
The Gut Microbiota-Derived Immune Response in Chronic Liver Disease

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Chronic Liver Disease
3. Gut–Liver Axis and Immune Response
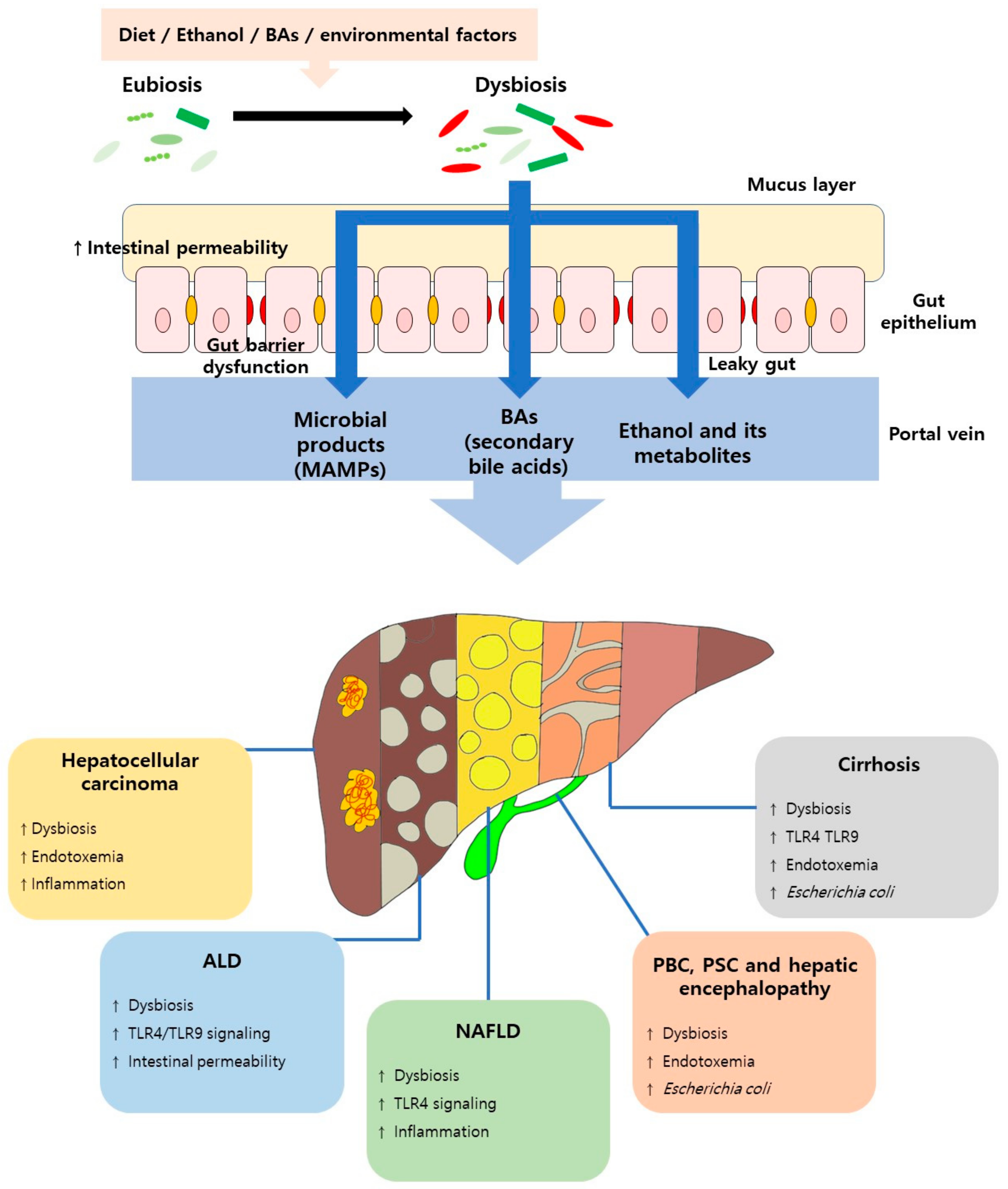
4. Gut Barrier Dysfunction
5. Immune Response Associated with Liver Disease
5.1. Nonalcoholic Fatty Liver Disease and the Immune Response
5.2. Alcoholic Liver Disease and the Immune Response
5.3. Liver Cirrhosis and the Immune Response
5.4. Hepatocellular Carcinoma and the Immune Response
5.5. Other Liver Disease and the Immune Response

{kind=link}
| Species | Study Type | Exposure | Main Results | Ref. |
|---|---|---|---|---|
| HCC | ||||
| Helicobacter-free C3H/HeN female mice | Animal | AFB1 and/or H. hepaticus | Intestinal colonization by H. hepaticus promoted aflatoxin and HCV transgene-induced HCC. H. hepaticus activated the nuclear factor-kappaB regulatory signaling pathway. | [130] |
| C3H/HeOuJ, C3H/HeJ, TLR2-deficient mice, TLR4-deficient mice, TNFR1-/IL-1R1-double deficient, and C57Bl/6 mice | Animal | Intraperitoneal injection of DEN or CCl4 | Activation of gut microbiota and TLR4 contributes to the development of cancer in chronically damaged livers. Intestinal microbiota and TLR4 contribute to promotion of HCC, proliferation of cancer, expression of hepatomitogen epiregulin and prevention of apoptosis. In the late stages of liver cancer, limited enteric sterilization reduced hepatocellular carcinoma. | [132] |
| Controls (n = 15), HCC patients (n = 15) | Human | The presence of HCC was associated with an increased number of E. coli in the patient’s stool. Intestinal E. coli overgrowth contributes to the development of liver cancer. | [128] | |
| Controls (n = 16), patients with primary liver carcinoma (n = 20) | Human | Helicobacter spp. DNA was found in liver cancer samples from patients with primary liver carcinoma. By bacterial translocation, H. pylori may be present in the liver of liver carcinoma patients and may be related to hepatic carcinogenesis. | [129] | |
| Hepatic Encephalopathy | ||||
| Male Sprague-Dawley rat | Animal | Bile duct ligation or Sham operation or High protein/ammoniagenic diet injected with LPS (0.5 mg/kg) | After LPS injection, only the bile duct ligation group progressed to the pre-coma stage. TNF-α and IL-6 levels were significantly increased in LPS-treated animals. LPS injection in a cirrhosis model induces coma due to synergistic effects of hyperammonemia and inflammatory response. It also exacerbates cytotoxic edema. | [135] |
| Cirrhotic patients (n = 10) | Human | Oral administration of an amino acid solution mimicking hemoglobin composition | Hyperammonemia was similar before and after resolution of inflammation in patients. There was a significant decrease in the white blood cell count, nitrate/nitrite, IL-6, IL-1β, and TNF-α by infection treatment. Induced hyperammonemia significantly worsened neuropsychological test scores. | [136] |
| PSC and PBC | ||||
| Germ-free C57BL/6 male mice | Animal | PSC patients fecal sample inoculation | T helper 17 cell responses were shown in the livers of Gnotobiotic mice inoculated with PSC patient-derived microbiota and increased susceptibility to hepatobiliary injury. PSC-associated Klebsiella pneumoniae has an epithelial damaging effect and contributes to bacterial translocation and initiation of hepatic inflammatory responses. | [140] |
| Male Mdr2−/−, Mdr2−/− crossed with hepatocyte-specific deletion of caspase-8 (Mdr2−/−/ casp8∆hepa) and wild-type (Wt) control mice | Animal | Administration of pan-caspase inhibitor (iDn-7314) | Abnormalities in the gut microbiome in Mdr2−/− mice caused intestinal barrier dysfunction and increased bacterial translocation, which amplifies the hepatic nlrP3-mediated innate immune response. Transfer of the Mdr2−/− microbiota to healthy wildtype control mice induced significant liver damage in recipient mice. MDr2-associated cholestasis causes intestinal bacterial imbalance. Translocation of endotoxin into the portal vein and subsequent nlrP3 inflammasome activation contributes to higher liver damage. | [141] |
| C57BL/6 and A/J mice onto the NOD background | Animal | Infection by intravenous injection of N. aromaticivorans | N. aromaticivorans infection induced liver inflammation and PBC. | [146] |
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALD | Alcohol-related liver disease |
| ALT | Alanine transaminase |
| ASC | Apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain |
| CCL2 CC | chemokine ligand 2 |
| CCl4 | Carbon tetrachloride |
| ECE1 | Extent of cell elongation 1 |
| GALT | Gut-associated lymphoid tissue |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HV | Chronic viral hepatitis |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| MCDD | Methionine/choline-deficient diet |
| MyD88 | Myeloid differentiation factor 88 |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NK | Natural Killer |
