
The Development of Molecular Biology of Osteoporosis


1
Tangshan Key Laboratory of Green Speciality Chemicals, Department of Chemistry, Tangshan Normal University, Tangshan 063000, China
2
Lab for Bone Metabolism, School of Life Sciences, Northwestern Polytechnical University, Xi’an 710072, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2021, 22(15), 8182; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158182
Submission received: 4 July 2021
/
Revised: 25 July 2021
/
Accepted: 26 July 2021
/
Published: 30 July 2021
(This article belongs to the Special Issue Novel Extracellular and Intracellular Signalling Mechanisms in Bone 2.0)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Osteoporosis is one of the major bone disorders that affects both women and men, and causes bone deterioration and bone strength. Bone remodeling maintains bone mass and mineral homeostasis through the balanced action of osteoblasts and osteoclasts, which are responsible for bone formation and bone resorption, respectively. The imbalance in bone remodeling is known to be the main cause of osteoporosis. The imbalance can be the result of the action of various molecules produced by one bone cell that acts on other bone cells and influence cell activity. The understanding of the effect of these molecules on bone can help identify new targets and therapeutics to prevent and treat bone disorders. In this article, we have focused on molecules that are produced by osteoblasts, osteocytes, and osteoclasts and their mechanism of action on these cells. We have also summarized the different pharmacological osteoporosis treatments that target different molecular aspects of these bone cells to minimize osteoporosis.
1. Introduction
Osteoporosis is a well-known skeletal disease that is characterized by declined bone strength due to weakened bone microarchitecture and increased vulnerability to fractures [1]. Osteoporosis is a prominent cause of concern in the aging population, and the intensity of osteoporotic fractures becomes prevalent with age and becomes challenging. As a result, it affects the physical, psycho-social, and financial aspects of human life [2]. Osteoporosis and osteoporosis-related fractures are predominant in older adults and post-menopausal women, leading to morbidity. Post-menopausal women are predominantly disposed to suffering from common bone fractures such as vertebral and hip fractures [3,4]. Osteoporosis is classified into two types: primary and secondary osteoporosis, with up to 30% of post-menopausal women and 50 to 80% of men having factors contributing to osteoporosis. When osteoporosis is age-related and arises in women post-menopause and men in the absence of an underlying disease, it is called primary osteoporosis. When it occurs in the presence of an underlying disease or medication, it is called secondary osteoporosis [5].
The most common methods of diagnosis include the measurement of bone mineral density (BMD) using dual-energy X-ray absorptiometry (DEXA) and quantitative computed tomography (QCT) [6], and the fracture risk calculation by using the Garvan fracture risk calculator, QFracture®, and fracture risk assessment tool (FRAX®) [7]. The available treatment guidelines provide recommendations on when to treat patients but do not specify the types of drugs to prescribe in various situations. Osteoporosis therapy needs to be based on individuals, and needs to consider the dosing regimen to achieve desired efficacy, safety, and cost [8]. As BMD, osteoporosis, and osteoporotic fractures are extremely heritable, understanding the molecular mechanisms of osteoporosis is important to develop effective and efficient therapeutics to reduce and treat osteoporosis. Bone remodeling is a coordinated process involving bone-forming osteoblasts and bone-resorbing osteoclasts to maintain bone mass, repair micro-damage, and mineral homeostasis [9]. Osteoporosis signifies an imbalance in the bone remodeling process because of the excessive osteoclast-mediated bone resorption or inadequate osteoblast-mediated bone formation. This leads to a loss in both trabecular and cortical bones, causing spinal and hip fractures, and long bone fractures, respectively. To sustain bone homeostasis, several molecules harmonizes osteoblast and osteoclast functions [10]. Many hormones, especially estrogens, are known to regulate bone remodeling by controlling cytokines and growth factors produced from bone marrow as well as bone cells [11]. In the present review, we have summarized the cellular and molecular mechanisms that control bone remodeling through osteoblasts, osteocytes, and osteoclasts and possible targets for osteoporosis treatment. Additionally, we have highlighted the anti-resorptive and anabolic osteoporosis treatment.
2. Bone Remodeling
Bone modeling is the process in which osteoclasts and osteoblasts work independently to degrade old bone and form a new one, respectively. This process is important in defining the progress of osteoporosis. Bone remodeling is crucial for adult bone homeostasis and involves balanced coordination in bone formation and resorption to support bone mass and systemic mineral homeostasis. Bone remodeling can be divided into five phases: activation phase, resorption phase, reversal phase, formation phase, and termination phase [12,13]. These steps take place simultaneously but asynchronously at multiple locations within the skeleton.
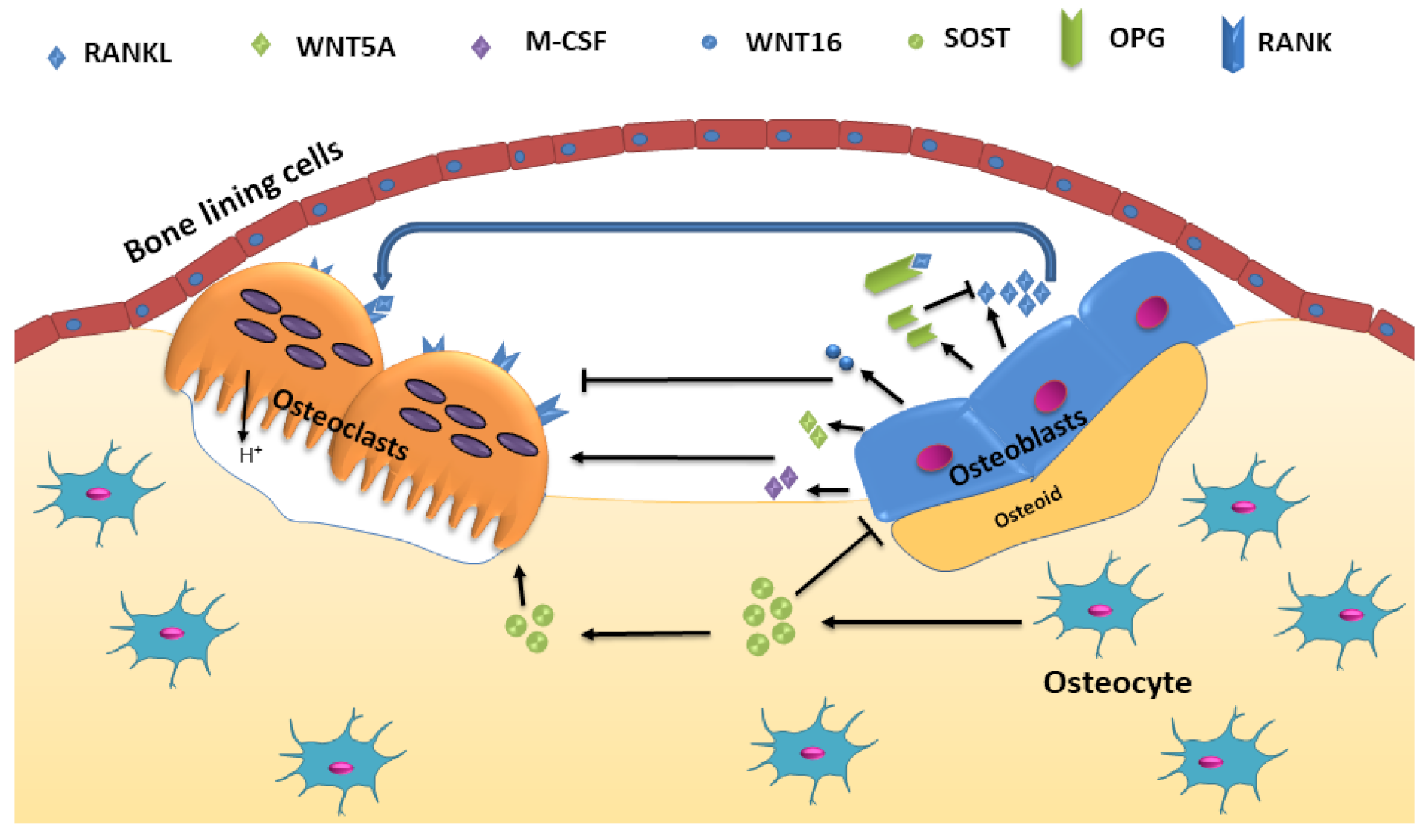
The significant signaling pathways that control osteoclastic bone resorption and osteoblastic bone formation include receptor activator of nuclear factor-κB (RANK)/RANK ligand (RANKL)/osteoprotegerin (OPG) and canonical Wnt signaling (Figure 1). The process is also regulated by paracrine regulators such as cytokines, growth factors, prostaglandins, and endocrine regulators such as parathyroid hormone (PTH), calcitonin, vitamin D, glucocorticoids, and growth hormone, and sex hormones [14]. The activation phase involves recruitment and activated osteoclast precursor cells and bone surface exposure by separating bone lining cells from the bone. This separation creates a canopy termed bone-remodeling compartment (BRC) over this site. Multiple mononuclear cells fuse to generate multinucleated preosteoclasts, forming sealing zones by binding to the bone matrix around bone-resorbing compartments, isolating the resorption pit from surrounding bone. Osteocytes provide initiating signal through their network of dendritic processes to signal other cells. Apoptosis of osteocytes also releases paracrine factors that increase the recruitment of osteoclast and osteoblast precursors [14]. Local as well as systemic regulators promote osteoclastogenesis. Transforming growth factor-beta (TGF-β), macrophage colony-stimulating factor (M-CSF) and RANKL are constituted as local regulators, whereas vitamin D, calcium, PTH, and estrogen act as systematic regulators [12]. Osteoclast attachment to bone stimulates changes into the cytoskeleton leading to the formation of a filamentous actin ring or sealing zone, which surrounds the ruffled border. This forms a separate resorptive environment between the osteoclast and the underlying bone matrix. The ruffled border is produced because of the fusion of secretory vesicles with the bone-apposing plasma membrane. A calcium sensor protein, Synaptotagmin VII is essential for the formation of the ruffled border in osteoclasts [15].
Bone resorption is determined by the formation and stability of the sealing zone, which is constituted of actin and integrin-based podosomes. Actin ring formation in the sealing zone consists of contractile proteins and is stabilized by actin and tubulin microtubules [16]. Vacuolar ATPases (V-ATPases) interaction with actin via the N-terminal domain and accumulation at the ruffled membrane leads to the release of H+ into resorption lacuna. The acid and proteases, such as matrix metalloproteinases (MMPs), dissolve crystalline hydroxyapatite and the organic matrix, respectively [17].
After bone resorption, the osteogenic environment is prepared at remodeling sites for bone formation by osteoclasts by releasing growth factors from the resorbed matrix, producing secreted proteins, and expressing membrane-bound factors that facilitate bone formation [18]. Once mesenchymal stem cells or osteoblast progenitors are returned to the resorption lacunae, they differentiate and secrete collagen type I, a primary component of bone. Non-collagenous proteins, such as proteoglycans, alkaline phosphatase, small integrin-binding ligand (SIBLING) proteins, Gla-containing proteins (matrix Gla protein and osteocalcin), and lipids then form the remaining organic material and hydroxylapatite is deposited into new bone, replacing the resorbed bone [19]. When bone loss is compensated, osteoblasts terminally differentiate into osteocytes and bone remodeling is terminated.
3. Molecular Biology of Osteoporosis
3.1. Osteoblasts
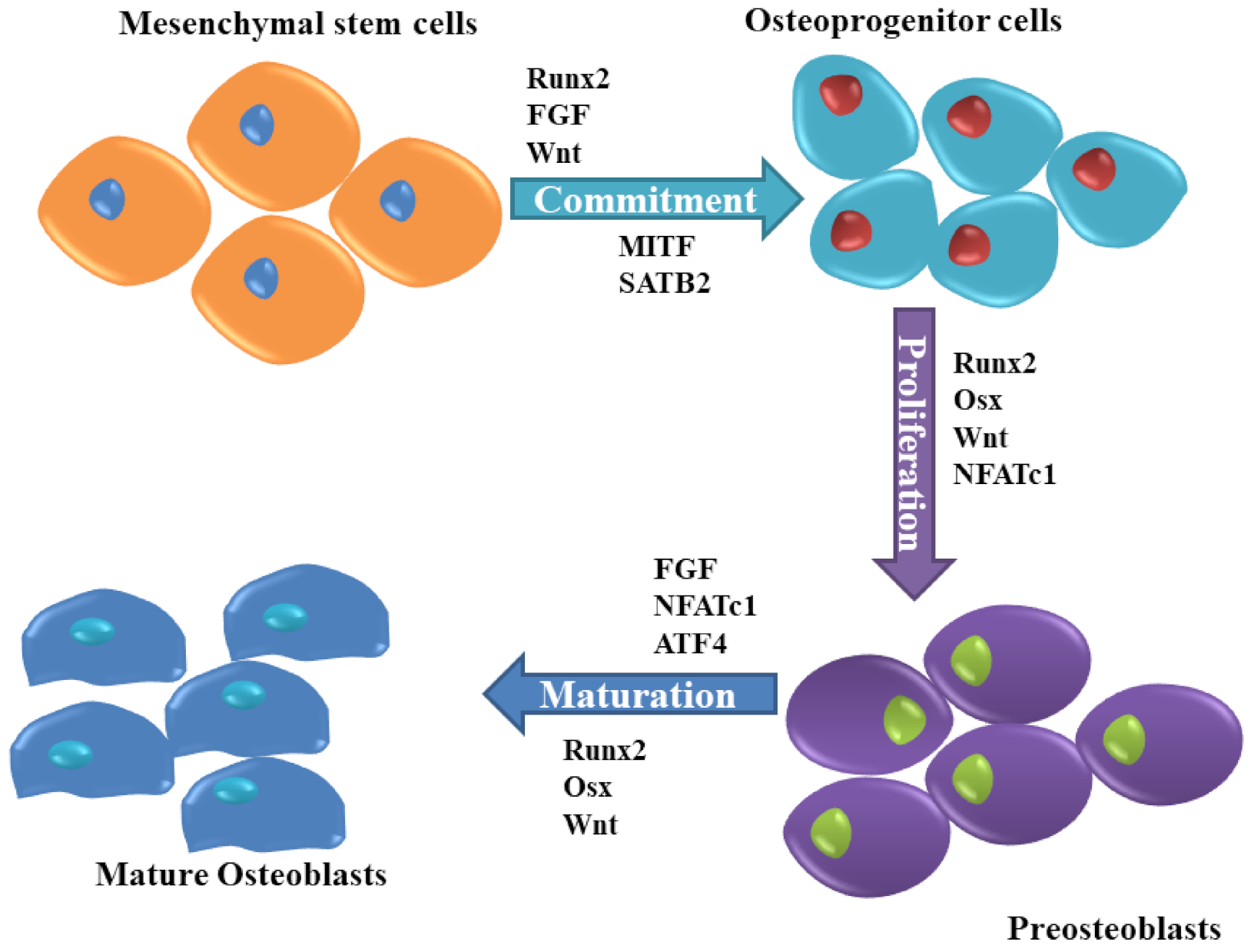
Due to the rapid developments in cellular metabolism research, many investigators have focused on the understanding of the pathophysiology of chronic diseases to investigate novel therapeutic targets in bone cells such as osteoblasts. Osteoblasts, derived from undifferentiated mesenchymal cells, are essential bone-forming cells required for the growth and maintenance of the skeleton, are regulated by various molecules (Figure 2). Dysfunction in osteoblast due to disorders of substrate availability such as diabetes mellitus, anorexia nervosa, and aging eventually leads to skeletal fragility and osteoporotic fractures [20].
