2 in cell migration and endocytosis. Curr. Opin. Cell Biol. 2019, 56, 7–13. [Google Scholar] [CrossRef]
- Murakami, N.; Elzinga, M.; Singh, S.S.; Chauhan, V.P. Direct binding of myosin II to phospholipid vesicles via tail regions and phosphorylation of the heavy chains by protein kinase C. J. Biol. Chem. 1994, 269, 16082–16090. [Google Scholar] [CrossRef]
- Murakami, N.; Singh, S.S.; Chauhan, V.P.S.; Elzinga, M. Phospholipid binding, phosphorylation by protein kinase C, and filament assembly of the COOH terminal heavy chain fragments of nonmuscle myosin II isoforms MIIA and MIIB. Biochemistry 1995, 34, 16046–16055. [Google Scholar] [CrossRef]
- Li, D.; Miller, M.; Chantler, P.D. Association of a cellular myosin II with anionic phospholipids and the neuronal plasma membrane. Proc. Natl. Acad. Sci. USA 1994, 91, 853–857. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shu, S.; Billington, N.; Williamson, C.D.; Yu, S.; Brzeska, H.; Donaldson, J.G.; Sellers, J.R.; Korn, E.D. Mammalian Nonmuscle Myosin II Binds to Anionic Phospholipids with Concomitant Dissociation of the Regulatory Light Chain. J. Biol. Chem. 2016, 291, 24828–24837. [Google Scholar] [CrossRef] [Green Version]
- Koe, C.T.; Tan, Y.S.; Lönnfors, M.; Hur, S.K.; Low, C.S.L.; Zhang, Y.; Kanchanawong, P.; Bankaitis, V.A.; Wang, H. Vibrator and PI4KIIIα govern neuroblast polarity by anchoring non-muscle myosin II. eLife 2018, 7, e33555. [Google Scholar] [CrossRef]
- Alexander, S.; Mathie, A.; Peters, J. Ion channels. Br. J. Pharmacol. 2011, 164, S137–S174. [Google Scholar] [CrossRef]
- Jensen, C.S.; Watanabe, S.; Rasmussen, H.B.; Schmitt, N.; Olesen, S.-P.; Frost, N.A.; Blanpied, T.A.; Misonou, H. Specific Sorting and Post-Golgi Trafficking of Dendritic Potassium Channels in Living Neurons. J. Biol. Chem. 2014, 289, 10566–10581. [Google Scholar] [CrossRef] [Green Version]
- Dash, B.; Dib-Hajj, S.D.; Waxman, S.G. Multiple myosin motors interact with sodium/potassium-ATPase alpha 1 subunits. Mol. Brain 2018, 11, 45. [Google Scholar] [CrossRef]
- Marquèze-Pouey, B.; Martin-Moutot, N.; Sakkou-Norton, M.; Lévêque, C.; Ji, Y.; Cornet, V.; Hsiao, W.L.; Seagar, M. Toxicity and Endocytosis of Spinocerebellar Ataxia Type 6 Polyglutamine Domains: Role of Myosin IIB†. Traffic 2008, 9, 1088–1100. [Google Scholar] [CrossRef]
- Stritt, S.; Nurden, P.; Favier, R.; Favier, M.; Ferioli, S.; Gotru, S.K.; van Eeuwijk, J.M.M.; Schulze, H.; Nurden, A.T.; Lambert, M.P.; et al. Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg2+ homeostasis and cytoskeletal architecture. Nat. Commun. 2016, 7, 11097. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell adhesion by integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef]
- Häussinger, D.; Reinehr, R.; Schliess, F. The hepatocyte integrin system and cell volume sensing. Acta Physiol. 2006, 187, 249–255. [Google Scholar] [CrossRef] [PubMed]
- vom Dahl, S.; Schliess, F.; Reissmann, R.; Görg, B.; Weiergräber, O.; Kocalkova, M.; Dombrowski, F.; Häussinger, D. Involvement of Integrins in Osmosensing and Signaling toward Autophagic Proteolysis in Rat Liver. J. Biol. Chem. 2003, 278, 27088–27095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, S.F.; Kapus, A.; Hoffmann, E.K. Osmosensory Mechanisms in Cellular and Systemic Volume Regulation. J. Am. Soc. Nephrol. 2011, 22, 1587–1597. [Google Scholar] [CrossRef] [Green Version]