| NLRP | Nucleotide- binding oligomerization-domain protein-like receptors protein |
| Reg3b | Regenerating islet-derived protein 3-β |
| Reg3g | Regenerating islet-derived protein 3-γ |
| ROS | Reactive oxygen species |
| TAA | Thioacetamide |
| TGF-β | Transforming growth factor-β |
| TLRs | Toll-like receptors |
| TNF-α | Tumor necrosis factor-α |
References
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Quinn, R.A.; Debelius, J.; Xu, Z.Z.; Morton, J.; Garg, N.; Jansson, J.K.; Dorrestein, P.C.; Knight, R. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 2016, 535, 94–103. [Google Scholar] [CrossRef]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Gou, Y.K.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazmanian, S.K.; Cui, H.L.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uematsu, S.; Fujimoto, K.; Jang, M.H.; Yang, B.-G.; Jung, Y.-J.; Nishiyama, M.; Sato, S.; Tsujimura, T.; Yamamoto, M.; Yokota, Y.; et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 2008, 9, 769–776. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Stärkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simreń, M.; Barbara, G.; Flint, H.J.; Spiegel, B.M.R.; Spiller, R.C.; Vanner, S.; Verdu, E.F.; Whorwell, P.J.; Zoetendal, E.G. Intestinal microbiota in functional bowel disorders: A Rome foundation report. Gut 2013, 62, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Flavell, R.A. The intestinal microbiota in chronic liver disease. Adv. Immunol. 2013, 117, 73–97. [Google Scholar] [CrossRef]
- Cani, P.; Delzenne, N. The Role of the Gut Microbiota in Energy Metabolism and Metabolic Disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Nagalli, S. Chronic Liver Disease; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Heidelbaugh, J.J.; Bruderly, M. Cirrhosis and chronic liver failure: Part I Diagnosis and evaluation. Am. Fam. Physician 2006, 74, 756–762. [Google Scholar]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gressner, A.M.; Weiskirchen, R.; Breitkopf, K.; Dooley, S. Roles of TGF-beta in hepatic fibrosis. Front. Biosci. 2002, 7, d793–d807. [Google Scholar] [CrossRef] [Green Version]
- Lo, R.C.; Kim, H. Histopathological evaluation of liver fibrosis and cirrhosis regression. Clin. Mol. Hepatol. 2017, 23, 302–307. [Google Scholar] [CrossRef] [Green Version]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Jung, Y.K.; Yim, H.J. Reversal of liver cirrhosis: Current evidence and expectations. Korean J. Intern. Med. 2017, 32, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Interactions 2007, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neish, A.S. Mucosal immunity and the microbiome. Ann. Am. Thorac. Soc. 2014, 11, S28–S32. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lu, D.; Zhuo, J.; Lin, Z.; Yang, M.; Xu, X. The gut-liver axis in immune remodeling: New insight into liver diseases. Int. J. Biol. Sci. 2020, 16, 2357–2366. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Adams, D.H. Gut-liver immunity. J. Hepatol. 2016, 64, 1187–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, M.; Gentile, M.; Yeiser, J.R.; Walland, A.C.; Bornstein, V.U.; Chen, K.; He, B.; Cassis, L.; Bigas, A.; Cols, M.; et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 2013, 342, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- Su, G.L.; Klein, R.D.; Aminlari, A.; Zhang, H.Y.; Steinstraesser, L.; Alarcon, W.H.; Remick, D.G.; Wang, S.C. Kupffer cell activation by lipopolysaccharide in rats: Role for lipopolysaccharide binding protein and Toll-like receptor 4. Hepatology 2000, 31, 932–936. [Google Scholar] [CrossRef]
- Kaur, D.; Patiyal, S.; Sharma, N.; Usmani, S.S.; Raghava, G.P.S. PRRDB 2.0: A comprehensive database of pattern-recognition receptors and their ligands. Database 2019, 2019, baz076. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Jeong, W.I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729–736. [Google Scholar] [CrossRef]
- Li, F.; Hao, X.; Chen, Y.; Bai, L.; Gao, X.; Lian, Z.; Wei, H.; Sun, R.; Tian, Z. The microbiota maintain homeostasis of liver-resident γδ T-17 cells in a lipid antigen/CD1d-dependent manner. Nat. Commun. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Rodrigue-Gervais, I.G.; Breton, V.; Colpitts, S.L.; Beauchemin, N.; et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmer, M.L.; Slack, E.; de Gottardi, A.; Lawson, M.A.E.; Hapfelmeier, S.; Miele, L.; Grieco, A.; Van Vlierberghe, H.; Fahrner, R.; Patuto, N.; et al. The Liver May Act as a Firewall Mediating Mutualism Between the Host and Its Gut Commensal Microbiota. Sci. Transl. Med. 2014, 6, 237ra66. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Yu, L.X.; Yan, H.X.; Liu, Q.; Yang, W.; Wu, H.P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.Q.; Zou, S.S.; et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef]
- Saltzman, E.T.; Palacios, T.; Thomsen, M.; Vitetta, L. Intestinal microbiome shifts, dysbiosis, inflammation, and non-alcoholic fatty liver disease. Front. Microbiol. 2018, 9, 61. [Google Scholar] [CrossRef]