3.1.1. Wnt Signaling
The Wnt-β-catenin signaling pathway is an evolutionarily conserved system of cell-cell communication that plays an important role in stem cell renewal, cell proliferation, and cell differentiation. The Wnt signaling pathway can be categorized into two pathways: canonical β-catenin-dependent pathway and non-canonical β-catenin-independent pathways. Genetic or epigenetic changes that might lower or over-activate the Wnt-β-catenin signaling can increase human diseases, including osteoporosis [21]. Wnt proteins are the members of the secreted glycoproteins family and play critical roles during osteoblast differentiation. Their signals are transduced by a family of seven-pass transmembrane G-protein coupled receptors of the frizzled (Fzd) family, and a co-receptor of the low-density lipoprotein receptor-related protein (LRP) family (e.g., LRP5 and LRP6), or a Ryk or Ror transmembrane tyrosine kinase [22].
Wnt signaling regulates mesenchymal stem cells (MSC) differentiation into osteoblasts and inhibits adipogenesis by preventing the expression of the adipogenic transcription factors C/EBPα and PPARγ [23]. Wnt family members like Wnt10a, Wnt10b, and Wnt6 have been found to regulate the fate of MSC. The gain- and loss-of-function study showed how Wnt6, Wnt10a, and Wnt10b regulate mesenchymal cell fate. The overexpression of Wnt10a and Wnt10b greatly stabilizes β-catenin, suppresses adipogenesis, and stimulates osteoblastogenesis through a canonical β-catenin-dependent pathway, whereas Wnt6 has a weaker effect. However, knockdown of Wnt6 enhanced preadipocyte differentiation and weakened osteoblastogenesis compared to Wnt10a or Wnt10b [24]. LRP5 expressed by osteoblasts, encodes the low-density lipoprotein receptor-related protein 5, a co-receptor required for signal transduction that affects bone mass. The mutations in LRP5 can lead to osteoporosis-pseudoglioma syndrome (OPPG), exhibiting reduced bone mass [25]
Wnt Signaling is checked by the inhibitors such as secreted frizzled-related protein (sFRP), Dickkopf-1 (DKK1), and sclerostin. sFRP acts as a decoy receptor of Wnt and inhibits the binding of the ligand to the receptor complex and canonical as well as non-canonical Wnt signaling pathways. While DKK1 binds to the BP1 and BP3 domains of the LRP5/6 receptor and formulate a complex with Kremens to increase internalization of the LRP5/6 receptor and inhibits the canonical Wnt pathway, sclerostin suppresses bone formation by obstructing the canonical Wnt signaling pathway. Sclerostin binds to the BP1 domain of the LRP5/6 receptor as well as LRP4 to enhance its inhibitory effects on the canonical Wnt signaling pathway [26]. The WNT signaling pathway has been identified as a candidate for therapeutic intervention to increase bone mass and strength. Particularly, the inhibition of sclerostin has been confirmed to have a very efficient osteo-anabolic effect increasing bone formation and bone density and reducing fracture risk in treated patients [27].
3.1.2. Runt-Related Transcription Factor 2 (RUNX2)
RUNX2 is an upstream transcription factor involved in the regulation of osteoblast differentiation, chondrocyte maturation, as well as osteoclastogenesis. In osteoblasts, RUNX2 regulates the expression of Sp7 protein, which is a key transcription factor for osteoblast differentiation and bone matrix genes [28]. In pre-confluent G(0)/G(1) transition immature MC3T3 osteoblastic cells, RUNX2 levels were upregulated with maximum expression levels during early G(1). Interestingly, enforced expression of RUNX2 suppressed proliferation of MC3T3 preosteoblasts and C2C12 mesenchymal cells by delaying G(1) phase [29].
However, the reestablishment of RUNX2 into RUNX2−/− calvarial osteoblast cells restored cell growth control and regulated normal osteoblast proliferation [30]. Fibroblast growth factor receptor (FGFR) genes that are expressed in mesenchymal cells constitute the important FGF signaling to control proliferation, anti-apoptosis, angiogenesis, epithelial-to-mesenchymal transition, and crosstalk with the canonical Wnt signaling. RUNX2 directly regulates FGFR2 and FGFR3 to increase the proliferation of osteoblast progenitors [31]. While RUNX2 also controls the commitment of mesenchymal cells to the osteoblast lineage by enhancing the expression of Col1a1 and Bglap and activating their promoters, RUNX2 overexpression inhibits osteoblast maturation and decreases Col1a1 and Bglap expression leading to the arrest of osteoblasts in an immature stage [32]. The change in the expression or activity of RUNX2 affects osteoblast differentiation and RUNX2 can be regulated by post-translational modifications such as phosphorylation, ubiquitination, and acetylation [33]. The upregulated RUNX2 binds to the DNA element in the Osx promoter and activates promoter of Osterix (Osx) in mesenchymal cells to regulate bone and cartilage formation [34].
3.1.3. Osteocalcin
Osteocalcin, also known as bone γ-carboxyglutamic acid-containing protein (BGLAP) is a non-collagenous, vitamin K-dependent protein secreted by osteoblasts. The presence of the three residues of γ-carboxyglutamic acid allows osteocalcin to bind to calcium and subsequently hydroxyapatite. The levels of carboxylated and undercarboxylated forms of osteocalcin are often used in clinical practice and research as a marker of bone turnover to predict fracture or diabetes risks [35,36]. Osteocalcin maintains the structural properties of bone by interacting with hydroxyapatite and collagen I. The levels of glycation are associated with bone fragility and therefore, the addition of sugar on a protein can affect osteocalcin’s ability to interact with collagen I or osteopontin (OPN) but not with hydroxyapatite [37]. The knockdown of osteocalcin in MSCs delays the maturation of mineral species and lowers total hydroxyapatite.
Moreover, the expression of RUNX2, alkaline phosphatase, type I collagen, and osteonectin genes was reduced during osteogenic differentiation of MSC [38]. Osteocalcin promotes the absorption of calcium into the hydroxyapatite and leads to bone mineralization. The reduction in bone mineralization frees osteocalcin and becomes circulated in the blood [39]. Osteoporosis is described to have decreased hydroxyapatite crystal formation and therefore results in elevated levels of osteocalcin in serum. Hence, serum osteocalcin can be employed as a diagnostic biomarker to screen primary osteoporosis in women [40].
3.1.4. RANKL
RANKL is a type II transmembrane protein produced by the osteoblasts and other stromal cells with an extracellular domain at the C-terminus. [41]. It is present as a membrane-bound and soluble form, which is produced by the proteolytic cleavage of the ectodomain by metalloproteinases and released to the extracellular environment. Both forms of RANKL act as agonistic ligands for RANK, but the membrane-bound RANKL is more effective than the soluble form [42]. RANKL exerts its osteoclastic effect by binding to its corresponding receptor RANK to promote RANK signaling and subsequently osteoclastogenesis [43]. Mice deficient in soluble RANKL have no effect on bone mass or structure in growing mice but decreases osteoclast number and stimulates cancellous bone mass in adult mice. Additionally, estrogen deficiency-induced bone loss also remained unaffected by the absence of soluble RANKL. This suggests that the membrane-bound RANKL is adequate for physiological bone remodeling in adult mice [44]. RANKL addition in cultured RAW264.7 cells demonstrated increased bone resorption pit, F-actin, and levels of osteoclastogenesis specific marker genes, which suggested amplified differentiation of RAW264.7 cells into multinucleated osteoclastic cells [45]. Mice lacking RANKL in osteocytes show reduced osteoclast number and the bone loss resulted due to ovariectomy and no surge in bone marrow B lymphocytes caused by ovariectomy. This demonstrates that RANKL produced by osteocytes is responsible for the increase in B cells and bone loss that are resulted from estrogen deficiency in mice [46].
3.1.5. OPG
OPG, a soluble decoy receptor for RANKL is mainly produced by osteoblasts. OPG inhibits osteoclast formation and bone resorption by inhibiting the interaction of RANKL-RANK [47]. The deficiency of OPG (OPG−/−) induces osteoporosis in mice due to increased bone resorption but also enhances bone formation. The application of bone resorption inhibitor risedronate in OPG−/− mice significantly decreased mineral apposition rate and osteoblast surface/bone surface as well as lowered high serum alkaline phosphatase activity and osteocalcin concentration. Bone morphogenetic protein-2 (BMP-2) establishment into OPG−/− mice did not accelerate ectopic bone formation even with a high turnover rate of bone; however, ectopic bone mineral density reduction was higher. This indicates bone formation and bone resorption are coupled at local sites in OPG−/− mice [48]. According to Koide, et al., OPG−/− mice also had higher alveolar bone loss and severe bone resorption in cortical areas of the alveolar bone due to increased osteoclasts number in the area [49]. Alveolar bone loss can be restored by the treatment of WP9QY (W9) peptide, a RANKL blocker, in OPG−/− mice, which suppress osteoclastogenesis and increase osteoblastogenesis. The treatment of W9 in OPG−/− mice decreased the osteoclast number in the alveolar bone and enhanced Wnt/β-catenin signaling to induce alveolar bone formation in OPG−/− mice. OPG−/− mice also express sclerostin in a low amount in tibiae and W9 treatment further repressed sclerostin expression [50]. The site of OPG production also matters in regulating bone and immune homeostasis. In OPG-floxed mice, it showed that locally produced OPG is important for bone and immune homeostasis rather than circulating OPG. It suggests that OPG limits its role within the tissue where it was produced [51].
3.1.6. Osx
Osx, also known as transcription factor SP7 is a zinc finger-containing protein that belongs to the Sp/KLF (Kruppel like Factor) factors [52]. The importance of Osx in osteoblast differentiation and bone formation was demonstrated in Osx null mice. In these mice, mesenchymal cells could not deposit bone matrix, whereas cells in the periosteum failed to differentiate into osteoblasts [53]. In adult bone, inactivation of Osx in all osteoblasts reduced BMD and bone-forming rate in a lumbar vertebra, and reduced the length of the long bones making cortical bone thinner and more porous but increased immature trabecular bones [54]. Moreover, the inactivation of Osx in already formed bones produces a functional defect in osteoblasts reducing bone formation, though does not affect osteoblast proliferation or osteoclast formation [55]. Lee, et al. have shown that Osx targets the different genes and mechanisms that regulate osteoblast differentiation by knocking down Osx in MC3T3-E1 osteoblastic cells. Osteoblasts displayed a significantly reduced cell differentiation and nodule formation, and Osx-knockdown upregulated 15 and downregulated two genes associated with cell differentiation.
Of the 15 upregulated genes, the expression of fibrillin-2 and periostin was considerably higher, suggesting the possibility of fibrillin-2 and periostin as target candidates of Osx in osteoblast differentiation [56]. Osx also contributes to corticalization for longitudinal bone growth in long bones. In osteoblast-specific Osx-knockout mice (Col-OM), corticalization was delayed, and femoral length was decreased. Mice also displayed compromised matrix coalescence and osteoblast migration. Integrin-mediated focal adhesion composed of vinculin, which mediates cell adhesion, migration, and signaling, was also reduced in osteoblasts from Col-OMT mice. The protein levels of integrin β3 were diminished in Col-OMT mice [57]. Among the various gap junction protein in bone cells, connexin43 (Cx43) is the most abundant and contributes to osteoblast differentiation. The study demonstrates a proportional relationship between Osx and Cx43. When Osx was silenced using shRNA, Cx43 expression was also suppressed. By direct occupying the promoter region of Cx43, Osx subsequently improved bone morphogenetic protein 4-induced Cx43 promoter activity [58]. Osx also affects hedgehog (Hh) signaling during bone development. Knockdown of Osx expression in MC3T3-E1 cells completely blocks Sonic hedgehog-induced osteoblast differentiation indicating Osx is important in early osteoblast differentiation [59].
3.1.7. Core-Binding Factor Subunit Beta (CBFB)
Cbfa1, also known as Pebp2αA (polyoma enhancer-binding protein), is a transcription factor that belongs to the runt-domain gene family (Cbfa1/Pebp2αA, Cbfa2/Pebp2αB, and Cbfa3/Pebp2αC) [60]. Cbfa1 is expressed exclusively in a cell type that develops into both osteoblast and chondrocyte but later during development, its expression is limited to osteoblast progenitors and differentiated osteoblasts [61]. Cbfa1 promotes the development of osteoblasts and targeted deletion of Cbfa1 prevents osteoblasts and osteoclasts maturation resulting in a complete lack of bone formation [60]. The Cbfb deleted mice (Cbfbfl/fl/Cre mice) displayed dwarfism, stunted ossifications, and osteoblast differentiation. The molecular effect of Cbfb deletion in Cbfbfl/fl/Cre primary osteoblasts was that the protein levels of Runx1, RUNX2, and Runx3 were reduced and affected the stability of RUNX2. Thus, Cbfb modulates bone development by stabilizing Runx family proteins [62].
