
Inhibition of Cardiac RIP3 Mitigates Early Reperfusion Injury and Calcium-Induced Mitochondrial Swelling without Altering Necroptotic Signalling

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
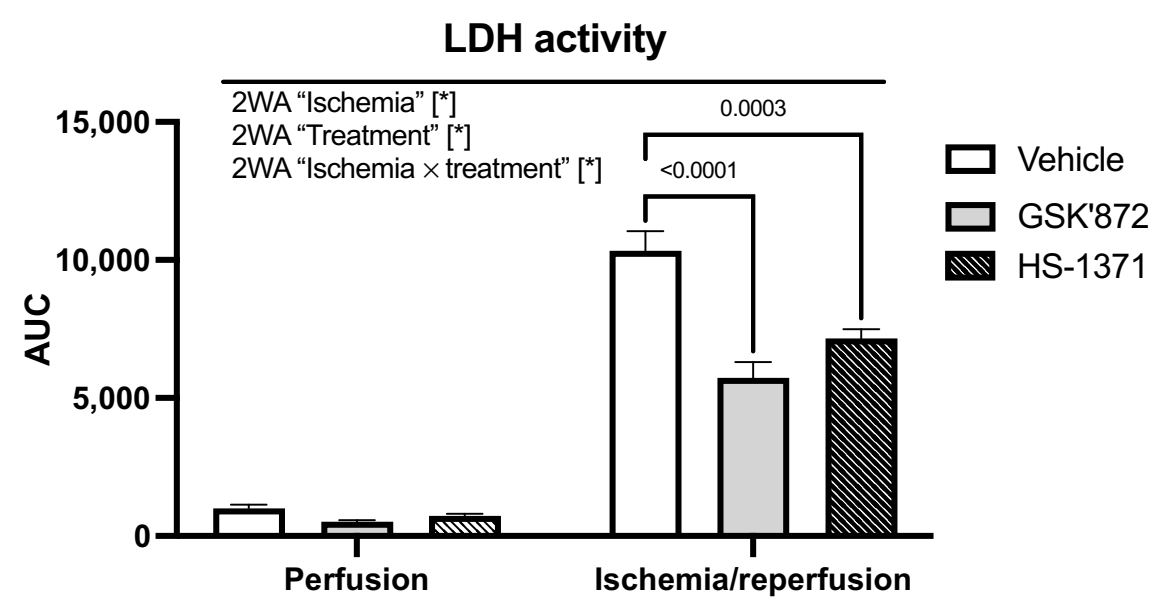
2.1. LDH Release
2.2. mPTP Opening
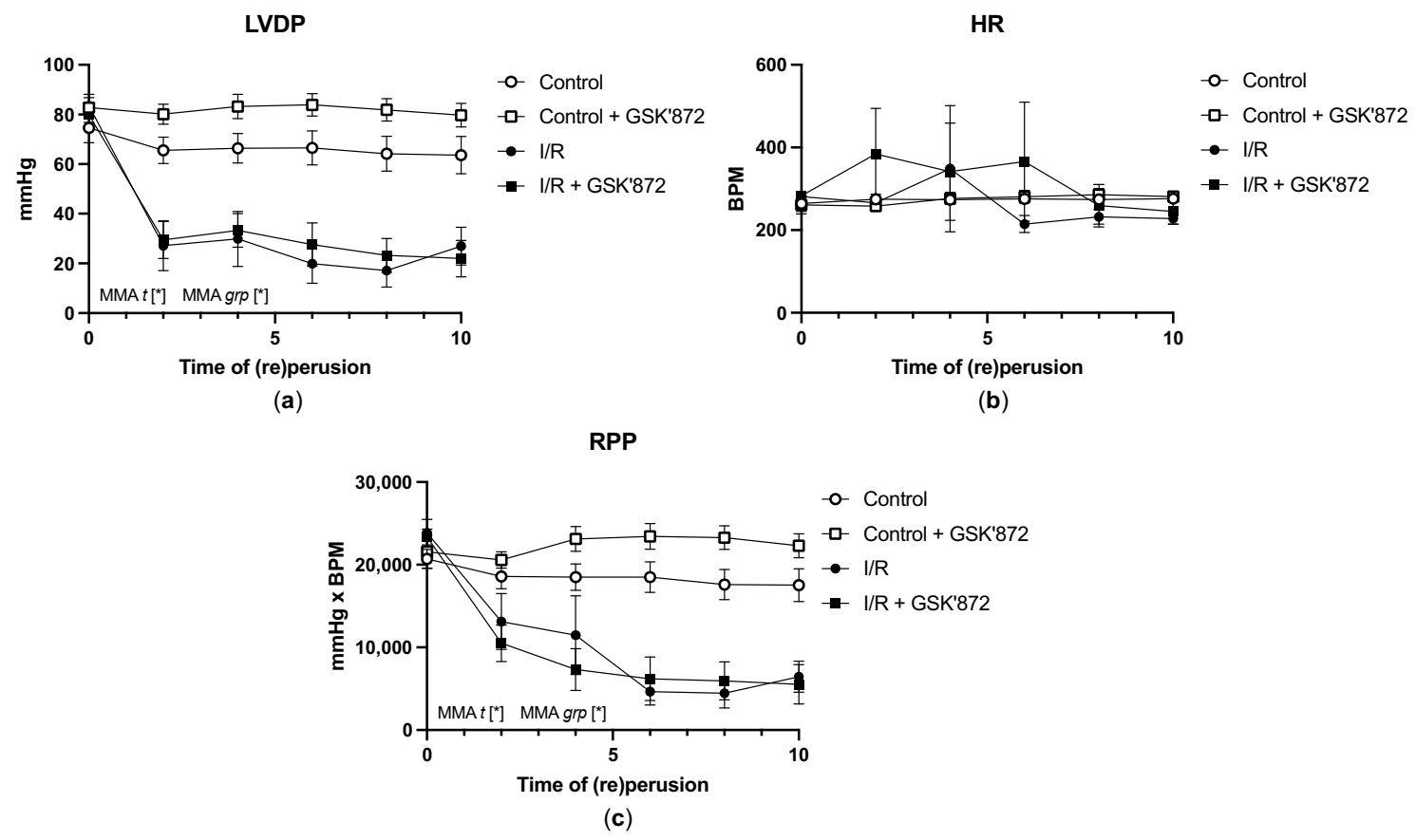
2.3. Hemodynamic Parameters of the Heart and Arrhythmia Triggering
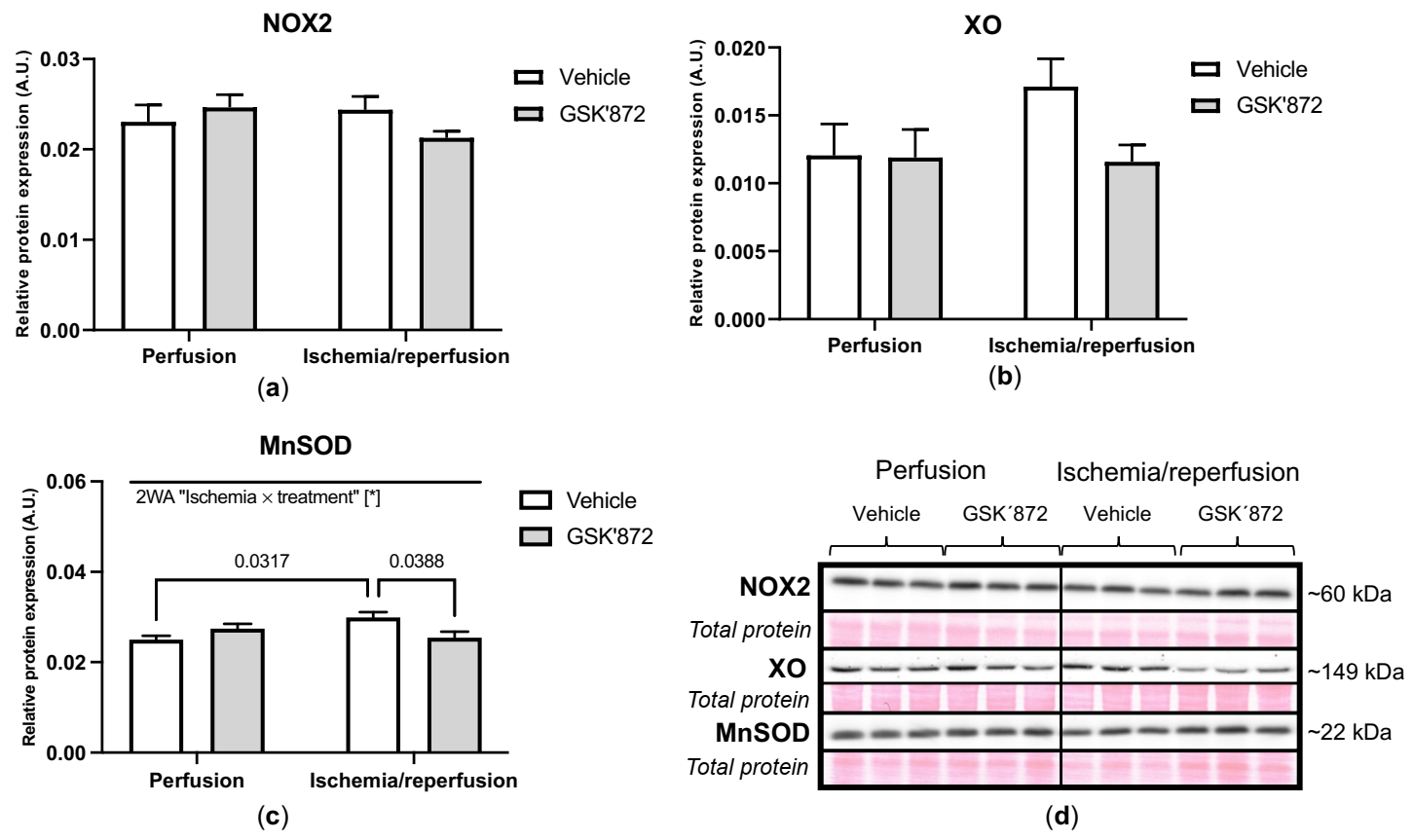
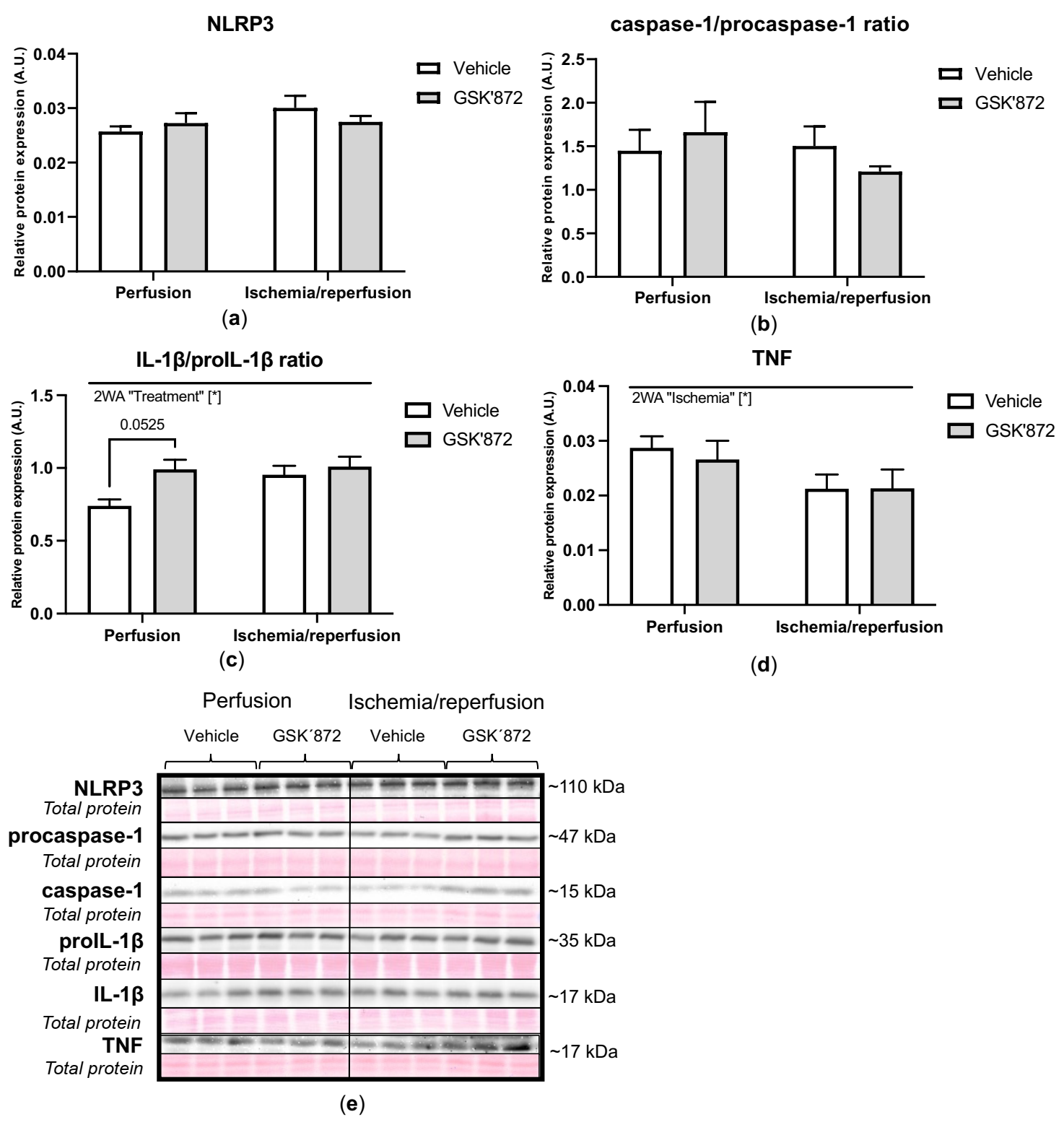
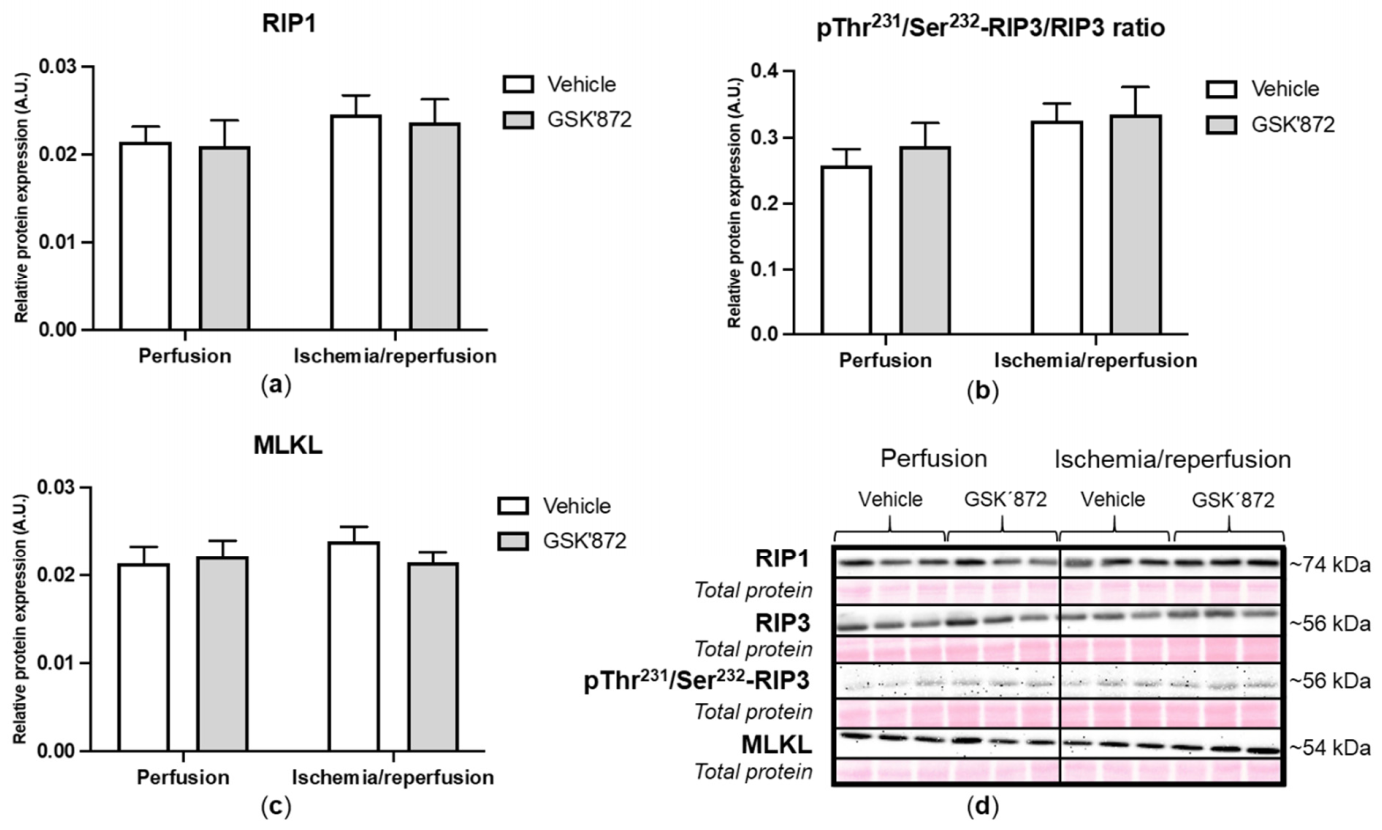
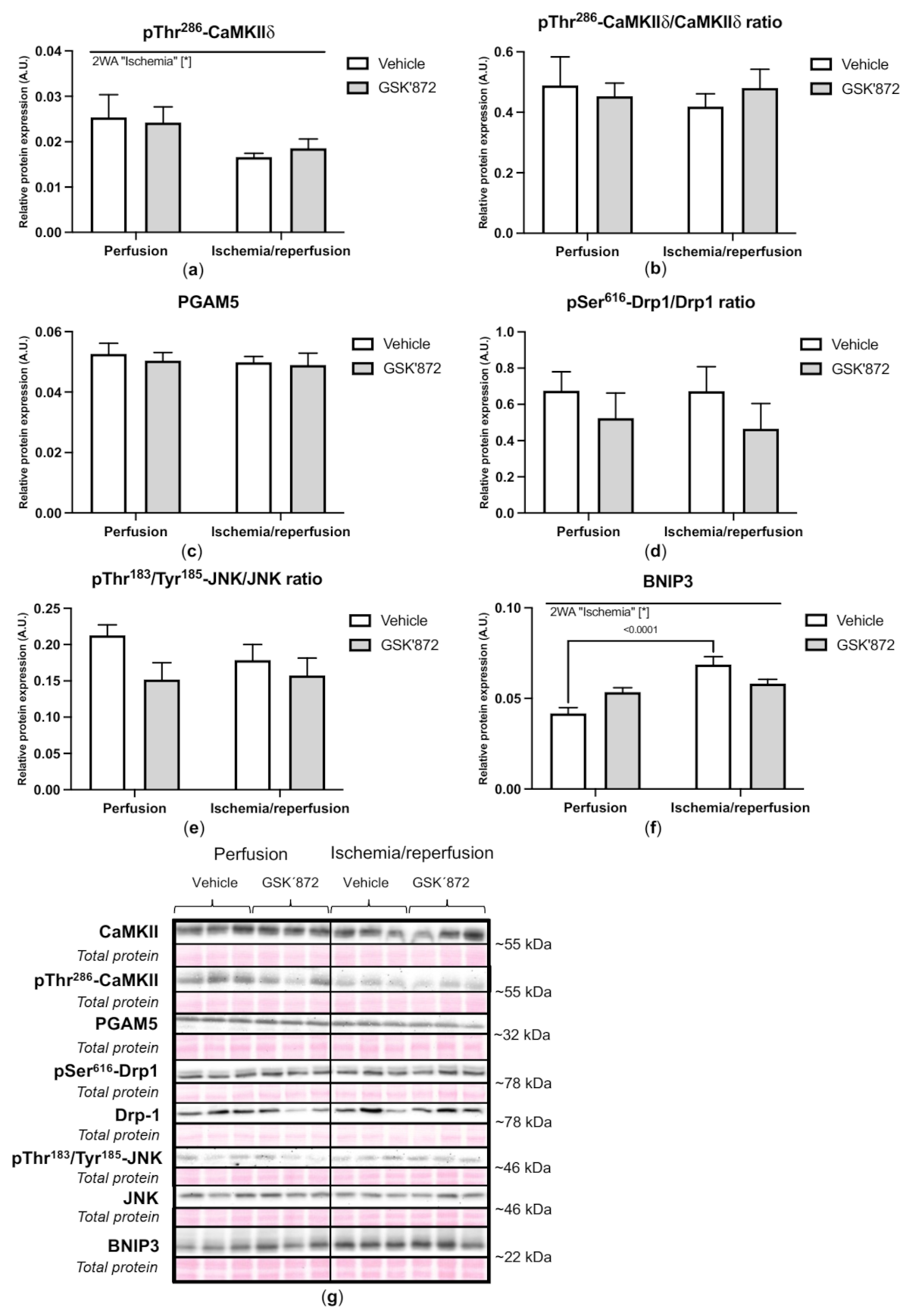
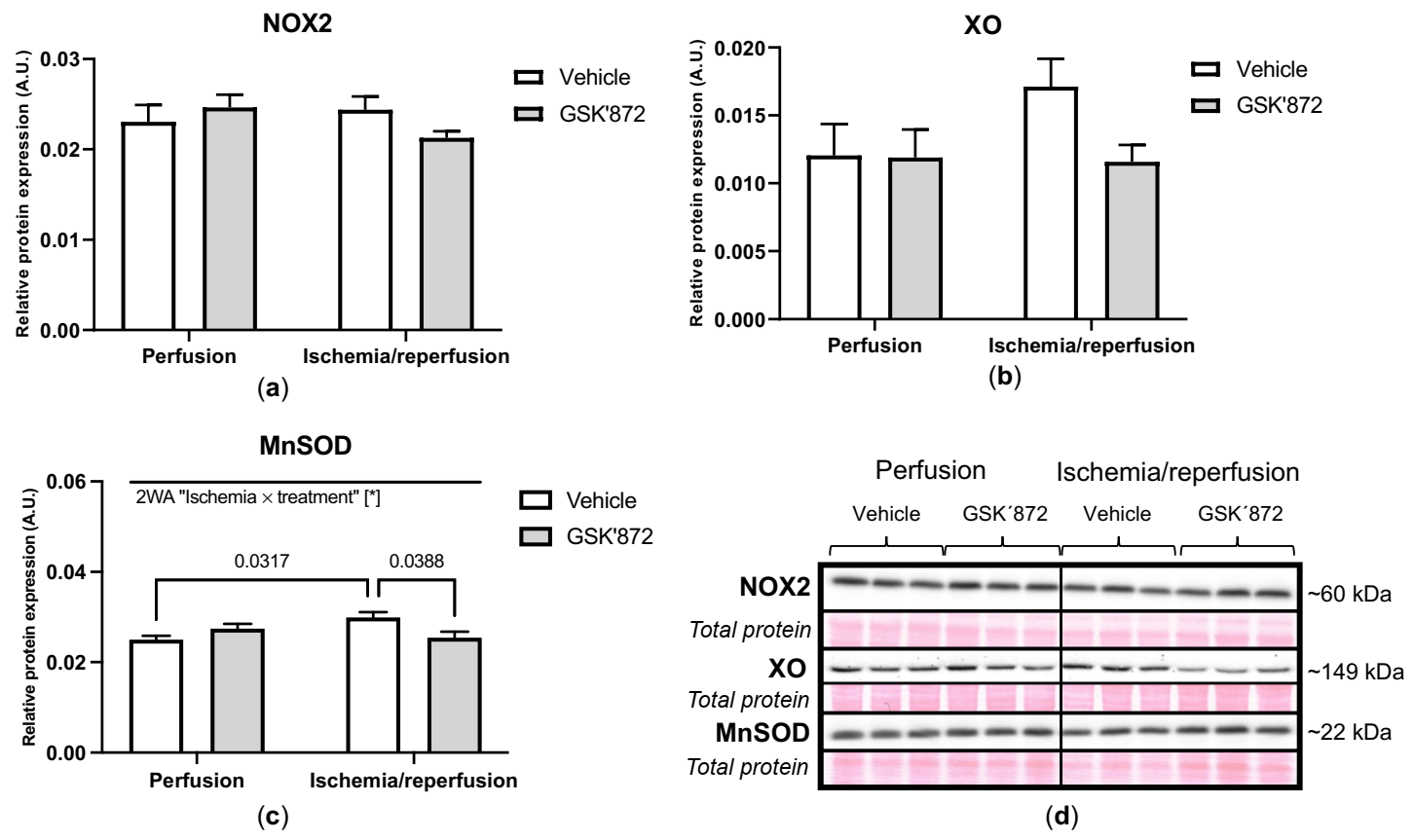
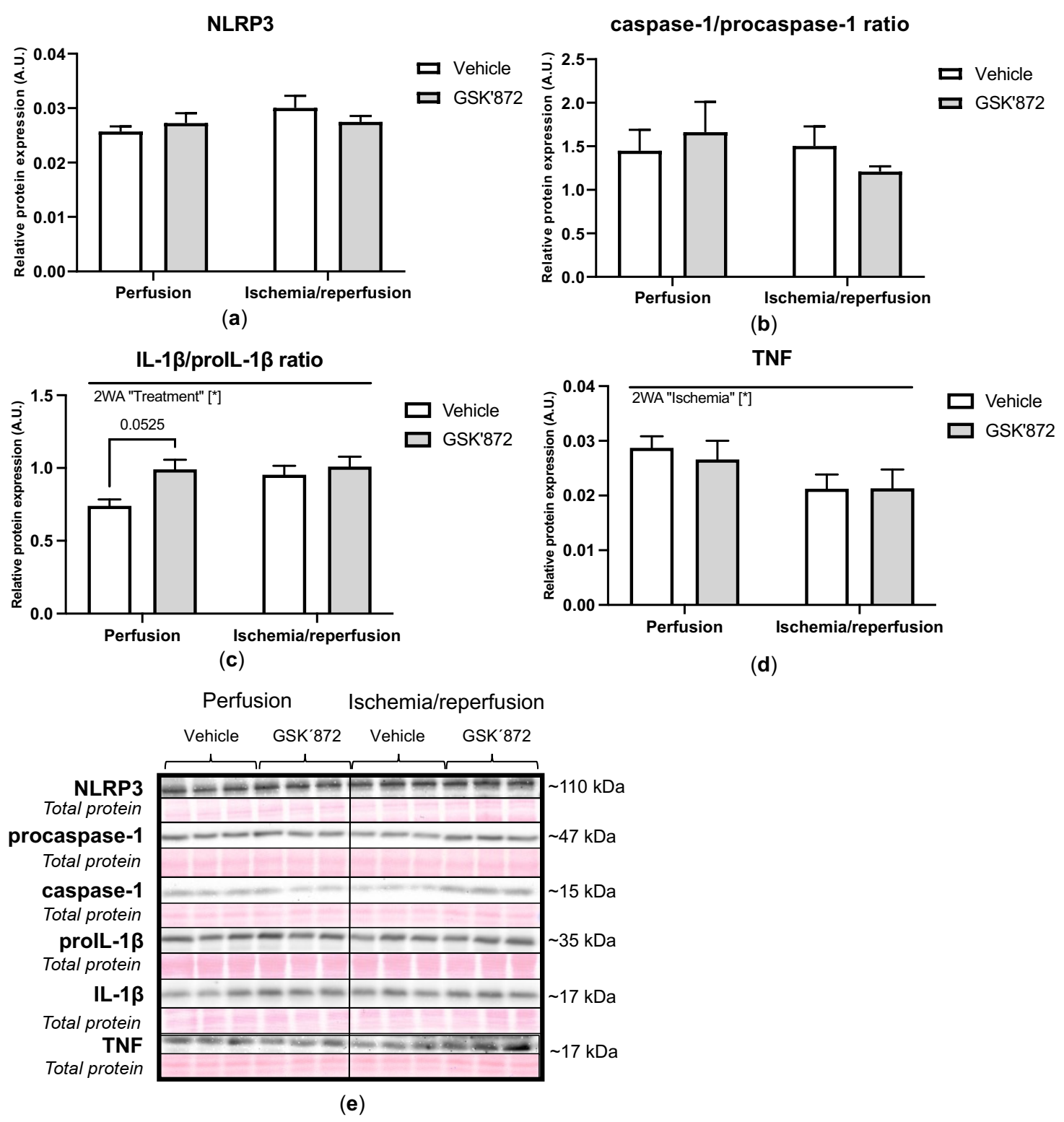
2.4. Molecular Analyses of Canonical and Non-Canonical Signalling Pathways of Necroptosis
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animals and Experimental Groups
4.3. Experimental Myocardial I/R Protocol
4.4. Determination of LDH Activity
4.5. Isolation of Mitochondrial Fractions
4.6. Determination of mPTP Opening
4.7. SDS-PAGE and Immunoblotting
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAMs | disintegrins and metalloproteinases |
| AMPK | 5’ adenosine monophosphate-activated protein kinase |
| BNIP3 | Bcl2 interacting protein 3 |
| CaMKIIδ | Ca2+/calmodulin-dependent protein kinase II delta |
| cMyBPc | cardiac myosin-binding protein c |
| Drp1 | dynamin-related protein 1 |
| FUNDC1 | FUN14 Domain Containing 1 |
| GSK2399872A | GSK’872 |
| HR | heart rate |
| I/R | ischemia/reperfusion |
| IL-1β | interleukin-1 beta |
| JNK | c-Jun N-terminal kinase |
| LDH | lactate dehydrogenase |
| LVDP | left ventricular developed pressure |
| LVEDP | LV end-diastolic pressure |
| MLKL | mixed lineage kinase domain-like pseudokinase |
| MnSOD | manganese superoxide dismutase |