- Flannigan, K.L.; Denning, T.L. Segmented filamentous bacteria-induced immune responses: A balancing act between host protection and autoimmunity. Immunology 2018, 154, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Parséus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Stahlman, M.; Greiner, T.U.; Perkins, R.; Bäckhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Birchenough, G.M.H.; Ståhlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Bäckhed, F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 2018, 23, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luck, H.; Tsai, S.; Chung, J.; Clemente-Casares, X.; Ghazarian, M.; Revelo, X.S.; Lei, H.; Luk, C.T.; Shi, S.Y.; Surendra, A.; et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab. 2015, 21, 527–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Wood, S.; Pithadia, R.; Rehman, T.; Zhang, L.; Plichta, J.; Radek, K.A.; Forsyth, C.; Keshavarzian, A.; Shafikhani, S.H. Chronic Alcohol Exposure Renders Epithelial Cells Vulnerable to Bacterial Infection. PLoS ONE 2013, 8, e54646. [Google Scholar] [CrossRef]
- Chen, P.; Stärkel, P.; Turner, J.R.; Ho, S.B.; Schnabl, B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 2015, 61, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, C.; Kugler, V.; Bode, J.C. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 1987, 4, 8–14. [Google Scholar] [CrossRef]
- Hartmann, P.; Chen, P.; Wang, H.J.; Wang, L.; Mccole, D.F.; Brandl, K.; Stärkel, P.; Belzer, C.; Hellerbrand, C.; Tsukamoto, H.; et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology 2013, 58, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Kalaitzakis, E. Gastrointestinal dysfunction in liver cirrhosis. World J. Gastroenterol. 2014, 20, 14686–14695. [Google Scholar] [CrossRef]
- Choi, Y.; Jeon, W.K.; Hwang, S.J.; Kim, B.I.; Sohn, C.I.; Park, D.I.; Cho, Y.K.; Kim, H.J.; Park, J.H. The role of the gut barrier function in the pathophysiology of viral liver cirrhosis. Hepatogastroenterology 2011, 58, 1244–1247. [Google Scholar] [CrossRef]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Tessari, R.; Zenari, L.; Day, C.; Arcaro, G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007, 30, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [Green Version]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z. The gut-liver axis: A busy two-way street. Hepatology 2012, 55, 1647–1649. [Google Scholar] [CrossRef]
- Li, D.-Y.; Yang, M.; Edwards, S.; Ye, S.-Q. Nonalcoholic Fatty Liver Disease. J. Parenter. Enter. Nutr. 2013, 37, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Paolella, G.; Mandato, C.; Pierri, L.; Poeta, M.; Di Stasi, M.; Vajro, P. Gut-liver axis and probiotics: Their role in non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 15518–15531. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Bode, J.C.; Bode, C.; Brenner, D.A.; Choudhry, M.A.; Hamilton, F.; Kang, Y.J.; Keshavarzian, A.; Rao, R.; Sartor, R.B.; et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: Summary of a symposium. Alcohol 2008, 42, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Xu, C.; Yu, C.; Li, Y. Role of NLRP3 Inflammasome in the Progression of NAFLD to NASH. Can. J. Gastroenterol. Hepatol. 2016, 2016, 6489012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8836–8847. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Shen, X.; O’Connell, R.; Gao, F.; Lassman, C.; Busuttil, R.W.; Cheng, G.; Kupiec-Weglinski, J.W. Cutting Edge: TLR4 Activation Mediates Liver Ischemia/Reperfusion Inflammatory Response via IFN Regulatory Factor 3-Dependent MyD88-Independent Pathway. J. Immunol. 2004, 173, 7115–7119. [Google Scholar] [CrossRef] [Green Version]
- Abu-Shanab, A.; Quigley, E.M.M. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Han, J.H.; Dzierlenga, A.L.; Lu, Z.; Billheimer, D.D.; Torabzadeh, E.; Lake, A.D.; Li, H.; Novak, P.; Shipkova, P.; Aranibar, N.; et al. Metabolomic profiling distinction of human nonalcoholic fatty liver disease progression from a common rat model. Obesity 2017, 25, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Gogiashvili, M.; Edlund, K.; Gianmoena, K.; Marchan, R.; Brik, A.; Andersson, J.T.; Lambert, J.; Madjar, K.; Hellwig, B.; Rahnenführer, J.; et al. Metabolic profiling of ob/ob mouse fatty liver using HR-MAS 1H-NMR combined with gene expression analysis reveals alterations in betaine metabolism and the transsulfuration pathway. Anal. Bioanal. Chem. 2017, 409, 1591–1606. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; Rooijen, N.; van Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; Tsutsui, H.; Nakano, H.; Tsuji, N.M.; Hoshino, K.; Adachi, O.; Adachi, K.; Futatsugi, S.; Kuida, K.; Takeuchi, O.; et al. Lipopolysaccharide-Induced IL-18 Secretion from Murine Kupffer Cells Independently of Myeloid Differentiation Factor 88 That Is Critically Involved in Induction of Production of IL-12 and IL-1β. J. Immunol. 