3.1.8. BMP-2
BMPs are growth factors that belong to the TGF-β superfamily. Over 20 BMPs have currently been identified and play vital roles in cardiogenesis, neurogenesis, eye formation, as well as osteogenesis, adipogenesis, chondrogenesis. BMP-2 is the first BMP that was identified in bone and since then, its functions in bone have been deeply elucidated in development and osteogenesis [63]. BMPs are secreted as cytokines that signal through three transmembrane type II receptors and four transmembrane type I receptors. The binding of BMP promotes phosphorylation of the type I receptor by the type II receptor and subsequently activates intracellular signaling, which is initiated by phosphorylation of receptor-regulated SMADs (R-SMADs). The formation of heteromeric complexes by activated R-SMADs with SMAD4 triggers transcriptional responses [64].
As bone fractures are common in the population with osteoporosis, BMPs have been suggested to have therapeutic potential to improve fracture healing in such patients [65]. It is known that mechanical stimulation enhances bone formation, and mechanical loading together with BMP-2 regulates early signaling events in the BMP pathway. Mechanical and BMP-2 stimulation increases phosphorylation of R-Smads and maintains its phosphorylation and regulates osteogenesis [66]. BMP-2 induces canonical Smad signaling and non-canonical pathways by activating MAPK cascades through Smad. BMP-2 stimulation upregulates the expression of multiple types of αvβ-integrins in human osteoblasts, which in turn mediates Smad signaling via the Cdc4-Src-FAK-ILK cascade. These interactions of BMP-2 signaling and mechanotransduction ultimately leads to the translocation of YAP/TAZ and activate osteogenic genes and drives osteogenic differentiation.
Moreover, in the absence of BMP-2, the Smad complexes remain bound and active on target genes [67]. The study conducted in osteoporosis rats showed that the overexpression of BMP-2 in rat bone marrow mesenchymal stem cells (rBMSCs) activates the BMP signaling pathway promoting osteoblastic differentiation, while the low expression of BMP-2 suppresses the BMP signaling pathway in rBMSCs [68]. The addition of BMP-2 in osteoporosis rats significantly improved bone repair and enhanced new bone formation in the margins of the defect as well as intramembranous ossification zones by reducing mineralization of the newly formed bone [69].
3.1.9. Forkhead Box Class O Family Member Proteins (FoxOs)
FoxOs are transcription factors that convert environmental signaling into gene expression to regulate cell survival, proliferation, differentiation, and oxidative stress resistance. It has been reported that loss- or gain-of-function of FoxOs can significantly alter the bone cell function and intercellular signaling, and dysfunction can lead to osteoporosis or other bone diseases [70]. FoxO activities in osteoblast significantly impact bone formation. In osteoblast progenitors, FoxOs inhibit Wnt signaling and bone formation, while increases osteoprotegerin expression in cells of the osteoblast lineage and decrease bone resorption by osteoclasts [71]. The previous study has demonstrated that by deleting FoxO1, 3, and 4 in osteoblast progenitor; cancellous bone mass loss can be mitigated in a mouse model of type 1 diabetes [72]. Under the influence of osteogenic stimulants, mouse mesenchymal cells exhibit increased FoxO1 expression and activity. This increased expression stimulates the expression of osteogenic markers such as RUNX2, alkaline phosphatase, and osteocalcin to promote mesenchymal cell differentiation into osteoblasts [73]. Additionally, FoxO1 acts as a negative regulator of RUNX2, osteoblast-specific transcription factor, and controls IGF1/insulin-dependent regulation of osteocalcin expression in osteoblasts [74]. The study has shown that downregulation of FoxO1, reduces mRNA levels of RUNX2, type 1 collagen, OCN, and MMP13, as well as the formation of mineralized nodules. However, overexpression of FoxO1 reduces the proliferation of MC3T3-E1 cells [75].
3.1.10. Nuclear Factor E2 p45-Related Factor 2 (Nrf2)
Nrf2 is an essential transcription factor required for the regulation of detoxifying and antioxidative genes and is an important modulator of carbohydrate, lipid, heme, and iron metabolisms. Nrf2 promotes survival of irradiated osteoblasts in hematopoietic compartments Nrf2 to support long-term hematopoietic stem cell niches [76]. In osteoblasts of cancellous bone in the femur of ovariectomized (OVX) mice, high expression of Nrf2 mRNA and protein was detected. In vitro, the nrf2 expression vector inhibited an increase in alkaline phosphatase activity and the mineralized matrix formation in MC3T3-E1 cells because of the impaired RUNX2-dependent stimulation of osteocalcin promoter activity. The recruitment of RUNX2 on osteocalcin promoter was also impaired without affecting the expression of RUNX2 mRNA. This indicates that Nrf2 acts as a negative regulator of cellular differentiation by inhibiting RUNX2-dependent transcriptional activity in osteoblasts [77].
3.2. Osteocytes
Osteocytes are mature osteoblasts that have a more differentiated morphology and, in time, become embedded in the matrix that they are actively synthesizing during the process of bone formation. Osteocytes maintain contact with adjacent osteocytes, osteoblasts, and endothelial cells and communicate via gap junctions [78]. The osteocyte cell and its dendritic processes are present in a complex lacuno-canalicular system and are important mechanosensing cells in bone. Osteocytes translate mechanical force on the bone to cellular signaling to maintain bone homeostasis. Various proteins and signaling molecules such as sclerostin, cathepsin K, Wnts, DKK1, DMP1, IGF1, and RANKL/OPG are produced by osteocytes to regulate osteoblast and osteoclast activity [79]. An altered expression of these molecules can lead to certain pathogenic conditions, including osteoporosis. Therefore, it is important to understand their function in the bone.
3.2.1. Stimulatory Subunit of G-Protein (Gsα)
In osteocytes, the stimulatory subunit of G-protein (Gsα) acts as a secondary messenger of G protein-coupled receptors (GPCRs). The absence of Gsα in osteocytes in mice induces high expression of sclerostin, which suppresses osteoblast activity, and decreases trabecular and cortical bone. Moreover, the deletion of Gsα also affects osteocyte morphology. Mice lacking GsαKO were deficient in dendrites and were randomly distributed throughout the bone matrix [80]. Additionally, deficiency of Gsα in osteocytes alters myelopoiesis and osteopenia. Osteocytes secrete Gsα-dependent factor(s), neuropilin-1 (Nrp-1), and granulin (Grn), and regulate myeloid cells proliferation by promoting myeloid and macrophage cells proliferation and differentiation into bone-resorbing osteoclasts [81].
3.2.2. Sclerostin
Sclerostin is highly expressed and secreted by osteocytes. It binds to LRP 5/6 and inhibits Wnt signaling, suppressing osteoblast differentiation [82,83]. The increased levels of circulating sclerostin in humans as well as in animal models have been reported during immobilization and prolonged bed rest. The mechanical unloading showed that it increases SOST/sclerostin expression in osteocytes [84]. Sclerostin regulates the expression of osteocyte-specific proteins, such as RANKL, OPG, and proteins encoded by DMP1, PHEX, and possibly FGF23. The unloading increases sclerostin levels which antagonize Wnt-canonical-β-catenin signaling pathways in osteocytes and osteoclasts, while mechanical loading lowers sclerostin levels and activates Wnt-canonical signaling and bone formation [85].
3.2.3. Integrins
Integrins in osteocytes are required for cell adhesion and signaling and are vital in facilitating mechanotransduction and, therefore, in load-induced bone formation. Osteocytes express β1 and β3 integrins subunits, and antagonism of these integrins result in osteoporosis, osteoarthritis, and bone metastases [86]. Osteocyte processes are more sensitive to mechanical loading than their cell bodies. The study showed that the membrane proteins involved in osteocyte mechanotransduction, such as purinergic channel pannexin1, the ATP-gated purinergic receptor P2 × 7R, and the low voltage transiently opened T-type calcium channel CaV3.2-1 all were preferentially localized close to β3 integrin attachment foci on osteocyte processes [87]. The integrin αvβ3 is important in maintaining osteocyte morphology and for mechanosensation and mechanotransduction. The integrin αvβ3 antagonism has shown that it alters osteocyte cell processes, reduces spread area, and process retractions. Moreover, in response to fluid shear stress, the blocking of integrin αvβ3 also interrupts COX-2 expression and PGE2 release [88].
3.3. Osteoclasts
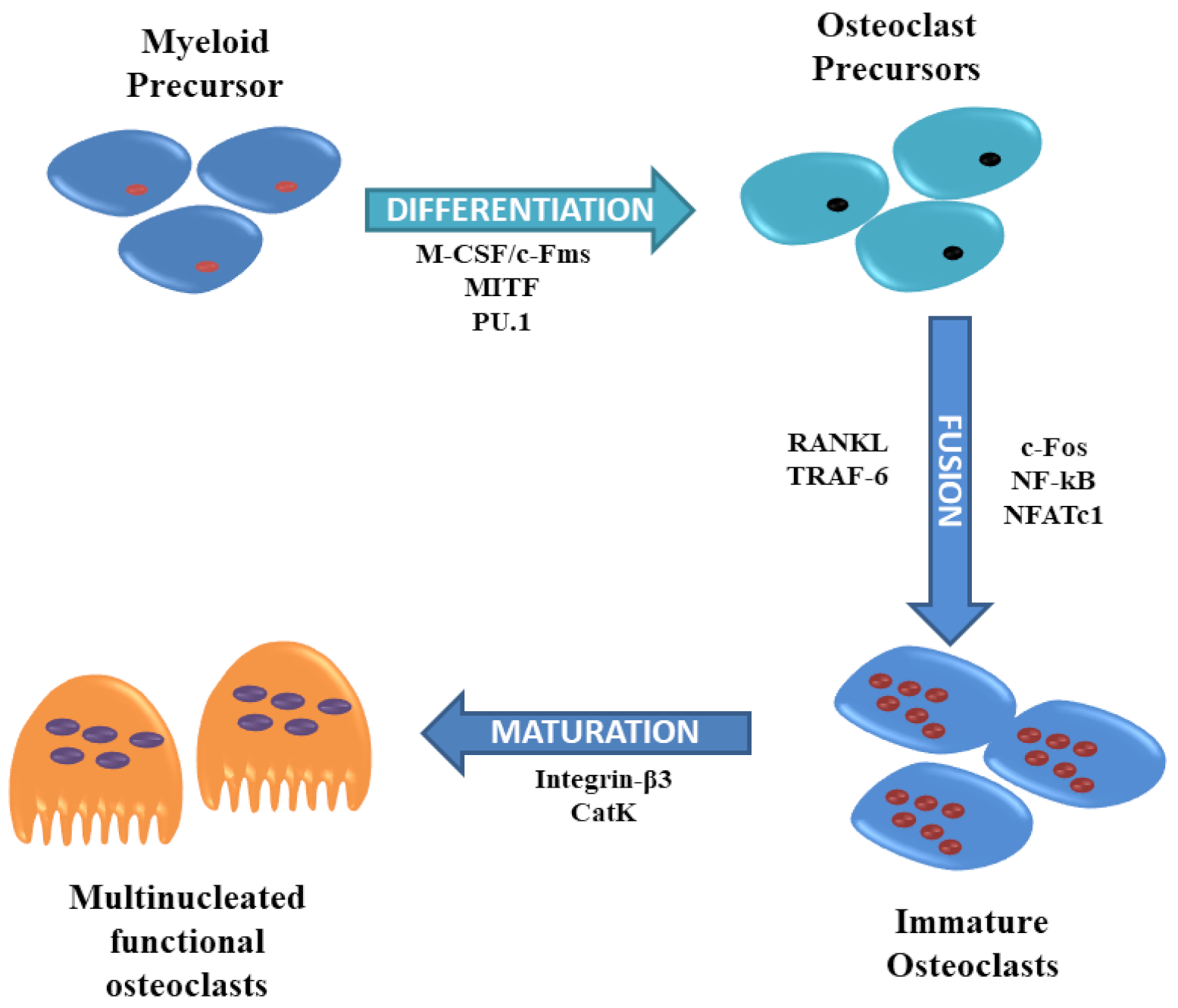
Osteoclasts are the primary bone-resorbing cells that contribute to the remodeling of the bone and therefore, defects in osteoclasts can result in unbalanced bone remodeling, causing pathological disorders such as bone metastasis, osteoporosis, and inflammatory bone erosion [89]. These are multinucleated giant cells produced by fusion of hematopoietic stem cells-derived monocytic precursors in the presence of colony-stimulating factor 1 (CSF1) and RANKL [90]. Osteoclasts remove damaged bone during the remodeling of the bone to maintain a healthy skeleton by releasing acid into the resorption lacunae. Several cytokines such as PU.1, M-CSF, c-Fos, CCAAT/enhancer-binding protein α (C/EBPα), RANK, nuclear factor of activated T-cells cytoplasmic 1 (NFATc1), and microphthalmia-associated transcription factor (MITF) regulates osteoclastogenesis and osteoclast differentiation and by regulating the expression of RANKL and its inhibitory decoy receptor OPG [91,92] (Figure 3).
PU.1 is a predominantly expressed member of the Ets family protein. PU.1 is involved in promoting the expression of NFATc1 and binding to the NFATc1 promoter to increase osteoclast-specific gene expression [93]. During bone marrow-derived macrophages (BMMs) differentiation into osteoclast, NFATc1 and interferon regulatory factor (IRF) 8 play positive and negative roles, respectively. The study has shown that PU.1, during RANKL-induced osteoclastogenesis, favors NFATc1 over IRF8 and modifies the binding regions that are associated with changes in epigenetic profiles, and thus controls cell type-specific gene expression [94]. Moreover, PU.1, in association with MITF coordinates a transcription factor network essential for osteoclast differentiation. Many of those are regulated by the binding of PU.1 with BRD4, forming superenhancers. The chromatin conformational changes in the superenhancer region of NFATc1 are also dependent on PU.1 [95].