| mPTP | mitochondrial permeability transition pore |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NOX2 | NADPH oxidase 2 |
| PGAM5 | phosphoglycerate mutase family member 5 |
| PLN | phospholamban |
| RIP1 | receptor-interacting protein kinase 1 |
| RIP3 | receptor-interacting protein kinase 3 |
| ROS | reactive oxygen species |
| RPP | rate pressure product |
| TNF | tumour necrosis factor |
| XO | xanthin oxidase |
References
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-E-Silva, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell Death in the Pathogenesis of Heart Disease: Mechanisms and Significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. 2011, 2, e244. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kroemer, G. Necroptosis: A Specialized Pathway of Programmed Necrosis. Cell 2008, 135, 1161–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moquin, D.; Chan, F.K.-M. The molecular regulation of programmed necrotic cell injury. Trends Biochem. Sci. 2010, 35, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, M.I.F.J.; Liu, J.; Arslan, F.; den Ouden, K.; van Middelaar, B.J.; Doevendans, P.A.; Sluijter, J.P.G. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia–reperfusion in vivo. Basic Res. Cardiol. 2012, 107, 270. [Google Scholar] [CrossRef]
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling aftermyocardial infarction. Cardiovasc. Res. 2014, 103, 206–216. [Google Scholar] [CrossRef]
- Koudstaal, S.; Oerlemans, M.I.F.J.; Van der Spoel, T.I.G.; Janssen, A.W.F.; Hoefer, I.E.; Doevendans, P.A.; Sluijter, J.P.G.; Chamuleau, S.A.J. Necrostatin-1 alleviates reperfusion injury following acute myocardial infarction in pigs. Eur. J. Clin. Investig. 2015, 45, 150–159. [Google Scholar] [CrossRef]