- Schedlbauer, A.; Tamma, G.; Rodighiero, S.; Civello, D.A.; Tamplenizza, M.; Ledolter, K.; Nofziger, C.; Patsch, W.; Konrat, R.; Paulmichl, M.; et al. Binding of the protein ICln to α-integrin contributes to the activation of IClswell current. Sci. Rep. 2019, 9, 12195. [Google Scholar] [CrossRef]
- Girault, A.; Chebli, J.; Privé, A.; Trinh, N.T.N.; Maillé, E.; Grygorczyk, R.; Brochiero, E. Complementary roles of KCa3.1 channels and β1-integrin during alveolar epithelial repair. Respir. Res. 2015, 16, 100. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, B.H.; Rasmussen, L.J.H.; Broberg, B.S.; Klausen, T.K.; Sauter, D.P.R.; Lambert, I.H.; Aspberg, A.; Hoffmann, E.K. Integrin β1, Osmosensing, and Chemoresistance in Mouse Ehrlich Carcinoma Cells. Cell. Physiol. Biochem. 2015, 36, 111–132. [Google Scholar] [CrossRef] [Green Version]
- Artym, V.V.; Petty, H.R. Molecular Proximity of Kv1.3 Voltage-gated Potassium Channels and β1-Integrins on the Plasma Membrane of Melanoma Cells: Effects of Cell Adherence and Channel Blockers. J. Gen. Physiol. 2002, 120, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Levite, M.; Cahalon, L.; Peretz, A.; Hershkoviz, R.; Sobko, A.; Ariel, A.; Desai, R.; Attali, B.; Lider, O. Extracellular K+ and Opening of Voltage-Gated Potassium Channels Activate T Cell Integrin Function: Physical and Functional Association between Kv1.3 Channels and β1 Integrins. J. Exp. Med. 2000, 191, 1167–1176. [Google Scholar] [CrossRef]
- Sajid, M.; Hu, Z.; Lele, M.; Stouffer, G. Protein complexes involving alpha v beta 3 integrins, nonmuscle myosin heavy chain-A, and focal adhesion kinase from in thrombospondin-treated smooth muscle cells. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2000, 48, 190–197. [Google Scholar]
- Rosado, L.A.R.; Horn, T.A.; McGrath, S.C.; Cotter, R.J.; Yang, J.T. Association between α4 integrin cytoplasmic tail and non-muscle myosin IIA regulates cell migration. J. Cell Sci. 2011, 124, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Morin, N.A.; Oakes, P.W.; Hyun, Y.-M.; Lee, D.; Chin, Y.E.; King, M.R.; Springer, T.A.; Shimaoka, M.; Tang, J.X.; Reichner, J.S.; et al. Nonmuscle myosin heavy chain IIA mediates integrin LFA-1 de-adhesion during T lymphocyte migration. J. Exp. Med. 2008, 205, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994, 372, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-J.; Kim, S.-G.; Yoo, J.; Han, M.-Y.; Park, J.-C.; Kim, H.-J.; Kang, S.S.; Choi, B.-D.; Jeong, M.-J. Increased association of dynamin II with myosin II in ras transformed NIH3T3 cells. Acta Biochim. Biophys. Sin. 2006, 38, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Li, P.; Wei, D.; Wang, Z.; Bao, Y.; Sun, J.; Qu, L.; Wang, L. NMMHC-IIA-dependent nuclear location of CXCR4 promotes migration and invasion in renal cell carcinoma. Oncol. Rep. 2016, 36, 2681–2688. [Google Scholar] [CrossRef] [PubMed]
- Spano, J.-P.; Andre, F.; Morat, L.; Sabatier, L.; Besse, B.; Combadiere, C.; Deterre, P.; Martin, A.; Azorin, J.; Valeyre, D.; et al. Chemokine receptor CXCR4 and early-stage non-small cell lung cancer: Pattern of expression and correlation with outcome. Ann. Oncol. 2004, 15, 613–617. [Google Scholar] [CrossRef]