MITF is essential for terminal osteoclast differentiation and shift from the cytoplasm to the nucleus upon RANKL/CSF-1 action. 14-3-3 chaperone-like adaptor proteins engage different proteins and regulate DNA replication, cell proliferation, differentiation, and apoptosis, which also is a binding partner of MITF in osteoclast precursors. In the absence of signals that are required for osteoclast differentiation, 14-3-3 promotes cytosolic localization of MITF, thus regulating its activity [96].
The deficiency of the component of activator protein-1 (AP-1), transcription factor c-Fos, impairs osteoclast development and positively regulates osteoclastogenesis [97]. The downregulation of c-Fos using siRNA in OVX mice improved the thickness of the trabeculae, amplified trabecular number, and reduced osteolysis. Additionally, the interleukin (IL) -7/interleukin-7 receptor has been demonstrated to activate the c-Fos/c-Jun pathway to promote RANKL-mediated osteoclast formation, bone resorption, and bone loss in OVX mice [98]. The transcription factor MADS-box transcription enhancer factor 2, polypeptide C (MEF2C) positively regulates osteoclast differentiation by binding to Fos regulatory regions to encourage c-Fos expression, which activates NFATc1 and downstream osteoclastogenesis. Moreover, the deletion of MEF2C in mice increased bone mass and arrested bone erosion due to reduced osteoclast formation [99]. AP-1, MITF, and NFATc1 stimulate cathepsin K (CTSK) promoter activity independent of each other but a combination of any two has a higher positive effect on CTSK promoter activity. However, the combination of AP-1 (c-fos/c-jun) and NFATc1 leads to the largest expression [100].
3.3.1. Cathepsin K
Cathepsins are lysosomal cysteine proteases, and based on their structures, catalytic mechanisms, and target proteins, there are 11 members of cathepsins (cathepsin B, C, F, H, K, L, O, S, V, W, and Z). CTSK is primarily secreted by activated osteoclasts and also by osteoblasts and osteocytes [101]. CTSK is largely responsible degradation of collagen and other matrix proteins during bone resorption and its collagenolytic activity is higher towards matured bone collagen [102]. The study has shown that the deletion of CTSK not only reduces bone resorption, but also improves bone formation. The pharmacological inhibition or deletion of cathepsin K (Ctsk−/− mice) also stimulates bone formation. Cathepsin K directly targets periostin for degradation and deletion of cathepsin K increases periostin and β-catenin expression in vivo to improve cortical bone formation. However, periostin deletion selectively inhibits cortical bone formation but not trabecular bone formation in Ctsk−/− mice [103]. CTSK deletion in osteoclasts increases sphingosine kinase 1 (Sphk1) expression, which is responsible for the synthesis of sphingosine 1-phosphate (S1P) that forms a negative feedback loop during osteoclast differentiation. However, high levels of S1P in Ctsk-deficient osteoclasts enhance alkaline phosphatase and mineralized nodules formation in osteoblast cultures in vitro. Additionally, osteoblasts from CTSK-deficient osteoclasts mice exhibit a high RANKL/OPG ratio forming a positive feedback loop to increase the number of osteoclasts [104]. Walia et al. have also demonstrated that CTSK deletion increases osteoblast differentiation by stimulating osteoclast precursor- and osteoclast-mediated secretion of platelet-derived growth factor (PDGF)-BB and S1P. In Ctsk−/− mice, the number of periosteal osteoclast precursors was low under homeostatic conditions; while, after fracture, their number increased, and in fractured Ctsk−/− mice, greater expression of PDGF-BB was observed and contributed to fracture healing [105]. CTSK KO mice also showed enhanced callus mineralization, improved bone healing and remodeling, as well as improved mechanical strength [106].
3.3.2. Osteopontin
OPN belongs to the SIBLING family of cell-matrix proteins and contributes to bone metabolism and homeostasis by regulating adhesion, proliferation, and migration of bone marrow mesenchymal stem cells, osteoclasts, and osteoblasts. Therefore, it has been demonstrated to have a close relationships with the occurrence and development of various bone disorders, such as osteosarcoma, rheumatoid arthritis, and also osteoporosis [107]. Osteopontin acts by influencing the secretion levels of IL-10, IL-12, IL-3, interferon-γ, integrin αvβ3, nuclear factor kappa B (NF-kB), macrophage, and T cells, and affecting CD44 receptors [108]. Human osteoclasts release OPN into the resorption pit, which acts as a chemokine for succeeding bone formation. Both, α2,3- and α2,6-linked sialic acid play a role in osteoclast differentiation and desialylation can affect osteoclast differentiation in bone [109]. OPN promotes osteoclastogenesis and osteoclast activity via CD44- and αvβ3-mediated cell signaling and has a decisive role in bone remodeling by contributing to the formation of podosomes, osteoclast survival, and osteoclast motility [110]. However, the addition of exogenous OPN has been shown to inhibit macrophage-to-osteoclast differentiation [111].
3.3.3. Phosphatidylinositol 3-Kinase (PI3K)/Akt Pathway
PI3K/AKT facilitates the anti-apoptotic function in a variety of cell types, including osteoclasts and promotes survival and differentiation. Akt regulates glycogen synthase kinase-3β (GSK-3β)/NFATc1 signaling to induce osteoclast differentiation. While inhibition of the PI3K reduces osteoclasts formation and NFATc1 expression, Akt overexpression in BMMs favorably stimulates NFATc1 expression. But both of these events do not affect c-Fos expression, which suggests that during RANKL-induced osteoclastogenesis NFATc1 induction mediated by PI3K/Akt is not dependent on c-Fos. Moreover, the overexpression of Akt increases the formation of an inactive form of GSK-3β and NFATc1 nuclear localization, and overexpression of the active form of GSK-3β decreases osteoclast formation by downregulating NFATc1 [112].
Guanine nucleotide-binding protein subunit α13 (Gα13) belonging to the G protein superfamily controls cell cytoskeleton organization by regulating the RhoGEF-RhoGTPase signaling pathway and negatively regulates osteoclast formation and activity. Osteoclast-specific Gna13 conditional knockout mice have shown severe osteoporosis. Deficiency of Gna13 strongly enhances osteoclast number as well as activity by reducing RhoA activity and enhancing Akt/GSK-3β/NFATc1 signaling. Moreover, the inhibition of Akt or RhoA activation rescues the over-activation of Gna13-deficient osteoclasts. Gα13 gain-of-function has an opposite effect on Akt activation and osteoclastogenesis, and produces a protective effect in mice by reducing pathological bone loss in disease models [113]. Moreover, a genome-wide linkage study has identified ARHGEF3 as a strong positional candidate for BMD in women. The product of the ARHGEF3 gene encoding Rho guanine-nucleotide exchange factor (RhoGEF) 3 activates two members of the RhoGTPase family: RHOA, which is required for osteoclast motility and attachment in bone resorption and RHOB, plays a role in bone role in osteoarthritis. The study showed significant associations between single-nucleotide polymorphisms (SNPs) in ARHGEF3 and decreased BMD in post-menopausal women, suggesting the role of the RhoGTPase-RhoGEF pathway in osteoporosis [114].
Conversely, microtubule actin crosslinking factor 1 (MACF1), a spectraplakin protein implicated in cytoskeletal distribution, cell migration, cell survival, and cell differentiation acts as a positive regulator of osteoclast differentiation through Akt/GSK-3β/NFATc1 signaling. In primary BMMs derived from osteoporotic mice, MACF1 expression was increased. Though knockdown of MACF1 did not affect the survival of pre-osteoclasts and mature osteoclasts, it diminished Akt and GSK-3β phosphorylation and reduced the expression of NFATc1, and inhibited the formation of multinucleated osteoclasts. Furthermore, MACF1 knockdown interrupted actin ring formation in osteoclasts and reduced the area and depth of pits, thus blocking the bone resorption activity of osteoclasts [115]. PI3K/Akt signaling is negatively regulated by phosphatase and tensin homolog (PTEN). Silencing of PTEN expression leads to enhanced Akt and GSK-3β phosphorylation levels by RANKL and promotes osteoclasts formation. Inactivation of GSK-3β through Akt blocks PTEN phosphorylation and phosphatase activity and improves Akt phosphorylation leading to enhanced osteoclast differentiation [116].
3.4. Estrogen and Androgen
Estradiol (E2) in women is produced mainly in ovarian follicles and men, in testes, and from peripheral aromatization. Testosterone is the primary circulating androgen synthesized in the testicles. Estrogens and androgens exert their bone-protective effect through estrogen receptor (ER) α and β and the androgen receptor (AR), respectively. These sex-steroid receptor proteins form homodimers and bind to hormone response elements (EREs or AREs), which are DNA sequences [117]. Estrogen affects osteocytes, osteoclasts, and osteoblasts and inhibits bone remodeling, reduces bone resorption, and maintains bone formation, respectively, but the main consequence of estrogen loss is amplified bone resorption [118]. Estrogen is an important key regulator of bone metabolism in men as well as women. The loss of estrogen in menopausal women is accompanied by reduced BMD and increased hip, and vertebral fractures in older women and men [119]. Estrogen controls bone turnover by targeting the expression of RANKL in bone lining cells [120]. The study has shown that the estrogen deficiency in the OVX rat model increases the vascular porosity of cortical bone, decreases bone volume fraction and trabecular number, and enhances trabecular separation in the proximal tibia, diminishing bone strength [121].
The proinflammatory cytokines, such as IL-1, IL-6, TNF-α, GM-CSF, M-CSF, and prostaglandin-E2 (PGE2), increases the number of pre-osteoclasts in the bone marrow and consequently bone resorption. The estrogen downregulates their levels and protects bone from excessive resorption. Estrogen also upregulates TGF-β, which acts on osteoclast and decreases their activity, and increases apoptosis [122]. Selective deletion of ERα in differentiated osteoclasts in female mice causes trabecular bone loss but not in males. In wild-type mice, estrogen prompted apoptosis by upregulating Fas ligand in osteoclasts of the trabecular bones but not in ERα deleted osteoclasts mice [123].
The targeted deletion of ERα in mice in mesenchymal osteoblast progenitors and osteoblast precursors reduces periosteal bone apposition and cortical bone mass, whereas, in mature osteoblasts, it has negligible or no effect on bone mass and architecture [117]. Though mice with global deletion of ERα (ERαKO) present confounding systemic effects, mice with tissue-specific ERα deletion in osteoblasts, osteocytes, chondrocytes, or osteoclasts lack the systemic effects. Osteoblast-specific ERαKO female mice (pOC-ERαKO) showed a reduced cancellous bone mass and decreased cortical bone mass. Moreover, osteoblast activity was also reduced in the cancellous bone of the proximal tibia and lowered whole bone strength in the femora and vertebrae [124]. Conversely, conditional deletion of ERβ specifically in early osteoprogenitor cells (ERβ-CKO) in the mouse model enhanced the trabecular bone volume fraction. Still, it did not affect cortical bone or bone formation and resorption indices. However, the number of colony-forming unit-osteoblasts (CFU-OBs) was higher in bone marrow cultures from ERβ-CKO. Quantitative polymerase chain reaction evaluation showed downregulation of 11 out of 16 prespecified pathways in ERβ-CKO mice [125].
ERα also plays an important role in osteocytes for the formation of trabecular bone in male mice. Male mice lacking ERα protein specifically in osteocytes demonstrated a substantial reduction in trabecular bone volume and reduced rate of bone formation without affecting the number of osteoclasts per bone surface and cortical bone. Mice had reduced expression of RUNX2, Sp7, and DMP1 markers in bone [126]. Female mice lacking ERα in osteocytes (ERα (ΔOcy/ΔOcy)) had a significant reduction in trabecular bone mineral density and bone formation parameters. The potential targets of ERα identified by gene expression microarray and gene ontology analysis were revealed to be Mdk and Sostdc1, the Wnt inhibitors, both of which were highly expressed in osteocytes of ERα (ΔOcy/ΔOcy) mice. Besides, unloading experiments displayed more trabecular bone loss without cortical bone loss [127]. Estrogen deficiency also reduces focal adhesion area and αvβ3 localization at focal adhesion sites, which increases RANKL/OPG ratio in osteocytes [128].
Deletion of androgen receptor (ARKO) in male mice showed lower serum testosterone levels and reduced cancellous bone volume [129]. Moreover, the inactivation of AR in female mice does not affect bone loss. ARKO mice exhibited high bone turnover with amplified bone resorption, which reduced trabecular and cortical bone mass. Osteoblasts and osteoclasts analysis from ARKO mice has shown that AR was essential for the inhibitory effect of androgens on osteoclastogenesis. RANKL expression was also upregulated in osteoblasts from AR-deficient mice [130]. However, osteoclast-specific ARKO mice did not affect osteoclast surface or bone microarchitecture, suggesting AR in osteoclasts is dispensable for bone maintenance [131]. Male mice with targeted deletion of an AR or ERα in the mesenchymal or myeloid cell lineage displayed reduced bone volume and trabecular number, and the high osteoclast number in the cancellous compartment but no loss of cortical bone mass. This suggests that effect androgens on cortical bone do not require AR or ERα signaling in osteoblasts and osteoclasts [132]. However, AR in osteocytes is essential to maintain male skeletal integrity [133].
4. Osteoporosis Treatments
Osteoporosis, a highly prevalent bone disorder worldwide, is characterized by low BMD and an increased risk of osteoporotic fractures [134]. It has been predicted to become a global challenge impacting the quality of life of the affected individuals. Therefore, several treatment options such as medications are available to control disease progression in post-menopausal women and elderly men. Recent studies on the molecular mechanisms in bone have identified novel therapeutic targets for osteoporosis [135].