- Adameova, A.; Hrdlicka, J.; Szobi, A.; Farkasova, V.; Murarikova, M.; Neckar, J.; Kolar, F.; Ravingerova, T. Evidence of necroptosis in hearts subjected to various forms of ischemic insults. Can. J. Physiol. Pharmacol. 2017, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szobi, A.; Farkašová-Ledvényiová, V.; Lichý, M.; Muráriková, M.; Čarnická, S.; Ravingerová, T.; Adameová, A. Cardioprotection of ischaemic preconditioning is associated with inhibition of translocation of MLKL within the plasma membrane. J. Cell. Mol. Med. 2018, 22, 4183–4196. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, Y.; Liu, S.; Yu, X.; Li, L.; Shi, C.; He, W.; Li, J.; Xu, L.; Hu, Z.; et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. USA 2014, 111, 15438–15443. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.-N.; Yang, Z.-H.; Wang, X.-K.; Zhang, Y.; Wan, H.; Song, Y.; Chen, X.; Shao, J.; Han, J. Distinct roles of RIP1–RIP3 hetero- and RIP3–RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014, 21, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Zhang, A.; Choksi, S.; Li, W.; Li, T.; Zhang, X.M.; Liu, Z.G. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016, 26, 886–900. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Dayani, L.; Rosenberg, P.A.; Li, J. RIP1 kinase mediates arachidonic acid-induced oxidative death of oligodendrocyte precursors. Int. J. Physiol. Pathophysiol. Pharmacol. 2010, 2, 137–147. [Google Scholar]
- Davidson, S.M.; Adameová, A.; Barile, L.; Cabrera-fuentes, H.A.; Lazou, A. Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J. Cell. Mol. Med. 2020, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress–induced myocardial necroptosis. Nat. Med. 2016. [Google Scholar] [CrossRef]