2001, 166, 2651–2657. [Google Scholar] [CrossRef] [Green Version]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, Y.H.; Jung, Y.S.; Kim, K.S.; Kim, D.K.; Na, S.Y.; Lee, J.M.; Lee, C.H.; Choi, H.S. Hepatocyte toll-like receptor 4 mediates lipopolysaccharide-induced hepcidin expression. Exp. Mol. Med. 2017, 49, e408–e409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo, J.; Cayón, A.; Fernández-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor α and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Kaden-Volynets, V.; Basic, M.; Neumann, U.; Pretz, D.; Rings, A.; Bleich, A.; Bischoff, S.C. Lack of liver steatosis in germ-free mice following hypercaloric diets. Eur. J. Nutr. 2019, 58, 1933–1945. [Google Scholar] [CrossRef]
- Wang, R.; Li, H.; Yang, X.; Xue, X.; Deng, L.; Shen, J.; Zhang, M.; Zhao, L.; Zhang, C. Genetically obese human gut microbiota induces liver steatosis in germ-free mice fed on normal diet. Front. Microbiol. 2018, 9, 1602. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, W.W.; Burt, A.D. Alcoholic liver disease. Semin. Diagn. Pathol. 2006, 23, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Gustot, T.; Lemmers, A.; Moreno, C.; Nagy, N.; Quertinmont, E.; Nicaise, C.; Franchimont, D.; Louis, H.; Devière, J.; Le Moine, O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology 2006, 43, 989–1000. [Google Scholar] [CrossRef]
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, N.; Wang, L.; Mukhopadhyay, P.; Park, O.; Jeong, W.I.; Lafdil, F.; Osei-Hyiaman, D.; Moh, A.; Fu, X.Y.; Pacher, P.; et al. Cell Type-Dependent Pro- and Anti-Inflammatory Role of Signal Transducer and Activator of Transcription 3 in Alcoholic Liver Injury. Gastroenterology 2008, 134, 1148–1158. [Google Scholar] [CrossRef] [Green Version]
- Minagawa, M.; Deng, Q.; Liu, Z.X.; Tsukamoto, H.; Dennert, G. Activated Natural Killer T Cells Induce Liver Injury by Fas and Tumor Necrosis Factor-α during Alcohol Consumption. Gastroenterology 2004, 126, 1387–1399. [Google Scholar] [CrossRef]
- Wang, R.; Tang, R.; Li, B.; Ma, X.; Schnabl, B.; Tilg, H. Gut microbiome, liver immunology, and liver diseases. Cell. Mol. Immunol. 2021, 18, 4–17. [Google Scholar] [CrossRef]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef]
- Gao, B.; Seki, E.; Brenner, D.A.; Friedman, S.; Cohen, J.I.; Nagy, L.; Szabo, G.; Zakhari, S. Innate immunity in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, 516–525. [Google Scholar] [CrossRef]
- Bluemel, S.; Williams, B.; Knight, R.; Schnabl, B. Precision medicine in alcoholic and nonalcoholic fatty liver disease via modulating the gut microbiota. Am. J. Physiol. -Gastrointest. Liver Physiol. 2016, 311, G1018–G1036. [Google Scholar] [CrossRef]
- Brenner, D.A.; Paik, Y.-H.; Schnabl, B. Role of Gut Microbiota in Liver Disease. J. Clin. Gastroenterol. 2015, 49, S25–S27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandl, K.; Hartmann, P.; Jih, L.J.; Pizzo, D.P.; Ventura-cots, M.; Coulter, S.; Liddle, C.; Ling, L.; Stephen, J.; Depaoli, A.M.; et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. Dysregulation 2018, 69, 396–405. [Google Scholar] [CrossRef]
- López-Lázaro, M. A local mechanism by which alcohol consumption causes cancer. Oral Oncol. 2016, 62, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, K.K.; Shukla, P.K.; Mir, H.; Manda, B.; Gangwar, R.; Yadav, N.; McMullen, M.; Nagy, L.E.; Rao, R.K. Glutamine supplementation attenuates ethanol-induced disruption of apical junctional complexes in colonic epithelium and ameliorates gut barrier dysfunction and fatty liver in mice. J. Nutr. Biochem. 2016, 27, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Park, B.; Lee, H.R.; Lee, Y.J. Alcoholic liver disease: Focus on prodromal gut health. J. Dig. Dis. 2016, 17, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, V.; Moreau, R.; Kamath, P.S.; Jalan, R.; Ginès, P.; Nevens, F.; Fernández, J.; To, U.; García-Tsao, G.; Schnabl, B. Acute-on-chronic liver failure in cirrhosis. Nat. Rev. Dis. Prim. 2016, 2, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hendrikx, T.; Schnabl, B. Antimicrobial proteins: Intestinal guards to protect against liver disease. J. Gastroenterol. 2019, 54, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Moore, L.E.; Bradford, B.U.; Gao, W.; Thurman, R.G. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995, 108, 218–224. [Google Scholar] [CrossRef]
- Fujimoto, M.; Uemura, M.; Nakatani, Y.; Tsujita, S.; Hoppo, K.; Tamagawa, T.; Kitano, H.; Kikukawa, M.; Ann, T.; Ishii, Y.; et al. Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: Relation to severity of liver disturbance. Alcohol. Clin. Exp. Res. 2000, 24, 48S–54S. [Google Scholar] [CrossRef]