- Shrivastava, S.; Sarkar, P.; Preira, P.; Salomé, L.; Chattopadhyay, A. Role of Actin Cytoskeleton in Dynamics and Function of the Serotonin1A Receptor. Biophys. J. 2020, 118, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Lezama, R.; Díaz-Téllez, A.; Ramos-Mandujano, G.; Oropeza, L.; Pasantes-Morales, H. Epidermal Growth Factor Receptor is a Common Elementin the Signaling Pathways Activated by Cell Volume Changes in Isosmotic, Hyposmotic or Hyperosmotic Conditions. Neurochem. Res. 2005, 30, 1589–1597. [Google Scholar] [CrossRef]
- Di Blasio, L.; Gagliardi, P.A.; Puliafito, A.; Primo, L. Serine/threonine kinase 3-phosphoinositide-dependent protein kinase-1 (PDK1) as a key regulator of cell migration and cancer dissemination. Cancers 2017, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Lezama, R.; Ordaz, B.; Pasantes-Morales, H. Epidermal growth factor receptor is activated by hyposmolarity and is an early signal modulating osmolyte efflux pathways in Swiss 3T3 fibroblasts. Pflüg. Arch. 2004, 447, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Konishi, H.; Touhara, K.; Sakane, F.; Hirata, M.; Ono, Y.; Kikkawa, U. Identification of Myosin II as a Binding Protein to the PH Domain of Protein Kinase B. Biochem. Biophys. Res. Commun. 1999, 255, 169–174. [Google Scholar] [CrossRef]
- Milburn, C.C.; Deak, M.; Kelly, S.M.; Price, N.C.; Alessi, D.R.; van Aalten, D.M.F. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem. J. 2003, 375, 531–538. [Google Scholar] [CrossRef]
- Lowenstein, E.; Daly, R.; Batzer, A.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Koh, W.S.; Lee, M.K.; Park, Y.M.; Han, M.Y. Dynamin II Associates with Grb2 SH3 Domain in Ras Transformed NIH3T3 Cells. Biochem. Biophys. Res. Commun. 1997, 234, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O.; Moolenaar, W.H. Ras-MAP kinase signaling by lysophosphatidic acid and other G protein-coupled receptor agonists. Oncogene 2001, 20, 1540–1546. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-J.; Kim, Y.; Kim, M.S.; Ju, H.-M.; Choi, B.; Lee, H.; Jeoung, D.; Moon, K.-W.; Kang, D.; Choi, J.; et al. CD99–PTPN12 Axis Suppresses Actin Cytoskeleton-Mediated Dimerization of Epidermal Growth Factor Receptor. Cancers 2020, 12, 2895. [Google Scholar] [CrossRef] [PubMed]
- Kohlstedt, K.; Kellner, R.; Busse, R.; Fleming, I. Signaling via the Angiotensin-Converting Enzyme Results in the Phosphorylation of the Nonmuscle Myosin Heavy Chain IIA. Mol. Pharmacol. 2006, 69, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Fisher, S.K.; Cheema, T.A.; Foster, D.J.; Heacock, A.M. Volume-dependent osmolyte efflux from neural tissues: Regulation by G-protein-coupled receptors. J. Neurochem. 2008, 106, 1998–2014. [Google Scholar] [CrossRef] [Green Version]
- Erdogmus, S.; Storch, U.; Danner, L.; Becker, J.; Winter, M.; Ziegler, N.; Wirth, A.; Offermanns, S.; Hoffmann, C.; Gudermann, T.; et al. Helix 8 is the essential structural motif of mechanosensitive GPCRs. Nat. Commun. 2019, 10, 5784. [Google Scholar] [CrossRef]
- Zhou, Y.; Meng, J.; Xu, C.; Liu, J. Multiple GPCR Functional Assays Based on Resonance Energy Transfer Sensors. Front. Cell Dev. Biol. 2021, 9, 779. [Google Scholar] [CrossRef] [PubMed]
- Adjobo-Hermans, M.J.; Goedhart, J.; van Weeren, L.; Nijmeijer, S.; Manders, E.M.; Offermanns, S.; Gadella, T.W. Real-time visualization of heterotrimeric G protein Gq activation in living cells. BMC Biol. 2011, 9, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, D.M. SRC family kinases in cell volume regulation. Am. J. Physiol. Cell Physiol. 2005, 288, C483–C493. [Google Scholar] [CrossRef] [Green Version]