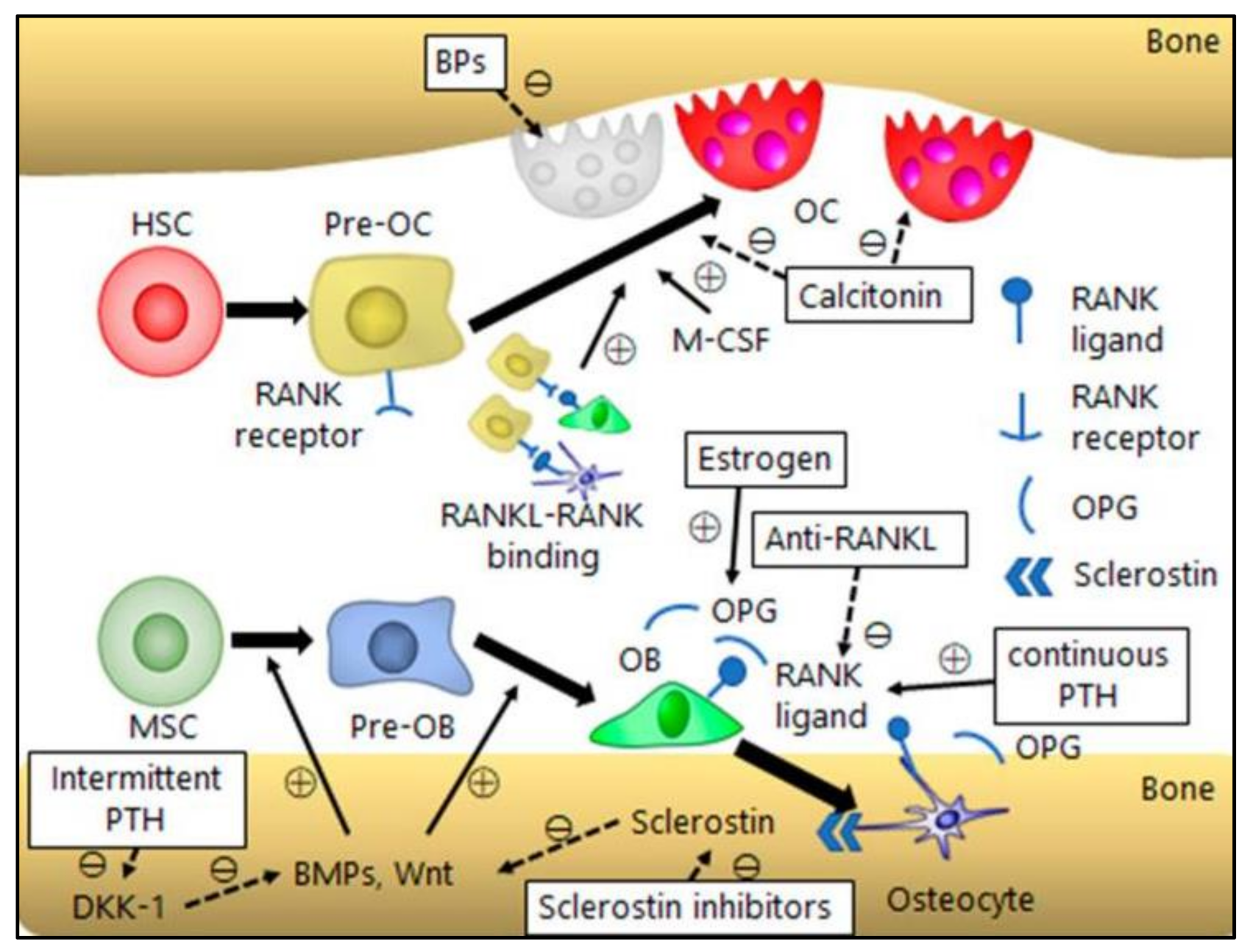
The proper maintenance of a healthy and active lifestyle is important for bone homeostasis. Physical exercise, a balanced diet, and calcium supplementation improve the quality of osteoporosis patients. Calcium supplementation decreases the rate of bone mineral density and, in combination with vitamin D, reduces the risk of total fractures and hip fractures. However, these methods are inadequate in preventing the progression of osteoporosis. Therefore, pharmacological therapies based on molecular targets have been developed to restore the normal bone balance. The most common therapies are the anti-resorptive and anabolic therapies [136] (Figure 4).
4.1. Antiresorptive Drugs
Antiresorptive drugs are aimed towards bone resorption and target osteoclasts. These include bisphosphonates, estrogen and selective estrogen receptor modulators (SERMs), and monoclonal antibodies, such as denosumab and odanacatib.
Bisphosphonates: The bisphosphonates as inhibitors of bone resorption were first identified in the late 1960s and have come to be the important treatment for various skeletal disorders, including osteoporosis [137]. Bisphosphonates bind to calcium exposed areas in the skeleton and cause osteoclast apoptosis. This leads to a reduction in the remodeling rate. Their treatment in hip fracture has shown the ability to increase lifespan [138]. BPs are categorized into two groups: non-nitrogen-containing and nitrogen-containing BPs. Non-nitrogen-containing BPs include clodronate, tiludronate, and etidronate reduces bone resorption through inhibition of ATP-dependent intracellular enzymes by integrating themselves into non-hydrolyzable adenosine triphosphate analogs via class II aminoacyl–transfer RNA synthetases. In comparison, nitrogen-containing BPs, such as alendronate, prevent farnesyl pyrophosphate synthase, which is an indispensable enzyme in the mevalonate pathway essential for the synthesis of cholesterol and isoprenoid compounds [92]. However, more than eight weeks of exposure to bisphosphonates to the bone in the jaw area is linked to a serious adverse event known as bisphosphonate-related osteonecrosis of the jaw (BRONJ) [139].
Estrogen and SERMs: Considering the important role of estrogen in women, deficiency of estrogen in menopausal women predisposes them to declined BMD and increased risks of fractures. Hormonal therapy (HT) has been shown to reduce the incidence of all osteoporosis-related fractures in post-menopausal women [140]. SERMs constitute a group of nonsteroidal compounds that function as ligands for ERs. But, contrasting estrogens that function as ER agonists, SERMs selectively act as agonists or antagonists in a target gene- and tissue-specific manner. SERM express tissue-specific ER subtypes, co-regulatory proteins in various tissues, and varies ER conformational changes by inducing ligand binding. Toremifene, Droloxifene, Idoxifene, and Raloxifene act as agonists and have anti-resoptive effect on bone [141]. The effect of SERMs on the ER downregulates osteoclast activity through TGF-β3 and reduces bone resorption [142].
Denosumab: Denosumab targets RANKL and blocks its interaction with RANK. This leads to inhibition in RANKL/RANK signaling and subsequently development and activity of osteoclasts, which reduces bone resorption [143]. The treatment with denosumab in post-menopausal osteoporosis women reduces the risk of vertebral, non-vertebral, and hip fractures and improves BMD [144].
Odanacatib: Odanacatib is a selective and reversible inhibitor of cathepsin K. Osteoclast-mediated bone resorption involves demineralization of inorganic bone mineral and degradation of organic bone matrix. CATK is a lysosomal cysteine protease and has strong collagenolytic activity. It degrades collagen type I and other bone matrix proteins [145]. Thus, cathepsin K inhibition reduces bone resorption without affecting bone formation and increases bone mineral density in post-menopausal women. Nevertheless, odanacatib was linked to an enhanced risk of cardiovascular events, especially stroke, in post-menopausal women with osteoporosis. Therefore, the development of odanacatib for treatment of osteoporosis was no longer pursued [146].
Strontium ranelate: Strontium ranelate consist of two cation atoms of stable strontium, which are closely related to calcium, and a ranelic acid. It has been proposed that strontium ranelate has a dual effect on bone in vitro, but in vivo in OVX rats and monkeys, it has been contradicted. However, in human studies, strontium ranelate showed a dual effect on bone; improved bone formation marker levels and decreased bone resorption markers [147]. The mechanism of action of strontium ranelate is not quite clear but is believed that strontium ranelate inhibits osteoclast function and osteoblast activity via a calcium-sensing receptor (CaSR) by strontium resulting in increased BMD and reduced fracture risks [148].
Calcitonin: Calcitonin is a polypeptide hormone that regulates calcium metabolism in the body and improves bone mineral density and reduces the fracture rate [149]. Calcitonin reduces calcium levels in the systemic circulation through the inhibition of bone resorption or prevention of calcium release from the bone. Therefore, calcitonin is used for the treatment of various bone disorders, including osteoporosis [150]. Salmon calcitonin is analogous to human calcitonin and is used in the treatment of post-menopausal osteoporosis. The treatment of post-menopausal osteoporosis with acupoint injection of salmon calcitonin showed stimulated osteoblast activity and reduced osteoclast-mediated bone absorption [151].
4.2. Anabolic Drugs
The anabolic drugs work by enhancing bone formation and include PTH and PTH-related peptides (PTHrP).
PTH and PTHrP: PTH is primarily secreted by the parathyroid glands and enhances trabecular and cortical bone formation, but in hypocalcemic conditions, it increases renal calcium reabsorption and activates calcium from cortical bone by enhancing turnover [152]. PTH or PTHrP can be administered daily and weekly to osteoporotic patients to improve bone mass and lower bone fracture risk. The continuous PTH treatment has catabolic effects on bone, whereas the intermittent PTH administration has anabolic action in osteoporotic patients, and stimulates the proliferation of pre-osteoblasts and osteoblast-mediated bone formation [153]. PTH and PTHrP facilitate osteoblast differentiation and proliferation through extracellular signal-related kinases, cyclin-dependent kinase inhibitor ERK, P27, and RANKL/OPG. Intermittent PTH acts as a Wnt/β-catenin signaling agonist and activates the canonical Wnt pathway and inhibits canonical Wnt antagonist, DKK1 mRNA levels in osteoblasts [154]. The PTH-1 receptor (PTH1R), a G-protein-coupled receptor, is the main receptor through which PTH and PTHrP exercise their action, and is essential for the proliferation and differentiation of mesenchymal cells into osteoblasts. PTH and PTHrP have both pro-and anti-apoptotic effects and depend on the cell status. The study showed when mesenchymal and osteoblastic cells were in the preconfluent stage, PTH and PTHrP improved cell viability, whereas in post-confluent stage had the opposite effect. This dual effect of PTH and PTHrP was attributed to the PKA pathway [155]. PTHrP, such as teriparatide and abaloparatide increases bone formation and reduces vertebral as well as non-vertebral fracture risk. Teriparatide and abaloparatide effectively improve BMD by boosting bone architecture and reduces the fracture risk in the upper arm, wrist, or hip [156]. Teriparatide in post-menopausal osteoporosis women has shown significant improvement in both cortical and cancellous bone by enhancing cancellous bone volume and connectivity, trabecular morphology, and cortical bone thickness [157].
Romosozumab: Romosozumab (Evenity®) is a human monoclonal sclerostin antibody that promotes bone formation and inhibits bone resorption and has been approved in the EU and the USA to treat severe osteoporosis [158]. In post-menopausal osteoporosis women subcutaneous romosozumab administration showed a reduction in risk of new vertebral and non-vertebral fractures [159].
5. Conclusions
Osteoblasts, osteocytes, and osteoclasts are the important bone cells that regulate bone remodeling. The signaling molecules are secreted by one cell act as stimulators or inhibitors for other cells and determine the fate of bone remodeling and, in turn, bone homeostasis. Some molecules affect bone formation or bone resorption, or both. The disruption in their levels either suppresses or promotes bone formation and/or resorption, which can lead to skeletal disorders, including osteoporosis. The insight into the molecular mechanisms behind bone cell function has helped understand the biology of osteoporosis to develop therapeutics by targeting different factors.
The current osteoporosis treatments involve targeting factors to reduce bone resorption or increase bone formation. Anti-resorptive treatments, such as BPs, estrogen and SERMs, denosumab, and odanacatib, target osteoclasts, whereas anabolic drugs, such as PTH and PTH-related peptide (PTHrP) are aimed towards improving bone formation. However, as the pathophysiology of osteoporosis differs among persons, treatments can produce unwanted side effects. The role of different non-coding RNAs, such as micro-, lnc-, and circ-RNA, in regulating bone cell survival, proliferation, differentiation, and apoptosis, has been extensively summarized and therefore can be employed not only as therapeutic, but also targets to improve bone health [160,161]. Different delivery systems developed for targeted gene delivery in different organs, including bone, has opened new opportunities in gene engineering and the preliminary works on microRNA and SiRNA-based gene delivery systems in bone have shown promising results in improving bone microarchitecture and BMD in osteoporosis mice [162]. However, the targeted delivery of therapeutic in vivo remains a challenging aspect in gene-based therapeutics and therefore, further research is warranted.
Author Contributions
Writing–Original Draft Preparation, S.P., Y.G.; Writing–Review & Editing, J.J., Y.G.; Funding Acquisition, Y.G. All authors have read and agreed to the published version of the manuscript.