- She, L.; Tu, H.; Zhang, Y.-Z.; Tang, L.; Li, N.; Ma, Q.-L.; Liu, B.; Li, Q.; Luo, X.-J.; Peng, J. Inhibition of Phosphoglycerate Mutase 5 Reduces Necroptosis in Rat Hearts Following Ischemia/Reperfusion Through Suppression of Dynamin-Related Protein 1. Cardiovasc. Drugs Ther. 2019, 33, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Vila-Petroff, M.; Salas, M.A.; Said, M.; Valverde, C.A.; Sapia, L.; Portiansky, E.; Hajjar, R.J.; Kranias, E.G.; Mundiña-Weilenmann, C.; Mattiazzi, A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia–reperfusion injury. Cardiovasc. Res. 2007, 73, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Szobi, A.; Rajtik, T.; Carnicka, S.; Ravingerova, T.; Adameova, A. Mitigation of postischemic cardiac contractile dysfunction by CaMKII inhibition: Effects on programmed necrotic and apoptotic cell death. Mol. Cell Biochem. 2014, 388, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Zhou, T.; Chuang, C.-C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. Biomed Res. Int. 2015, 2015, 864946. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.A.; Clarke, S.; Das, M.; Griffiths, E.J.; Lim, K.H.H.; Halestrap, A.P. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J. Physiol. 2003, 549, 513–524. [Google Scholar] [CrossRef]
- Khaliulin, I.; Parker, J.E.; Halestrap, A.P. Consecutive pharmacological activation of PKA and PKC mimics the potent cardioprotection of temperature preconditioning. Cardiovasc. Res. 2010, 88, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Feng, N.; Anderson, M.E. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. J. Mol. Cell. Cardiol. 2017, 103, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Hou, H.; Wang, Y.; Li, Q.; Li, Z.; Teng, Y.; Li, J.; Wang, X.; Chen, J.; Huang, N. The role of RIP3 in cardiomyocyte necrosis induced by mitochondrial damage of myocardial ischemia–reperfusion. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 1131–1140. [Google Scholar] [CrossRef]