- Schäfer, C.; Parlesak, A.; Schütt, C.; Bode, J.C.; Bode, C. Concentrations of lipopolysaccharide-binding protein, bactericidal/permeability-increasing protein, soluble CD14 and plasma lipids in relation to endotoxaemia in patients with alcoholic liver disease. Alcohol Alcohol. 2002, 37, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Szabo, G.; Dolganiuc, A.; Mandrekar, P. Pattern recognition receptors: A contemporary view on liver diseases. Hepatology 2006, 44, 287–298. [Google Scholar] [CrossRef]
- Quiroz, S.C.; Bucio, L.; Souza, V.; Hernández, E.; González, E.; Gómez-Quiroz, L.; Kershenobich, D.; Vargas-Vorackova, F.; Gutiérrez-Ruiz, M.C. Effect of endotoxin pretreatment on hepatic stellate cell response to ethanol and acetaldehyde. J. Gastroenterol. Hepatol. 2001, 16, 1267–1273. [Google Scholar] [CrossRef]
- Duryee, M.J.; Klassen, L.W.; Freeman, T.L.; Willis, M.S.; Tuma, D.J.; Thiele, G.M. Lipopolysaccharide is a cofactor for malondialdehyde-acetaldehyde adduct-mediated cytokine/chemokine release by rat sinusoidal liver endothelial and Kupffer cells. Alcohol. Clin. Exp. Res. 2004, 28, 1931–1938. [Google Scholar] [CrossRef]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef]
- Machida, K.; Tsukamoto, H.; Mkrtchyan, H.; Duan, L.; Dynnyk, A.; Liu, H.M.; Asahina, K.; Govindarajan, S.; Ray, R.; James, J.H.; et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc. Natl. Acad. Sci. USA 2009, 106, 1548–1553. [Google Scholar] [CrossRef] [Green Version]
- Von Montfort, C.; Beier, J.I.; Guo, L.; Kaiser, J.P.; Arteel, G.E. Contribution of the sympathetic hormone epinephrine to the sensitizing effect of ethanol on LPS-induced liver damage in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, 1227–1234. [Google Scholar] [CrossRef] [Green Version]
- Thakur, V. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-α production. J. Leukoc. Biol. 2006, 79, 1348–1356. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.B.; Barve, S.; Joshi-Barve, S.; McClain, C. Increased monocyte nuclear factor-κB activation and tumor necrosis factor production in alcoholic hepatitis. J. Lab. Clin. Med. 2000, 135, 387–395. [Google Scholar] [CrossRef]
- Chu, H.; Duan, Y.; Lang, S.; Jiang, L.; Wang, Y.; Llorente, C.; Liu, J.; Mogavero, S.; Bosques-Padilla, F.; Abraldes, J.G.; et al. The Candida albicans exotoxin candidalysin promotes alcohol-associated liver disease. J. Hepatol. 2020, 72, 391–400. [Google Scholar] [CrossRef]
- Canesso, M.C.C.; Lacerda, N.L.; Ferreira, C.M.; Gonçalves, J.L.; Almeida, D.; Gamba, C.; Cassali, G.; Pedroso, S.H.; Moreira, C.; Martins, F.S.; et al. Comparing the effects of acute alcohol consumption in germ-free and conventional mice: The role of the gut microbiota. BMC Microbiol. 2014, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.C.; Eckmann, L.; Schnabl, B. Microbiota Protects Mice against Acute Alcohol-Induced Liver Injury. Alcohol. Clin. Exp. Res. 2015, 39, 2313–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.; Arendt, B.M.; Bhat, V.; Renner, E.L.; Humar, A.; Allard, J.P. Implication of the intestinal microbiome in complications of cirrhosis. World J. Hepatol. 2016, 8, 1128–1136. [Google Scholar] [CrossRef]
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut–liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Albhaisi, S.A.M.; Bajaj, J.S.; Sanyal, A.J. Role of gut microbiota in liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G84–G98. [Google Scholar] [CrossRef] [Green Version]
- Fukui, H. Role of Gut Dysbiosis in Liver Diseases: What Have We Learned So Far? Diseases 2019, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Rai, R.; Saraswat, V.A.; Dhiman, R.K. Gut Microbiota: Its Role in Hepatic Encephalopathy. J. Clin. Exp. Hepatol. 2015, 5, S29–S36. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.S.; Lee, F.Y.; Lee, S.D.; Te Tsai, Y.; Lin, H.C.; Rei-Hwa, L.; Wan-Ching, H.; Cheng-Chun, H.; Sun-Sang, W.; Kwang-Juei, L. Endotoxemia in patients with chronic liver diseases: Relationship to severity of liver diseases, presence of esophaegeal varices, and hyperdynamic circulation. J. Hepatol. 1995, 22, 165–172. [Google Scholar] [CrossRef]
- Bellot, P.; García-Pagán, J.C.; Francés, R.; Abraldes, J.G.; Navasa, M.; Pérez-Mateo, M.; Such, J.; Bosch, J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology 2010, 52, 2044–2052. [Google Scholar] [CrossRef]
- Bauer, T.M.; Schwacha, H.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Aponte, J.J.; Pelz, K.; Berger, D.; Kist, M.; Blum, H.E. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am. J. Gastroenterol. 2002, 97, 2364–2370. [Google Scholar] [CrossRef]
- Pérez-Paramo, M.; Munoz, J.; Albillos, A.; Freile, I.; Portero, F.; Santos, M.; Ortiz-Berrocal, J. Effect of propranolol on the factors promoting bacterial translocation in cirrhotic rats with ascites. Hepatology 2000, 31, 43–48. [Google Scholar] [CrossRef]
- Zhang, W.; Gu, Y.; Chen, Y.; Deng, H.; Chen, L.; Chen, S.; Zhang, G.; Gao, Z. Intestinal flora imbalance results in altered bacterial translocation and liver function in rats with experimental cirrhosis. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1481–1486. [Google Scholar] [CrossRef]