- Vetter, M.L.; Zhang, Z.; Liu, S.; Wang, J.; Cho, H.; Zhang, J.; Zhang, W.; Gray, N.S.; Yang, P.L. Fluorescent Visualization of Src by Using Dasatinib-BODIPY. ChemBioChem 2014, 15, 1317–1324. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Singh, R.; Udayan, S.; Biswas, S.; Reddy, P.P.; Manmadhan, S.; George, G.; Kumar, S.; Das, R.; Rao, B.M.; et al. A Fyn biosensor reveals pulsatile, spatially localized kinase activity and signaling crosstalk in live mammalian cells. eLife 2020, 9, e50571. [Google Scholar] [CrossRef] [PubMed]
- Ham, T.R.; Collins, K.L.; Hoffman, B.D. Molecular tension sensors: Moving beyond force. Mol. Cell. Single Mol. Technol. Neural Eng. High Resolut. Cell Imaging 2019, 12, 83–94. [Google Scholar] [CrossRef]
- Lavrenyuk, K.; Conway, D.; Dahl, K.N. Imaging methods in mechanosensing: A historical perspective and visions for the future. Mol. Biol. Cell 2021, 32, 842–854. [Google Scholar] [CrossRef]
- Richards, M.A.; Simon, J.N.; Ma, R.; Loonat, A.A.; Crabtree, M.J.; Paterson, D.J.; Fahlman, R.P.; Casadei, B.; Fliegel, L.; Swietach, P. Nitric oxide modulates cardiomyocyte pH control through a biphasic effect on sodium/hydrogen exchanger-1. Cardiovasc. Res. 2020, 116, 1958–1971. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Surcel, A.; Schiffhauer, E.S.; Thomas, D.G.; Zhu, Q.; DiNapoli, K.T.; Herbig, M.; Otto, O.; West-Foyle, H.; Jacobi, A.; Kräter, M.; et al. Targeting Mechanoresponsive Proteins in Pancreatic Cancer: 4-Hydroxyacetophenone Blocks Dissemination and Invasion by Activating MYH14. Cancer Res. 2019, 79, 4665–4678. [Google Scholar] [CrossRef] [Green Version]
- Bryan, D.S.; Stack, M.; Krysztofiak, K.; Cichoń, U.; Thomas, D.G.; Surcel, A.; Schiffhauer, E.S.; Beckett, M.A.; Khodarev, N.N.; Xue, L.; et al. 4-Hydroxyacetophenone modulates the actomyosin cytoskeleton to reduce metastasis. Proc. Natl. Acad. Sci. USA 2020, 117, 22423–22429. [Google Scholar] [CrossRef]
- Naydenov, N.G.; Lechuga, S.; Huang, E.H.; Ivanov, A.I. Myosin Motors: Novel Regulators and Therapeutic Targets in Colorectal Cancer. Cancers 2021, 13, 741. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barvitenko, N.; Aslam, M.; Lawen, A.; Saldanha, C.; Skverchinskaya, E.; Uras, G.; Manca, A.; Pantaleo, A. Two Motors and One Spring: Hypothetic Roles of Non-Muscle Myosin II and Submembrane Actin-Based Cytoskeleton in Cell Volume Sensing. Int. J. Mol. Sci. 2021, 22, 7967. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157967
Barvitenko N, Aslam M, Lawen A, Saldanha C, Skverchinskaya E, Uras G, Manca A, Pantaleo A. Two Motors and One Spring: Hypothetic Roles of Non-Muscle Myosin II and Submembrane Actin-Based Cytoskeleton in Cell Volume Sensing. International Journal of Molecular Sciences. 2021; 22(15):7967. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157967
Chicago/Turabian StyleBarvitenko, Nadezhda, Muhammad Aslam, Alfons Lawen, Carlota Saldanha, Elisaveta Skverchinskaya, Giuseppe Uras, Alessia Manca, and Antonella Pantaleo. 2021. "Two Motors and One Spring: Hypothetic Roles of Non-Muscle Myosin II and Submembrane Actin-Based Cytoskeleton in Cell Volume Sensing" International Journal of Molecular Sciences 22, no. 15: 7967. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157967