- Elahi, M.M.; Kong, Y.X.; Matata, B.M. Oxidative stress as a mediator of cardiovascular disease. Oxid. Med. Cell. Longev. 2009, 2, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Wallach, D.; Kang, T.-B.; Dillon, C.P.; Green, D.R. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 2016, 352, 51–59. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Li, T. Ripk3 mediates cardiomyocyte necrosis through targeting mitochondria and the JNK-Bnip3 pathway under hypoxia-reoxygenation injury. J. Recept. Signal Transduct. 2019, 39, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, Y.V.; Minasian, S.M.; Demchenko, E.A.; Galagudza, M.M. Study of cardioprotective effects of necroptosis inhibitors on isolated rat heart subjected to global ischemia-reperfusion. Bull. Exp. Biol. Med. 2013, 155, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Koshinuma, S.; Miyamae, M.; Kaneda, K.; Kotani, J.; Figueredo, V.M. Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J. Anesth. 2014, 28, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Horvath, C.; Szobi, A.; Kindernay, L.; Ravingerova, T.; Adameova, A. Pleiotropic, non-cell death-associated effects of inhibitors of receptor-interacting protein kinase 1 in the heart. Mol. Cell. Biochem. 2021, 476, 3079–3087. [Google Scholar] [CrossRef]
- Yang, X.-S.; Yi, T.-L.; Zhang, S.; Xu, Z.-W.; Yu, Z.-Q.; Sun, H.-T.; Yang, C.; Tu, Y.; Cheng, S.-X. Hypoxia-inducible factor-1 alpha is involved in RIP-induced necroptosis caused by in vitro and in vivo ischemic brain injury. Sci. Rep. 2017, 7, 5818. [Google Scholar] [CrossRef]