- Seki, E.; Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalk between the liver and gut. J. Physiol. 2012, 590, 447–458. [Google Scholar] [CrossRef]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair 2010, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; de Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Mazagova, M.; Wang, L.; Anfora, A.T.; Wissmueller, M.; Lesley, S.A.; Miyamoto, Y.; Eckmann, L.; Dhungana, S.; Pathmasiri, W.; Sumner, S.; et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. 2015, 29, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Grąt, M.; Wronka, K.M.; Krasnodębski, M.; Masior, Ł.; Lewandowski, Z.; Kosińska, I.; Grąt, K.; Stypułkowski, J.; Rejowski, S.; Wasilewicz, M.; et al. Profile of Gut Microbiota Associated With the Presence of Hepatocellular Cancer in Patients With Liver Cirrhosis. Transplant. Proc. 2016, 48, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fan, X.G.; Wang, Z.M.; Zhou, J.H.; Tian, X.F.; Li, N. Identification of helicobacter species in human liver samples from patients with primary hepatocellular carcinoma. J. Clin. Pathol. 2004, 57, 1273–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.G.; Feng, Y.; Theve, E.J.; Raczynski, A.R.; Fiala, J.L.A.; Doernte, A.L.; Williams, M.; McFaline, J.L.; Essigmann, J.M.; Schauer, D.B.; et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut 2010, 59, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Nakagawa, S.; Sawayama, H.; Ishimoto, T.; Imai, K.; Iwatsuki, M.; Hashimoto, D.; Baba, Y.; Yamashita, Y.; Yoshida, N.; et al. The microbiome and hepatobiliary-pancreatic cancers. Cancer Lett. 2017, 402, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Pár, A.; Pár, G. Nem alkoholos zsírmáj és hepatocellularis carcinoma-2016. Orv. Hetil. 2016, 157, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Romero-Gómez, M.; Jover, M.; Galán, J.J.; Ruiz, A. Gut ammonia production and its modulation. Metab. Brain Dis. 2009, 24, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Davies, N.A.; Shawcross, D.L.; Hodges, S.J.; Zwingmann, C.; Brooks, H.F.; Mani, A.R.; Harry, D.; Stadlbauer, V.; Zou, Z.; et al. Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology 2007, 45, 1517–1526. [Google Scholar] [CrossRef]
- Shawcross, D.L.; Davies, N.A.; Williams, R.; Jalan, R. Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J. Hepatol. 2004, 40, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcovich, M.; Zocco, M.A.; Roccarina, D.; Ponziani, F.R.; Gasbarrini, A. Prevention and treatment of hepatic encephalopathy: Focusing on gut microbiota. World J. Gastroenterol. 2012, 18, 6693–6700. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.H. Primary sclerosing cholangitis: 50 years of a gut-liver relationship and still no love? Gut 2016, 65, 1579–1581. [Google Scholar] [CrossRef]
- Kummen, M.; Hov, J.R. The gut microbial influence on cholestatic liver disease. Liver Int. 2019, 39, 1186–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, N.; Sasaki, N.; Aoki, R.; Miyamoto, K.; Suda, W.; Teratani, T.; Suzuki, T.; Koda, Y.; Chu, P.S.; Taniki, N.; et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat. Microbiol. 2019, 4, 492–503. [Google Scholar] [CrossRef]
- Liao, L.; Schneider, K.M.; Galvez, E.J.C.; Frissen, M.; Marschall, H.U.; Su, H.; Hatting, M.; Wahlström, A.; Haybaeck, J.; Puchas, P.; et al. Intestinal dysbiosis augments liver disease progression via NLRP3 in a murine model of primary sclerosing cholangitis. Gut 2019, 68, 1477–1492. [Google Scholar] [CrossRef]
- Little, R.; Wine, E.; Kamath, B.M.; Griffiths, A.M.; Ricciuto, A. Gut microbiome in primary sclerosing cholangitis: A review. World J. Gastroenterol. 2020, 26, 2768–2780. [Google Scholar] [CrossRef]
- Kummen, M.; Thingholm, L.B.; Rühlemann, M.C.; Holm, K.; Hansen, S.H.; Moitinho-Silva, L.; Liwinski, T.; Zenouzi, R.; Storm-Larsen, C.; Midttun, Ø.; et al. Altered Gut Microbial Metabolism of Essential Nutrients in Primary Sclerosing Cholangitis. Gastroenterology 2021, 160, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- Mattner, J. Impact of microbes on the pathogenesis of primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC). Int. J. Mol. Sci. 2016, 17, 1864. [Google Scholar] [CrossRef] [PubMed]
- Padgett, K.A.; Selmi, C.; Kenny, T.P.; Leung, P.S.C.; Balkwill, D.L.; Ansari, A.A.; Coppel, R.L.; Gershwin, M.E. Phylogenetic and immunological definition of four lipoylated proteins from Novosphingobium aromaticivorans, implications for primary biliary cirrhosis. J. Autoimmun. 2005, 24, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, J.P.; Fusakio, M.E.; Rainbow, D.B.; Moule, C.; Fraser, H.I.; Clark, J.; Todd, J.A.; Peterson, L.B.; Savage, P.B.; Wills-Karp, M.; et al. Identification of Cd101 as a Susceptibility Gene for Novosphingobium aromaticivorans-Induced Liver Autoimmunity. J. Immunol. 2011, 187, 337–349. [Google Scholar] [CrossRef] [Green Version]

| Scheme | Study Type | Exposure | Main Results | Ref. |