Funding
The authors gratefully acknowledge the financial assistance provided by the Scientific Research Foundation of Tangshan Normal University (No. 2021A03), Key Project for Scientific Research in Colleges and Universities of Hebei Province (No. ZD2021408), Natural Science Foundation of Hebei Province (No. 2021105001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aspray, T.J.; Hill, T.R. Osteoporosis and the Ageing Skeleton. Subcell. Biochem. 2019, 91, 453–476. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, A.; Luyten, F.P.; Flamaing, J.; Gielen, E. Pharmacological treatment of osteoporosis in the oldest old. Clin. Interv. Aging 2017, 12, 1065–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, C.B.; Dagar, M. Osteoporosis in Older Adults. Med. Clin. N. Am. 2020, 104, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Nitta, K.; Yajima, A.; Tsuchiya, K. Management of Osteoporosis in Chronic Kidney Disease. Intern. Med. 2017, 56, 3271–3276. [Google Scholar] [CrossRef] [Green Version]
- Mirza, F.; Canalis, E. Management of endocrine disease: Secondary osteoporosis: Pathophysiology and management. Eur. J. Endocrinol. 2015, 173, R131–R151. [Google Scholar] [CrossRef] [Green Version]
- Johannesdottir, F.; Thrall, E.; Muller, J.; Keaveny, T.M.; Kopperdahl, D.L.; Bouxsein, M.L. Comparison of non-invasive assessments of strength of the proximal femur. Bone 2017, 105, 93–102. [Google Scholar] [CrossRef]
- Kanis, J.A.; Cooper, C.; Rizzoli, R.; Reginster, J.Y. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos. Int. 2019, 30, 3–44. [Google Scholar] [CrossRef] [Green Version]
- Silverman, S.; Christiansen, C. Individualizing osteoporosis therapy. Osteoporos. Int. 2012, 23, 797–809. [Google Scholar] [CrossRef]
- Lerner, U.H. Bone remodeling in post-menopausal osteoporosis. J. Dent. Res. 2006, 85, 584–595. [Google Scholar] [CrossRef]
- Ukon, Y.; Makino, T.; Kodama, J.; Tsukazaki, H.; Tateiwa, D.; Yoshikawa, H.; Kaito, T. Molecular-Based Treatment Strategies for Osteoporosis: A Literature Review. Int. J. Mol. Sci. 2019, 20, 2557. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, Z. Ovarian Aging and Osteoporosis. Advances in experimental medicine and biology. Aging Aging Relat. Dis. 2018, 1086, 199–215. [Google Scholar] [CrossRef]
- Li, H.; Xiao, Z.; Quarles, L.D.; Li, W. Osteoporosis: Mechanism, Molecular Target and Current Status on Drug Development. Curr. Med. Chem. 2021, 28, 1489–1507. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Partridge, N.C. Physiological Bone Remodeling: Systemic Regulation and Growth Factor Involvement. Physiology 2016, 31, 233–245. [Google Scholar] [CrossRef]
- Kenkre, J.S.; Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef]
- Zhao, H.; Ito, Y.; Chappel, J.; Andrews, N.W.; Teitelbaum, S.L.; Ross, F.P. Synaptotagmin VII Regulates Bone Remodeling by Modulating Osteoclast and Osteoblast Secretion. Dev. Cell 2008, 14, 914–925. [Google Scholar] [CrossRef] [Green Version]
- Batsir, S.; Geiger, B.; Kam, Z. Dynamics of the sealing zone in cultured osteoclasts. Cytoskeleton 2017, 74, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, M.; Blair, H.C.; Moonga, B.S.; Abe, E.; Huang, C.L.H. Osteoclastogenesis, Bone Resorption, and Osteoclast-Based Therapeutics. J. Bone Miner. Res. 2003, 18, 599–609. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells Through Multiple Mechanisms. Annu. Rev. Physiol. 2020, 82, 507–529. [Google Scholar] [CrossRef]
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.C.; Guntur, A.R.; Long, F.; Rosen, C.J. Energy Metabolism of the Osteoblast: Implications for Osteoporosis. Endocr. Rev. 2017, 38, 255–266. [Google Scholar] [CrossRef]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karner, C.M.; Long, F. Wnt signaling and cellular metabolism in osteoblasts. Cell. Mol. Life Sci. 2017, 74, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cawthorn, W.P.; Bree, A.J.; Yao, Y.; Du, B.; Hemati, N.; Martinez-Santibañez, G.; MacDougald, O.A. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone 2012, 50, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef] [Green Version]
- Baron, R.; Gori, F. Targeting WNT signaling in the treatment of osteoporosis. Curr. Opin. Pharmacol. 2018, 40, 134–141. [Google Scholar] [CrossRef]
- Komori, T. Roles of Runx2 in Skeletal Development. Adv. Exp. Med. Biol. 2017, 962, 83–93. [Google Scholar] [CrossRef]
- Galindo, M.; Pratap, J.; Young, D.W.; Hovhannisyan, H.; Im, H.J.; Choi, J.Y.; Lian, J.B.; Stein, J.L.; Stein, G.S.; van Wijnen, A.J. The bone-specific expression of Runx2 oscillates during the cell cycle to support a G1-related antiproliferative function in osteoblasts. J. Biol. Chem. 2005, 280, 20274–20285. [Google Scholar] [CrossRef] [Green Version]
- Pratap, J.; Galindo, M.; Zaidi, S.K.; Vradii, D.; Bhat, B.M.; Robinson, J.A.; Choi, J.Y.; Komori, T.; Stein, J.L.; Lian, J.B.; et al. Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Res. 2003, 63, 5357–5362. [Google Scholar]
- Kawane, T.; Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, H.; Yoshida, C.A.; Matsuura-Kawata, V.; Sakane, C.; Matsuo, Y.; Nagai, K.; et al. Runx2 is required for the proliferation of osteoblast progenitors and induces proliferation by regulating Fgfr2 and Fgfr3. Sci. Rep. 2018, 8, 13551. [Google Scholar] [CrossRef]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Gomathi, K.; Akshaya, N.; Srinaath, N.; Moorthi, A.; Selvamurugan, N. Regulation of Runx2 by post-translational modifications in osteoblast differentiation. Life Sci. 2020, 245, 117389. [Google Scholar] [CrossRef]
- Nishio, Y.; Dong, Y.; Paris, M.; O’Keefe, R.J.; Schwarz, E.M.; Drissi, H. Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene 2006, 372, 62–70. [Google Scholar] [CrossRef]
- Smith, C.; Voisin, S.; Al Saedi, A.; Phu, S.; Brennan-Speranza, T.; Parker, L.; Eynon, N.; Hiam, D.; Yan, X.; Scott, D.; et al. Osteocalcin and its forms across the lifespan in adult men. Bone 2020, 130, 115085. [Google Scholar] [CrossRef]
- Neve, A.; Corrado, A.; Cantatore, F.P. Osteocalcin: Skeletal and extra-skeletal effects. J. Cell Physiol. 2013, 228, 1149–1153. [Google Scholar] [CrossRef]
- Thomas, C.J.; Cleland, T.P.; Zhang, S.; Gundberg, C.M.; Vashishth, D. Identification and characterization of glycation adducts on osteocalcin. Anal. Biochem. 2017, 525, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Tsao, Y.T.; Huang, Y.J.; Wu, H.H.; Liu, Y.A.; Liu, Y.S.; Lee, O.K. Osteocalcin Mediates Biomineralization during Osteogenic Maturation in Human Mesenchymal Stromal Cells. Int. J. Mol. Sci. 2017, 18, 159. [Google Scholar] [CrossRef]
- Soroush, M.; Khabbazi, A.; Malek Mahdavi, A. Serum osteocalcin levels in postmenopausal osteoporotic women receiving alendronate. Rheumatol. Res. 2018, 3, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Kumar, D.; Lal, A.K. Serum Osteocalcin as a Diagnostic Biomarker for Primary Osteoporosis in Women. J. Clin. Diagn. Res. 2015, 9, Rc04-07. [Google Scholar] [CrossRef]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef]
- Pérez-Sayáns, M.; Somoza-Martín, J.M.; Barros-Angueira, F.; Rey, J.M.; García-García, A. RANK/RANKL/OPG role in distraction osteogenesis. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2010, 109, 679–686. [Google Scholar] [CrossRef]
- Xiong, J.; Cawley, K.; Piemontese, M.; Fujiwara, Y.; Zhao, H.; Goellner, J.J.; O’Brien, C.A. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat. Commun. 2018, 9, 2909. [Google Scholar] [CrossRef]
- Song, C.; Yang, X.; Lei, Y.; Zhang, Z.; Smith, W.; Yan, J.; Kong, L. Evaluation of efficacy on RANKL induced osteoclast from RAW264.7 cells. J. Cell Physiol. 2019, 234, 11969–11975. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Piemontese, M.; Liu, Y.; Thostenson, J.D.; Xiong, J.; O’Brien, C.A. RANKL (Receptor Activator of NFκB Ligand) Produced by Osteocytes Is Required for the Increase in B Cells and Bone Loss Caused by Estrogen Deficiency in Mice. J. Biol. Chem. 2016, 291, 24838–24850. [Google Scholar] [CrossRef] [Green Version]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Nakamura, M.; Udagawa, N.; Matsuura, S.; Mogi, M.; Nakamura, H.; Horiuchi, H.; Saito, N.; Hiraoka, B.Y.; Kobayashi, Y.; Takaoka, K.; et al. Osteoprotegerin regulates bone formation through a coupling mechanism with bone resorption. Endocrinology 2003, 144, 5441–5449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, M.; Kobayashi, Y.; Ninomiya, T.; Nakamura, M.; Yasuda, H.; Arai, Y.; Okahashi, N.; Yoshinari, N.; Takahashi, N.; Udagawa, N. Osteoprotegerin-deficient male mice as a model for severe alveolar bone loss: Comparison with RANKL-overexpressing transgenic male mice. Endocrinology 2013, 154, 773–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, Y.; Koide, M.; Furuya, Y.; Ninomiya, T.; Yasuda, H.; Nakamura, M.; Kobayashi, Y.; Takahashi, N.; Yoshinari, N.; Udagawa, N. Treatment of OPG-deficient mice with WP9QY, a RANKL-binding peptide, recovers alveolar bone loss by suppressing osteoclastogenesis and enhancing osteoblastogenesis. PLoS ONE 2017, 12, e0184904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukasaki, M.; Asano, T.; Muro, R.; Huynh, N.C.; Komatsu, N.; Okamoto, K.; Nakano, K.; Okamura, T.; Nitta, T.; Takayanagi, H. OPG Production Matters Where It Happened. Cell Rep. 2020, 32, 108124. [Google Scholar] [CrossRef]
- Yoshida, C.A.; Komori, H.; Maruyama, Z.; Miyazaki, T.; Kawasaki, K.; Furuichi, T.; Fukuyama, R.; Mori, M.; Yamana, K.; Nakamura, K.; et al. SP7 inhibits osteoblast differentiation at a late stage in mice. PLoS ONE 2012, 7, e32364. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Baek, W.Y.; Lee, M.A.; Jung, J.W.; Kim, S.Y.; Akiyama, H.; de Crombrugghe, B.; Kim, J.E. Positive regulation of adult bone formation by osteoblast-specific transcription factor osterix. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2009, 24, 1055–1065. [Google Scholar] [CrossRef]
- Baek, W.Y.; de Crombrugghe, B.; Kim, J.E. Postnatally induced inactivation of Osterix in osteoblasts results in the reduction of bone formation and maintenance. Bone 2010, 46, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Lee, E.H.; Park, S.Y.; Kim, J.E. Induction of fibrillin-2 and periostin expression in Osterix-knockdown MC3T3-E1 cells. Gene 2017, 596, 123–129. [Google Scholar] [CrossRef]
- Moon, Y.J.; Yun, C.Y.; Choi, H.; Kim, J.R.; Park, B.H.; Cho, E.S. Osterix regulates corticalization for longitudinal bone growth via integrin β3 expression. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Cho, D.H.; Chung, D.J.; Lee, K.Y. Osterix plays a critical role in BMP4-induced promoter activity of connexin43. Biochem. Biophys. Res. Commun. 2016, 478, 683–688. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, Y.; Fu, Q.; Dong, Y. Osterix is required for Sonic hedgehog-induced osteoblastic MC3T3-E1 cell differentiation. Cell Biochem. Biophys. 2012, 64, 169–176. [Google Scholar] [CrossRef]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Karsenty, G. Role of Cbfa1 in osteoblast differentiation and function. Semin. Cell Dev. Biol. 2000, 11, 343–346. [Google Scholar] [CrossRef]
- Qin, X.; Jiang, Q.; Matsuo, Y.; Kawane, T.; Komori, H.; Moriishi, T.; Taniuchi, I.; Ito, K.; Kawai, Y.; Rokutanda, S.; et al. Cbfb regulates bone development by stabilizing Runx family proteins. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2015, 30, 706–714. [Google Scholar] [CrossRef]
- Halloran, D.; Durbano, H.W.; Nohe, A. Bone Morphogenetic Protein-2 in Development and Bone Homeostasis. J. Dev. Biol. 2020, 8, 19. [Google Scholar] [CrossRef]
- Gomez-Puerto, M.C.; Iyengar, P.V.; García de Vinuesa, A.; Ten Dijke, P.; Sanchez-Duffhues, G. Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 2019, 247, 9–20. [Google Scholar] [CrossRef]
- Dumic-Cule, I.; Peric, M.; Kucko, L.; Grgurevic, L.; Pecina, M.; Vukicevic, S. Bone morphogenetic proteins in fracture repair. Int. Orthop. 2018, 42, 2619–2626. [Google Scholar] [CrossRef] [Green Version]
- Kopf, J.; Petersen, A.; Duda, G.N.; Knaus, P. BMP2 and mechanical loading cooperatively regulate immediate early signalling events in the BMP pathway. BMC Biol. 2012, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Holle, A.; Li, J.; Posa, F.; Biagioni, F.; Croci, O.; Benk, A.S.; Young, J.; Noureddine, F.; Deng, J.; et al. BMP-2 Signaling and Mechanotransduction Synergize to Drive Osteogenic Differentiation via YAP/TAZ. Adv. Sci. 2020, 7, 1902931. [Google Scholar] [CrossRef]
- Zhao, B.; Xing, G.; Wang, A. The BMP signaling pathway enhances the osteoblastic differentiation of bone marrow mesenchymal stem cells in rats with osteoporosis. J. Orthop. Surg. Res. 2019, 14, 462. [Google Scholar] [CrossRef] [PubMed]