- Hu, W.; Wu, X.; Yu, D.; Zhao, L.; Zhu, X.; Li, X.; Huang, T.; Chu, Z.; Xu, Y. Regulation of JNK signaling pathway and RIPK3/AIF in necroptosis-mediated global cerebral ischemia/reperfusion injury in rats. Exp. Neurol. 2020, 331, 113374. [Google Scholar] [CrossRef]
- Yang, F.; Shang, L.; Wang, S.; Liu, Y.; Ren, H.; Zhu, W.; Shi, X. TNFα-Mediated Necroptosis Aggravates Ischemia-Reperfusion Injury in the Fatty Liver by Regulating the Inflammatory Response. Oxid. Med. Cell. Longev. 2019, 2019, 2301903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichý, M.; Szobi, A.; Hrdlička, J.; Horváth, C.; Kormanová, V.; Rajtík, T.; Neckář, J.; Kolář, F.; Adameová, A. Different signalling in infarcted and non-infarcted areas of rat failing hearts: A role of necroptosis and inflammation. J. Cell. Mol. Med. 2019, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.-B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Halestrap, A.P. Protection by Cyclosporin A of Ischemia/Reperfusion-Induced Damage in Isolated Rat Hearts. J. Mol. Cell. Cardiol. 1993, 25, 1461–1469. [Google Scholar] [CrossRef]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Zhu, P.; Guo, J.; Hu, N.; Wang, S.; Li, D.; Hu, S.; Ren, J.; Cao, F.; Chen, Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017, 13, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wan, K.; Yin, M.; Hu, P.; Que, Y.; Zhou, X.; Zhang, L.; Li, T.; Du, Y.; Xu, G.; et al. RIPK3 Induces Cardiomyocyte Necroptosis via Inhibition of AMPK-Parkin-Mitophagy in Cardiac Remodelling after Myocardial Infarction. Oxid. Med. Cell. Longev. 2021, 2021, 6635955. [Google Scholar] [CrossRef]
- Smith, C.C.T.; Davidson, S.M.; Lim, S.Y.; Simpkin, J.C.; Hothersall, J.S.; Yellon, D.M. Necrostatin: A potentially novel cardioprotective agent? Cardiovasc. Drugs Ther. 2007, 21, 227–233. [Google Scholar] [CrossRef]