|---|---|---|---|---|
| Germ-free C57BL/6J female mice | Animal | Western-style diet and high-fructose diet | Although intestinal barrier damage was observed in the germ-free mouse group, hepatic steatosis did not occur due to the absence of aseptically induced LPS translocation. Required for commensal bacteria in the gut microbiota to induce hepatic steatosis by factors, such as diet | [80] |
| Germ-free C57BL/6J male mice | Animal | Normal chow diet and FMT in genetically obese human donor | The gut microbiota of genetically obese humans influences the hepatic transcriptional profile of lipid metabolism such as PPAR α in mice, promoting the pathogenesis of hepatic steatosis. High serum LPS levels in the obese group can suppress the expression of PPAR α. | [81] |
| Male C57BL/6, C3H/HouJ and TLR4 mutant C3H/HeJ mice | Animal | Methionine/choline-deficient diet and weekly intravenous injections of clodronate liposomes | (↑): Steatohepatitis histological condition, portal endotoxemia and TLR4 expression in control mice fed MCDD. (↓): Liver injury and lipid accumulation marker in TLR4 mutant mice. Intravenous injections of clodronate liposomes: depleting liver Kupffer cells → changes in histological condition of steatohepatitis and prevented increases in TLR4 expression. | [72] |
| TLR4 mutant C3H/HeJ mice and wildtype C3H/HouJ mice | Animal | Water enriched with 30% fructose | (↑): Hepatic steatosis and plasma ALT levels in wildtype mice fed fructose. (↓): Hepatic triglyceride accumulation in TLR4 mutant mice fed fructose. Hepatic lipid peroxidation, MyD88, and TNF-α levels were significantly decreased in TLR4 mutant mice fed fructose group in comparison to wildtype mice fed fructose. | [74] |
| Inflammasome-deficient mice and Asc and Il18-deficient mice | Animal | NASH model: methionine-choline-deficient diet for 24 days High fat diet model: 60% calories from fat for 10–12 weeks | (↑): Severity of NASH in inflammasome-deficient mice, Asc and Il18-deficient mice. Co-housing of inflammasome-deficient animals to wild type mice: exacerbation of hepatic steatosis and metabolic dysfunctions, alteration of gut microbiota configuration. | [77] |
| Obese patients (n = 52) | Human | (↑): Expression of mRNA of TNF-α and TNF receptors p55 in hepatic tissue and peripheral fat of patients with NASH. | [79] |
| Species | Study Types | Exposure | Main Results | Ref. |
|---|---|---|---|---|
| Germ-free NIH Swiss female mice | Animal | Oral gavage with alcohol (5 mg/kg) | (↓): Alcohol-induced liver injury, neutrophil infiltration, and levels of pro-inflammatory cytokines were lower in the germ-free mice group than in the other alcohol-fed mice groups. Gut microbiota plays a key role in liver injury through alcohol-induced dysbiosis | [111] |
| Germ-free C57BL/6 mice | Animal | Oral gavage with acute alcohol (3 g/kg) | (↑): The absence of gut microbiome increases alcohol susceptibility to binge drinking and increases ethanol metabolism in the liver. Acute alcohol supply increased liver inflammation in the sterile mice group due to binge-induced liver damage. In acute alcoholic liver disease, the gut microbiota may play a protective role in inflammation and hepatic steatosis. | [112] |
| Male Wistar rats | Animal | Continuous ethanol supply for 3 weeks. gut sterilization with polymyxin B and neomycin | (↓): Plasma endotoxin levels (80–90 pg/mL → <25 pg/mL), average hepatic pathological score in ethanol-fed and antibiotic-treated rats Antibiotic treatment prevented elevated aspartate aminotransferase levels and hepatic surface hypoxia. | [99] |
| Alcohol-fed NS5A Tg mice | Animal | Lieber–DeCarli diet containing 3.5% ethanol or isocaloric dextrin for long-term alcohol feeding, repetitive LPS injection | (↑): Ethanol-induced endotoxemia, liver injury and tumorigenesis after TLR4 induction through hepatocyte-specific transgenic expression of the HCV nonstructural protein NS5A. | [106] |
| Male C57BL/6J mice | Animal | Administered epinephrine for 5 days (2 mg/kg per day) or bolus ethanol for 3 days (6 g/kg per day), 24 h later, inject LPS (10 mg/kg) | (↑): Severity of liver damage and inflammation due to LPS through prior exposure to epinephrine and ethanol. (↓): Sensitivity of ethanol to liver damage due to co-administration of ethanol and propranolol. Sympathetic nerves influence the progression of ALD. | [107] |
| Male Wistar rats | Animal | Chronic ethanol diet fed | (↑): ROS production by LPS in Kupffer cells isolated from ethanol-fed mice. ROS production in Kupffer cells by LPS stimulation is increased NADPH oxidase-dependently. ERK1/2 contributes to the increase of TNF-α production in Kupffer cells by LPS stimulation. | [108] |
| Patients (n = 14: alcoholic hepatitis 8, cirrhotic with alcoholic hepatitis 5, severe alcoholic hepatitis 1) | Human | (↑): Plasma endotoxin levels and Serum IL-6 and IL-8 levels of patients compared to healthy subjects. Serum LBP was positively correlated with white blood cell and neutrophil counts as an indicator of an inflammatory response. | [100] | |