- Segredo-Morales, E.; García-García, P.; Reyes, R.; Pérez-Herrero, E.; Delgado, A.; Évora, C. Bone regeneration in osteoporosis by delivery BMP-2 and PRGF from tetronic-alginate composite thermogel. Int. J. Pharm. 2018, 543, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Su, P.; Yin, C.; Lin, X.; Wang, X.; Gao, Y.; Patil, S.; War, A.R.; Qadir, A.; Tian, Y.; et al. The Roles of FoxO Transcription Factors in Regulation of Bone Cells Function. Int. J. Mol. Sci. 2020, 21, 692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.N.; Iyer, S.; Ring, R.; Almeida, M. The Role of FoxOs in Bone Health and Disease. Curr. Top. Dev. Biol. 2018, 127, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Han, L.; Ambrogini, E.; Yavropoulou, M.; Fowlkes, J.; Manolagas, S.C.; Almeida, M. Deletion of FoxO1, 3, and 4 in Osteoblast Progenitors Attenuates the Loss of Cancellous Bone Mass in a Mouse Model of Type 1 Diabetes. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, C.C.; Liu, Y.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a novel regulator of osteoblast differentiation and skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Xu, H.; Yu, S.; Cao, H.; Fan, J.; Ge, C.; Fransceschi, R.T.; Dong, H.H.; Xiao, G. Foxo1 mediates insulin-like growth factor 1 (IGF1)/insulin regulation of osteocalcin expression by antagonizing Runx2 in osteoblasts. J. Biol. Chem. 2011, 286, 19149–19158. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, M.F.; Flowers, S.; Bhattacharya, R.; Faibish, D.; Behl, Y.; Kotton, D.N.; Gerstenfeld, L.; Moran, E.; Graves, D.T. FOXO1 modulates osteoblast differentiation. Bone 2011, 48, 1043–1051. [Google Scholar] [CrossRef] [Green Version]
- Cameron, B.D.; Sekhar, K.R.; Ofori, M.; Freeman, M.L. The Role of Nrf2 in the Response to Normal Tissue Radiation Injury. Radiat. Res. 2018, 190, 99–106. [Google Scholar] [CrossRef]
- Hinoi, E.; Fujimori, S.; Wang, L.; Hojo, H.; Uno, K.; Yoneda, Y. Nrf2 negatively regulates osteoblast differentiation via interfering with Runx2-dependent transcriptional activation. J. Biol. Chem. 2006, 281, 18015–18024. [Google Scholar] [CrossRef] [Green Version]
- Uda, Y.; Azab, E.; Sun, N.; Shi, C.; Pajevic, P.D. Osteocyte Mechanobiology. Curr. Osteoporos. Rep. 2017, 15, 318–325. [Google Scholar] [CrossRef]
- Pathak, J.L.; Bravenboer, N.; Klein-Nulend, J. The Osteocyte as the New Discovery of Therapeutic Options in Rare Bone Diseases. Front. Endocrinol. 2020, 11, 405. [Google Scholar] [CrossRef]
- Fulzele, K.; Dedic, C.; Lai, F.; Bouxsein, M.; Lotinun, S.; Baron, R.; Divieti Pajevic, P. Loss of Gsα in osteocytes leads to osteopenia due to sclerostin induced suppression of osteoblast activity. Bone 2018, 117, 138–148. [Google Scholar] [CrossRef]
- Azab, E.; Chandler, K.B.; Uda, Y.; Sun, N.; Hussein, A.; Shuwaikan, R.; Lu, V.; Costello, C.E.; McComb, M.E.; Divieti Pajevic, P. Osteocytes control myeloid cell proliferation and differentiation through Gsα-dependent and -independent mechanisms. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 10191–10211. [Google Scholar] [CrossRef]
- Weivoda, M.M.; Youssef, S.J.; Oursler, M.J. Sclerostin expression and functions beyond the osteocyte. Bone 2017, 96, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Matsumoto, T. Sclerostin: From bench to bedside. J. Bone Miner. Metab. 2021, 39, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Spatz, J.M.; Wein, M.N.; Gooi, J.H.; Qu, Y.; Garr, J.L.; Liu, S.; Barry, K.J.; Uda, Y.; Lai, F.; Dedic, C.; et al. The Wnt Inhibitor Sclerostin Is Up-regulated by Mechanical Unloading in Osteocytes in Vitro. J. Biol. Chem. 2015, 290, 16744–16758. [Google Scholar] [CrossRef] [Green Version]
- Sapir-Koren, R.; Livshits, G. Osteocyte control of bone remodeling: Is sclerostin a key molecular coordinator of the balanced bone resorption-formation cycles? Osteoporos. Int. 2014, 25, 2685–2700. [Google Scholar] [CrossRef]
- Geoghegan, I.P.; Hoey, D.A.; McNamara, L.M. Integrins in Osteocyte Biology and Mechanotransduction. Curr. Osteoporos. Rep. 2019, 17, 195–206. [Google Scholar] [CrossRef]
- Cabahug-Zuckerman, P.; Stout, R.F., Jr.; Majeska, R.J.; Thi, M.M.; Spray, D.C.; Weinbaum, S.; Schaffler, M.B. Potential role for a specialized β3 integrin-based structure on osteocyte processes in bone mechanosensation. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2018, 36, 642–652. [Google Scholar] [CrossRef]
- Haugh, M.G.; Vaughan, T.J.; McNamara, L.M. The role of integrin αvβ3 in osteocyte mechanotransduction. J. Mech. Behav. Biomed. Mater. 2015, 42, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Park-Min, K.H. Metabolic reprogramming in osteoclasts. Semin. Immunopathol. 2019, 41, 565–572. [Google Scholar] [CrossRef]
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 2019, 568, 541–545. [Google Scholar] [CrossRef]
- Lerner, U.H. Osteoclasts in Health and Disease. Pediatr. Endocrinol. Rev. 2019, 17, 84–99. [Google Scholar] [CrossRef]
- Zhao, X.; Patil, S.; Xu, F.; Lin, X.; Qian, A. Role of Biomolecules in Osteoclasts and Their Therapeutic Potential for Osteoporosis. Biomolecules 2021, 11, 747. [Google Scholar] [CrossRef]
- Ishiyama, K.; Yashiro, T.; Nakano, N.; Kasakura, K.; Miura, R.; Hara, M.; Kawai, F.; Maeda, K.; Tamura, N.; Okumura, K.; et al. Involvement of PU.1 in NFATc1 promoter function in osteoclast development. Allergol. Int. Off. J. Jpn. Soc. Allergol. 2015, 64, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Izawa, N.; Kurotaki, D.; Nomura, S.; Fujita, T.; Omata, Y.; Yasui, T.; Hirose, J.; Matsumoto, T.; Saito, T.; Kadono, Y.; et al. Cooperation of PU.1 With IRF8 and NFATc1 Defines Chromatin Landscapes During RANKL-Induced Osteoclastogenesis. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2019, 34, 1143–1154. [Google Scholar] [CrossRef]
- Carey, H.A.; Hildreth, B.E., 3rd; Geisler, J.A.; Nickel, M.C.; Cabrera, J.; Ghosh, S.; Jiang, Y.; Yan, J.; Lee, J.; Makam, S.; et al. Enhancer variants reveal a conserved transcription factor network governed by PU.1 during osteoclast differentiation. Bone Res. 2018, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Bronisz, A.; Sharma, S.M.; Hu, R.; Godlewski, J.; Tzivion, G.; Mansky, K.C.; Ostrowski, M.C. Microphthalmia-associated transcription factor interactions with 14-3-3 modulate differentiation of committed myeloid precursors. Mol. Biol. Cell 2006, 17, 3897–3906. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Ray, N. Osteoclasts, mononuclear phagocytes, and c-Fos: New insight into osteoimmunology. Keio J. Med. 2004, 53, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.J.; Wu, Z.F.; Yu, Y.H.; Wang, L.; Cheng, L. Effects of interleukin-7/interleukin-7 receptor on RANKL-mediated osteoclast differentiation and ovariectomy-induced bone loss by regulating c-Fos/c-Jun pathway. J. Cell Physiol. 2018, 233, 7182–7194. [Google Scholar] [CrossRef]
- Fujii, T.; Murata, K.; Mun, S.H.; Bae, S.; Lee, Y.J.; Pannellini, T.; Kang, K.; Oliver, D.; Park-Min, K.H.; Ivashkiv, L.B. MEF2C regulates osteoclastogenesis and pathologic bone resorption via c-FOS. Bone Res. 2021, 9, 4. [Google Scholar] [CrossRef]
- Pang, M.; Rodríguez-Gonzalez, M.; Hernandez, M.; Recinos, C.C.; Seldeen, K.L.; Troen, B.R. AP-1 and Mitf interact with NFATc1 to stimulate cathepsin K promoter activity in osteoclast precursors. J. Cell Biochem. 2019, 120, 12382–12392. [Google Scholar] [CrossRef]
- Dai, R.; Wu, Z.; Chu, H.Y.; Lu, J.; Lyu, A.; Liu, J.; Zhang, G. Cathepsin K: The Action in and Beyond Bone. Front. Cell Dev. Biol. 2020, 8, 433. [Google Scholar] [CrossRef] [PubMed]
- Borel, O.; Gineyts, E.; Bertholon, C.; Garnero, P. Cathepsin K preferentially solubilizes matured bone matrix. Calcif. Tissue Int. 2012, 91, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, N.; Brun, J.; Rousseau, J.C.; Duong, L.T.; Ferrari, S.L. Cathepsin K Controls Cortical Bone Formation by Degrading Periostin. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Lotinun, S.; Kiviranta, R.; Matsubara, T.; Alzate, J.A.; Neff, L.; Lüth, A.; Koskivirta, I.; Kleuser, B.; Vacher, J.; Vuorio, E.; et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J. Clin. Investig. 2013, 123, 666–681. [Google Scholar] [CrossRef]
- Walia, B.; Lingenheld, E.; Duong, L.; Sanjay, A.; Drissi, H. A novel role for cathepsin K in periosteal osteoclast precursors during fracture repair. Ann. N. Y. Acad. Sci. 2018, 1415, 57–68. [Google Scholar] [CrossRef]
- Gentile, M.A.; Soung do, Y.; Horrell, C.; Samadfam, R.; Drissi, H.; Duong, L.T. Increased fracture callus mineralization and strength in cathepsin K knockout mice. Bone 2014, 66, 72–81. [Google Scholar] [CrossRef]
- Si, J.; Wang, C.; Zhang, D.; Wang, B.; Zhou, Y. Osteopontin in Bone Metabolism and Bone Diseases. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e919159. [Google Scholar] [CrossRef]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef]
- Luukkonen, J.; Hilli, M.; Nakamura, M.; Ritamo, I.; Valmu, L.; Kauppinen, K.; Tuukkanen, J.; Lehenkari, P. Osteoclasts secrete osteopontin into resorption lacunae during bone resorption. Histochem. Cell Biol. 2019, 151, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Gill, G.; Kaur, H.; Amhmed, M.; Jakhu, H. Role of osteopontin in bone remodeling and orthodontic tooth movement: A review. Prog. Orthod. 2018, 19, 18. [Google Scholar] [CrossRef]
- Ge, Q.; Ruan, C.C.; Ma, Y.; Tang, X.F.; Wu, Q.H.; Wang, J.G.; Zhu, D.L.; Gao, P.J. Osteopontin regulates macrophage activation and osteoclast formation in hypertensive patients with vascular calcification. Sci. Rep. 2017, 7, 40253. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.B.; Kim, J.H.; Kim, K.; Youn, B.U.; Ko, A.; Lee, S.Y.; Kim, N. Akt induces osteoclast differentiation through regulating the GSK3β/NFATc1 signaling cascade. J. Immunol. 2012, 188, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Chen, W.; Lu, Y.; Zhu, G.; Hao, L.; Li, Y.P. Gα13 negatively controls osteoclastogenesis through inhibition of the Akt-GSK3β-NFATc1 signalling pathway. Nat. Commun. 2017, 8, 13700. [Google Scholar] [CrossRef] [Green Version]
- Mullin, B.H.; Prince, R.L.; Dick, I.M.; Hart, D.J.; Spector, T.D.; Dudbridge, F.; Wilson, S.G. Identification of a role for the ARHGEF3 gene in postmenopausal osteoporosis. Am. J. Hum. Genet. 2008, 82, 1262–1269. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Xiao, Y.; Chen, Z.; Ma, J.; Qiu, W.; Zhang, K.; Xu, F.; Dang, K.; Qian, A. Microtubule actin crosslinking factor 1 (MACF1) knockdown inhibits RANKL-induced osteoclastogenesis via Akt/GSK3β/NFATc1 signalling pathway. Mol. Cell. Endocrinol. 2019, 494, 110494. [Google Scholar] [CrossRef]
- Jang, H.D.; Noh, J.Y.; Shin, J.H.; Lin, J.J.; Lee, S.Y. PTEN regulation by the Akt/GSK-3β axis during RANKL signaling. Bone 2013, 55, 126–131. [Google Scholar] [CrossRef]
- Almeida, M.; Laurent, M.R.; Dubois, V.; Claessens, F.; O’Brien, C.A.; Bouillon, R.; Vanderschueren, D.; Manolagas, S.C. Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol. Rev. 2017, 97, 135–187. [Google Scholar] [CrossRef]
- Khosla, S.; Oursler, M.J.; Monroe, D.G. Estrogen and the skeleton. Trends Endocrinol. Metab. 2012, 23, 576–581. [Google Scholar] [CrossRef] [Green Version]
- Cauley, J.A. Estrogen and bone health in men and women. Steroids 2015, 99, 11–15. [Google Scholar] [CrossRef]
- Streicher, C.; Heyny, A.; Andrukhova, O.; Haigl, B.; Slavic, S.; Schüler, C.; Kollmann, K.; Kantner, I.; Sexl, V.; Kleiter, M.; et al. Estrogen Regulates Bone Turnover by Targeting RANKL Expression in Bone Lining Cells. Sci. Rep. 2017, 7, 6460. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Larriera, A.I.; Palacio-Mancheno, P.E.; Gatti, V.; Fritton, J.C.; Bromage, T.G.; Cardoso, L.; Doty, S.B.; Fritton, S.P. The effects of estrogen deficiency on cortical bone microporosity and mineralization. Bone 2018, 110, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Riggs, B.L. The mechanisms of estrogen regulation of bone resorption. J. Clin. Investig. 2000, 106, 1203–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Imai, Y.; Matsumoto, T.; Sato, S.; Takeuchi, K.; Igarashi, K.; Harada, Y.; Azuma, Y.; Krust, A.; Yamamoto, Y.; et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 2007, 130, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Melville, K.M.; Kelly, N.H.; Khan, S.A.; Schimenti, J.C.; Ross, F.P.; Main, R.P.; van der Meulen, M.C. Female mice lacking estrogen receptor-alpha in osteoblasts have compromised bone mass and strength. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2014, 29, 370–379. [Google Scholar] [CrossRef]