- Lim, S.Y.; Davidson, S.M.; Mocanu, M.M.; Yellon, D.M.; Smith, C.C.T. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc. Drugs Ther. 2007, 21, 467–469. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.-M.; Chen, X.; Pang, H.-Y.; Liu, H.-H.; Chen, P.-P.; Shi, J.-L.; Tang, S.; Wu, Z.-H.; Zhang, S.-Y. Plasma levels of receptor interacting protein kinase-3 correlated with coronary artery disease. Chin. Med. J. (Engl.) 2019, 132, 1400–1405. [Google Scholar] [CrossRef]
- Hu, X.; Li, H.; Chen, X.; Liu, H.; Zuo, W.; Zhang, Y.; Zhang, S. Plasma concentration of receptor-interacting protein kinase-3 as a potential biomarker for diagnosis and prognosis in heart failure. Clin. Chim. Acta 2020, 509, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Hofmans, S.; Declercq, W.; Augustyns, K.; Vandenabeele, P. Inhibitors Targeting RIPK1/RIPK3: Old and New Drugs. Trends Pharmacol. Sci. 2020, 41, 209–224. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef]
- Moritz, C.P. Tubulin or Not Tubulin: Heading Toward Total Protein Staining as Loading Control in Western Blots. Proteomics 2017, 17, 1600189. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stabilization | 10 min (re) Perfusion | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LVDP (mmHg) | LVEDP (mmHg) | HR (BPM) | +dP/dt (mmHg/s) | −dP/dt (mmHg/s) | RPP (mmHg × BPM) | LVDP (mmHg) | LVEDP (mmHg) | HR (BPM) | +dP/dt (mmHg/s) | −dP/dt (mmHg/s) | RPP (mmHg × BPM) | |
| Control | 74.6 ± 6.1 | 5.5 ± 1.1 | 264.0 ± 19.0 | 1591.6 ± 121.3 | 1225.9 ± 76.7 | 20,724.5 ± 1156.9 | 63.6 ± 7.6 | 3.4 ± 1.3 | 276.1 ± 7.2 | 1387.6 ± 163.4 | 1047.7 ± 128.6 | 17,529.6 ± 1978.9 |