| Controls (n = 11), Alcoholics (n = 30: minimal patients: 10, intermediate patients: 9, cirrhotic alcoholic liver disease patients: 11) | Human | (↑): Endotoxin levels and endotoxin activity-related binding factors concentration in alcoholic groups | [101] | |
| Controls (n = 6), patients with alcoholic hepatitis (n = 6) | Human | (↑): nuclear factor-κB activity in the monocytes of 6 patients with alcoholic hepatitis as compared with normal subjects. (↑): Nuclear factor-kB activity, TNF-α RNA expression and TNF-α release by endotoxin in alcoholic hepatitis patients. | [109] | |
| Controls (n = 11), patients with alcohol use disorder (n = 42) and alcoholic hepatitis (n = 91) | Human | (↑): Retention levels of ECE1 in individuals according to alcoholic patient severity Genetically engineered C. albicans strain exacerbates ethanol-induced liver disease in mice and increases mortality in mice. Candidalysin can exacerbate ethanol-induced liver disease and damage hepatocytes independently of the β-glucan receptor. | [110] |
| Species | Study Type | Exposure | Main Results | Ref. |
|---|---|---|---|---|
| Germ-free C57BL/6 male mice | Animal | TAA or CCl4 | (↑): Liver fibrosis was increased in the germ-free mice group compared to the control mice. More toxin-induced oxidative stress and cell death were observed. The commensal gut microbiota prevents liver fibrosis in conditions of chronic liver injury. | [127] |
| Male Sprague-Dawley rats | Animal | Administration of CCl4 and fed phenobarbital in drinking water (35 mg/dL) | Bacterial translocation was seen in 48% of cirrhosis rat models. Cirrhosis rat model with small intestinal bacterial overgrowth had a significantly higher bacterial translocation rate and slower intestinal transit rate compared to the control group. | [122] |
| Male Sprague–Dawley rats | Animal | Subcutaneous injection of an equal mixture of CCl4 and olive oil. antibiotic (norfloxacin) and different probiotic treatments | (↑): Levels of Enterobacteriaceae compared to controls in a cirrhosis rat model. (↑): Levels of Lactobacillus in the cirrhotic rat group treated with Bifidobacteria compared to the saline treated group. (↓): Levels of Enterobacteriaceae in the cirrhotic rat group treated with Bifidobacteria compared to the saline treated group. (↓): Levels of endotoxin in the cirrhotic rat group respectively treated with Bifidobacteria and Enterococcus compared to the saline treated group. | [123] |
| Male C3H/HeOuJ mice (TLR4 wild type), C3H/HeJ mice (TLR4 mutant), Tlr2 deficient mice, TrifLps2/Lps2 mice, C57BL/6 mice and MyD88 deficient mice | Animal | Underwent bile duct ligation. fed CCl4 or TAA | TLR4 and the gut microbiota play an essential role in liver fibrogenesis. (↑): TGFβ-mediated activation of hepatic stellate cells and collagen production. (↓): Regulation of TGFβ pseudo-receptor Bambi in quiescent hepatic stellate cells. | [126] |
| Controls (n = 45), Patients (n = 169) | Human | (↑): Plasma endotoxin levels of chronic hepatitis patients and cirrhosis patients compared with healthy subjects. Endotoxemia was identified in chronic hepatitis patients (27%), chronic hepatitis patients with acute exacerbation (85%) and cirrhosis patients (41%), respectively. In cirrhosis patients, plasma endotoxin levels increased progressively in association with the severity of liver dysfunction. | [119] | |
| Non-infected cirrhosis patients (n = 75: 55 ascites and 20 no ascites) | Human | Bacterial DNA detection only in patients with ascites. Presence of bacterial DNA in plasma contributed to systemic hemodynamic impairment in patients with ascites cirrhosis and exacerbated intrahepatic endothelial dysfunction in cirrhosis. | [120] | |
| Cirrhosis cohort patients (n = 53) | Human | Small intestinal bacterial overgrowth was seen in 59% of patients with cirrhosis and was significantly related to systemic endotoxemia. | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, S.-M.; Park, E.; Jeong, J.-J.; Ganesan, R.; Gupta, H.; Gebru, Y.A.; Sharma, S.; Kim, D.-J.; Suk, K.-T. The Gut Microbiota-Derived Immune Response in Chronic Liver Disease. Int. J. Mol. Sci. 2021, 22, 8309. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158309
Won S-M, Park E, Jeong J-J, Ganesan R, Gupta H, Gebru YA, Sharma S, Kim D-J, Suk K-T. The Gut Microbiota-Derived Immune Response in Chronic Liver Disease. International Journal of Molecular Sciences. 2021; 22(15):8309. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158309
Chicago/Turabian StyleWon, Sung-Min, Eunju Park, Jin-Ju Jeong, Raja Ganesan, Haripriya Gupta, Yoseph Asmelash Gebru, SatyaPriya Sharma, Dong-Joon Kim, and Ki-Tae Suk. 2021. "The Gut Microbiota-Derived Immune Response in Chronic Liver Disease" International Journal of Molecular Sciences 22, no. 15: 8309. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158309