- Nicks, K.M.; Fujita, K.; Fraser, D.; McGregor, U.; Drake, M.T.; McGee-Lawrence, M.E.; Westendorf, J.J.; Monroe, D.G.; Khosla, S. Deletion of Estrogen Receptor Beta in Osteoprogenitor Cells Increases Trabecular but Not Cortical Bone Mass in Female Mice. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2016, 31, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Windahl, S.H.; Börjesson, A.E.; Farman, H.H.; Engdahl, C.; Movérare-Skrtic, S.; Sjögren, K.; Lagerquist, M.K.; Kindblom, J.M.; Koskela, A.; Tuukkanen, J.; et al. Estrogen receptor-α in osteocytes is important for trabecular bone formation in male mice. Proc. Natl. Acad. Sci. USA 2013, 110, 2294–2299. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, S.; Inoue, K.; Igarashi, K.; Sugizaki, H.; Shirode-Fukuda, Y.; Inoue, E.; Yu, T.; Takeuchi, J.K.; Kanno, J.; Bonewald, L.F.; et al. Estrogen receptor α in osteocytes regulates trabecular bone formation in female mice. Bone 2014, 60, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Geoghegan, I.P.; Hoey, D.A.; McNamara, L.M. Estrogen deficiency impairs integrin αvβ3-mediated mechanosensation by osteocytes and alters osteoclastogenic paracrine signalling. Sci. Rep. 2019, 9, 4654. [Google Scholar] [CrossRef] [Green Version]
- Yeh, S.; Tsai, M.Y.; Xu, Q.; Mu, X.M.; Lardy, H.; Huang, K.E.; Lin, H.; Yeh, S.D.; Altuwaijri, S.; Zhou, X.; et al. Generation and characterization of androgen receptor knockout (ARKO) mice: An in vivo model for the study of androgen functions in selective tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 13498–13503. [Google Scholar] [CrossRef] [Green Version]
- Kawano, H.; Sato, T.; Yamada, T.; Matsumoto, T.; Sekine, K.; Watanabe, T.; Nakamura, T.; Fukuda, T.; Yoshimura, K.; Yoshizawa, T.; et al. Suppressive function of androgen receptor in bone resorption. Proc. Natl. Acad. Sci. USA 2003, 100, 9416–9421. [Google Scholar] [CrossRef] [Green Version]
- Sinnesael, M.; Jardi, F.; Deboel, L.; Laurent, M.R.; Dubois, V.; Zajac, J.D.; Davey, R.A.; Carmeliet, G.; Claessens, F.; Vanderschueren, D. The androgen receptor has no direct antiresorptive actions in mouse osteoclasts. Mol. Cell. Endocrinol. 2015, 411, 198–206. [Google Scholar] [CrossRef]
- Ucer, S.; Iyer, S.; Bartell, S.M.; Martin-Millan, M.; Han, L.; Kim, H.N.; Weinstein, R.S.; Jilka, R.L.; O’Brien, C.A.; Almeida, M.; et al. The Effects of Androgens on Murine Cortical Bone Do Not Require AR or ERα Signaling in Osteoblasts and Osteoclasts. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2015, 30, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Sinnesael, M.; Claessens, F.; Laurent, M.; Dubois, V.; Boonen, S.; Deboel, L.; Vanderschueren, D. Androgen receptor (AR) in osteocytes is important for the maintenance of male skeletal integrity: Evidence from targeted AR disruption in mouse osteocytes. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2012, 27, 2535–2543. [Google Scholar] [CrossRef]
- Yang, T.L.; Shen, H.; Liu, A.; Dong, S.S.; Zhang, L.; Deng, F.Y.; Zhao, Q.; Deng, H.W. A road map for understanding molecular and genetic determinants of osteoporosis. Nature reviews. Endocrinology 2020, 16, 91–103. [Google Scholar] [CrossRef]
- Noh, J.Y.; Yang, Y.; Jung, H. Molecular Mechanisms and Emerging Therapeutics for Osteoporosis. Int. J. Mol. Sci. 2020, 21, 7623. [Google Scholar] [CrossRef]
- Macías, I.; Alcorta-Sevillano, N.; Rodríguez, C.I.; Infante, A. Osteoporosis and the Potential of Cell-Based Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 1653. [Google Scholar] [CrossRef] [Green Version]
- Cremers, S.; Drake, M.T.; Ebetino, F.H.; Bilezikian, J.P.; Russell, R.G.G. Pharmacology of bisphosphonates. Br. J. Clin. Pharmacol. 2019, 85, 1052–1062. [Google Scholar] [CrossRef]
- Center, J.R.; Lyles, K.W.; Bliuc, D. Bisphosphonates and lifespan. Bone 2020, 141, 115566. [Google Scholar] [CrossRef]
- Rollason, V.; Laverrière, A.; MacDonald, L.C.; Walsh, T.; Tramèr, M.R.; Vogt-Ferrier, N.B. Interventions for treating bisphosphonate-related osteonecrosis of the jaw (BRONJ). Cochrane Database Syst. Rev. 2016, 2, Cd008455. [Google Scholar] [CrossRef]
- Levin, V.A.; Jiang, X.; Kagan, R. Estrogen therapy for osteoporosis in the modern era. Osteoporos. Int. 2018, 29, 1049–1055. [Google Scholar] [CrossRef]
- Martinkovich, S.; Shah, D.; Planey, S.L.; Arnott, J.A. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 9, 1437–1452. [Google Scholar] [CrossRef] [Green Version]
- An, K.C. Selective Estrogen Receptor Modulators. Asian Spine J. 2016, 10, 787–791. [Google Scholar] [CrossRef] [Green Version]
- Cummings, S.R.; San Martin, J.; McClung, M.R.; Siris, E.S.; Eastell, R.; Reid, I.R.; Delmas, P.; Zoog, H.B.; Austin, M.; Wang, A.; et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 2009, 361, 756–765. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D. Denosumab: A Review in Postmenopausal Osteoporosis. Drugs Aging 2018, 35, 163–173. [Google Scholar] [CrossRef]
- Stone, J.A.; McCrea, J.B.; Witter, R.; Zajic, S.; Stoch, S.A. Clinical and translational pharmacology of the cathepsin K inhibitor odanacatib studied for osteoporosis. Br. J. Clin. Pharmacol. 2019, 85, 1072–1083. [Google Scholar] [CrossRef]
- McClung, M.R.; O’Donoghue, M.L.; Papapoulos, S.E.; Bone, H.; Langdahl, B.; Saag, K.G.; Reid, I.R.; Kiel, D.P.; Cavallari, I.; Bonaca, M.P.; et al. Odanacatib for the treatment of postmenopausal osteoporosis: Results of the LOFT multicentre, randomised, double-blind, placebo-controlled trial and LOFT Extension study. Lancet Diabetes Endocrinol. 2019, 7, 899–911. [Google Scholar] [CrossRef]
- O’Donnell, S.; Cranney, A.; Wells, G.A.; Adachi, J.D.; Reginster, J.Y. Strontium ranelate for preventing and treating postmenopausal osteoporosis. Cochrane Database Syst. Rev. 2006, Cd005326. [Google Scholar] [CrossRef]
- Tabatabaei-Malazy, O.; Salari, P.; Khashayar, P.; Larijani, B. New horizons in treatment of osteoporosis. DARU J. Pharm. Sci. 2017, 25, 2. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, J.; Khiavi, R.K.; Ghavimi, M.A.; Mortazavi, A.; Hagh, E.J.; Ahmadpour, F. Calcitonin as an analgesic agent: Review of mechanisms of action and clinical applications. Braz. J. Anesthesiol. 2019, 69, 594–604. [Google Scholar] [CrossRef]
- Xie, J.; Guo, J.; Kanwal, Z.; Wu, M.; Lv, X.; Ibrahim, N.A.; Li, P.; Buabeid, M.A.; Arafa, E.A.; Sun, Q. Calcitonin and Bone Physiology: In Vitro, In Vivo, and Clinical Investigations. Int. J. Endocrinol. 2020, 2020, 3236828. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, N.; Ding, C.; Zhou, X.; Zhou, J. Postmenopausal osteoporosis treated with acupoint injection of salmon calcitonin:a randomized controlled trial. Zhongguo Zhen Jiu Chin. Acupunct. Moxibustion 2016, 36, 705–708. [Google Scholar] [CrossRef]
- Goltzman, D. Physiology of Parathyroid Hormone. Endocrinol. Metab. Clin. N. Am. 2018, 47, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Miyamoto, Y.; Yamamoto, T.; Amizuka, N. Anabolic action of teriparatide to osteoporotic patients. Nihon yakurigaku zasshi. Folia Pharmacol. Jpn. 2019, 153, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Osagie-Clouard, L.; Sanghani, A.; Coathup, M.; Briggs, T.; Bostrom, M.; Blunn, G. Parathyroid hormone 1-34 and skeletal anabolic action: The use of parathyroid hormone in bone formation. Bone Jt. Res. 2017, 6, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.L.; Demiralp, B.; Schneider, A.; Koh, A.J.; Silve, C.; Wang, C.Y.; McCauley, L.K. Parathyroid hormone and parathyroid hormone-related protein exert both pro- and anti-apoptotic effects in mesenchymal cells. J. Biol. Chem. 2002, 277, 19374–19381. [Google Scholar] [CrossRef] [Green Version]
- Anagnostis, P.; Gkekas, N.K.; Potoupnis, M.; Kenanidis, E.; Tsiridis, E.; Goulis, D.G. New therapeutic targets for osteoporosis. Maturitas 2019, 120, 1–6. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, J.J.; Mitlak, B.H.; Wang, O.; Genant, H.K.; Eriksen, E.F. Recombinant human parathyroid hormone (1-34) [teriparatide] improves both cortical and cancellous bone structure. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2003, 18, 1932–1941. [Google Scholar] [CrossRef]
- Paik, J.; Scott, L.J. Romosozumab: A Review in Postmenopausal Osteoporosis. Drugs Aging 2020, 37, 845–855. [Google Scholar] [CrossRef]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or Alendronate for Fracture Prevention in Women with Osteoporosis. N. Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Patil, S.; Qian, A. The Role of MicroRNAs in Bone Metabolism and Disease. Int. J. Mol. Sci. 2020, 21, 6081. [Google Scholar] [CrossRef]
- Patil, S.; Dang, K.; Zhao, X.; Gao, Y.; Qian, A. Role of LncRNAs and CircRNAs in Bone Metabolism and Osteoporosis. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Patil, S.; Gao, Y.G.; Lin, X.; Li, Y.; Dang, K.; Tian, Y.; Zhang, W.J.; Jiang, S.F.; Qadir, A.; Qian, A.R. The Development of Functional Non-Viral Vectors for Gene Delivery. Int. J. Mol. Sci. 2019, 20, 5491. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Cellular and molecular interplay during bone remodeling. M-CSF, RANKL produced by osteoblasts positively regulates while WNT5A/16 and OPG negatively regulates osteoclast function. SOST produced by osteocytes embedded in bone matrix positively and negatively regulates the osteoclast and osteoblast functions, respectively.
Figure 1.
Cellular and molecular interplay during bone remodeling. M-CSF, RANKL produced by osteoblasts positively regulates while WNT5A/16 and OPG negatively regulates osteoclast function. SOST produced by osteocytes embedded in bone matrix positively and negatively regulates the osteoclast and osteoblast functions, respectively.

Figure 2.
Molecules responsible for osteoblast differentiation. The commitment of mesenchymal stem cells to osteogenic progenitor cells is influenced by Runx2, FGF, Wnt, as well as MITF and SATB2, while Osx, Runx2, NFATc1 promote osteogenic progenitor cell proliferation to preosteoblasts, which is then muture into mature and functional osteoblasts by the action of Runx2, FGF, Wnt, NFATc1, MITF as well as ATF4.
Figure 2.
Molecules responsible for osteoblast differentiation. The commitment of mesenchymal stem cells to osteogenic progenitor cells is influenced by Runx2, FGF, Wnt, as well as MITF and SATB2, while Osx, Runx2, NFATc1 promote osteogenic progenitor cell proliferation to preosteoblasts, which is then muture into mature and functional osteoblasts by the action of Runx2, FGF, Wnt, NFATc1, MITF as well as ATF4.

Figure 3.
Molecules involved in the process of osteoclast differentiation. M-CSF/c-Fms, MITF, and PU.1 promote differentiation of myeloid precursors into osteoclasts precursor which fuse together to form immature osteoclasts. The maturation of immature osteoclasts to mature osteoclasts is facilitated by integrin-β3 and CatK.
Figure 3.
Molecules involved in the process of osteoclast differentiation. M-CSF/c-Fms, MITF, and PU.1 promote differentiation of myeloid precursors into osteoclasts precursor which fuse together to form immature osteoclasts. The maturation of immature osteoclasts to mature osteoclasts is facilitated by integrin-β3 and CatK.

Figure 4.
The mechanism of action of osteoporosis therapeutics. The antiresorptive drugs such as bisphosphonate (BP), estrogen, anti-RANKLs (denosumab) and calcitonin; anabolic drugs such as PTH and anti-sclerostins (romosozumab) affecting different bone cells. + mean positive effect; − mean negative effect [10].
Figure 4.
The mechanism of action of osteoporosis therapeutics. The antiresorptive drugs such as bisphosphonate (BP), estrogen, anti-RANKLs (denosumab) and calcitonin; anabolic drugs such as PTH and anti-sclerostins (romosozumab) affecting different bone cells. + mean positive effect; − mean negative effect [10].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gao, Y.; Patil, S.; Jia, J. The Development of Molecular Biology of Osteoporosis. Int. J. Mol. Sci. 2021, 22, 8182. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158182
AMA Style
Gao Y, Patil S, Jia J. The Development of Molecular Biology of Osteoporosis. International Journal of Molecular Sciences. 2021; 22(15):8182. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158182
Chicago/Turabian StyleGao, Yongguang, Suryaji Patil, and Jingxian Jia. 2021. "The Development of Molecular Biology of Osteoporosis" International Journal of Molecular Sciences 22, no. 15: 8182. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158182
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.