| Control + GSK’872 | 82.8 ± 3.7 | 4.0 ± 0.4 | 261.1 ± 21.9 | 1683.2 ± 78.3 | 1296.7 ± 59.5 | 21,595 ± 2009.8 | 79.7 ± 4.4 | 1.3 ± 0.6 | 281.0 ± 11.3 | 1655.9 ± 100.0 | 1343.0 ± 80.5 | 22,324.2 ± 1449.2 |
| IR | 83.4 ± 4.7 | 3.5 ± 1.0 | 281.3 ± 3.4 | 1749.5 ± 106.6 | 1397.4 ± 66.6 | 23,873.7 ± 1519.0 | 26.9 ± 6.4 * | 31.7 ± 7.8 * | 228.1 ± 12.7 * | 376.6 ± 110.9 * | 342.9 ± 111.1 * | 6469.1 ± 1596.7 * |
| IR + GSK’872 | 79.9 ± 3.1 | 3.2 ± 0.9 | 283.1 ± 7.2 | 1742.5 ± 70.0 | 1355.6 ± 48.5 | 23,352.9 ± 958.9 | 22.0 ± 7.4 * | 34.6 ± 9.5 * | 245.1 ± 30.6 | 376.8 ± 154.8 * | 319.5 ± 129.6 * | 5549.5 ± 2391.8 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horvath, C.; Young, M.; Jarabicova, I.; Kindernay, L.; Ferenczyova, K.; Ravingerova, T.; Lewis, M.; Suleiman, M.S.; Adameova, A. Inhibition of Cardiac RIP3 Mitigates Early Reperfusion Injury and Calcium-Induced Mitochondrial Swelling without Altering Necroptotic Signalling. Int. J. Mol. Sci. 2021, 22, 7983. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157983
Horvath C, Young M, Jarabicova I, Kindernay L, Ferenczyova K, Ravingerova T, Lewis M, Suleiman MS, Adameova A. Inhibition of Cardiac RIP3 Mitigates Early Reperfusion Injury and Calcium-Induced Mitochondrial Swelling without Altering Necroptotic Signalling. International Journal of Molecular Sciences. 2021; 22(15):7983. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157983
Chicago/Turabian StyleHorvath, Csaba, Megan Young, Izabela Jarabicova, Lucia Kindernay, Kristina Ferenczyova, Tanya Ravingerova, Martin Lewis, M. Saadeh Suleiman, and Adriana Adameova. 2021. "Inhibition of Cardiac RIP3 Mitigates Early Reperfusion Injury and Calcium-Induced Mitochondrial Swelling without Altering Necroptotic Signalling" International Journal of Molecular Sciences 22, no. 15: 7983. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157983