
Evidence for Protein–Protein Interaction between Dopamine Receptors and the G Protein-Coupled Receptor 143


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
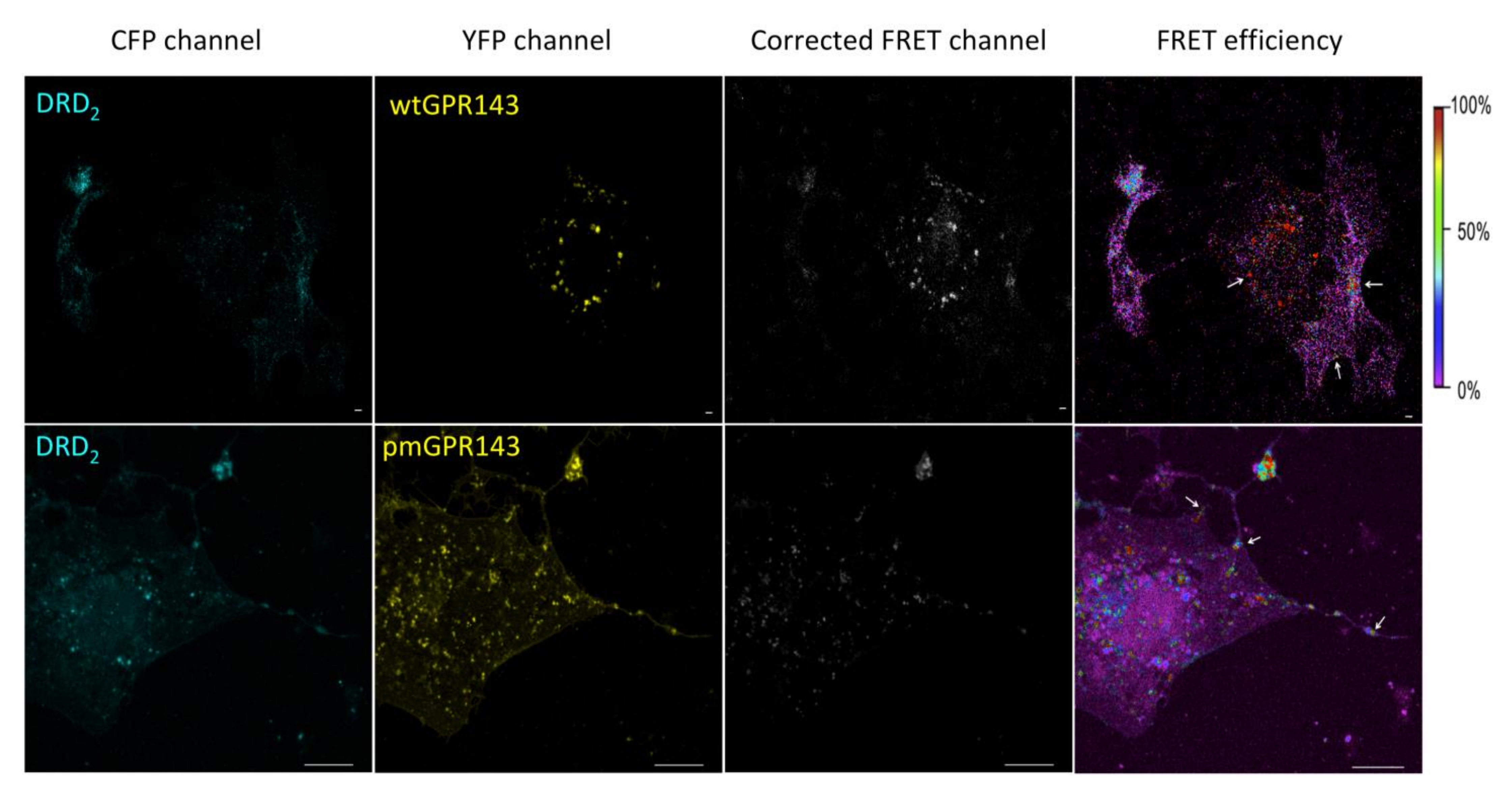
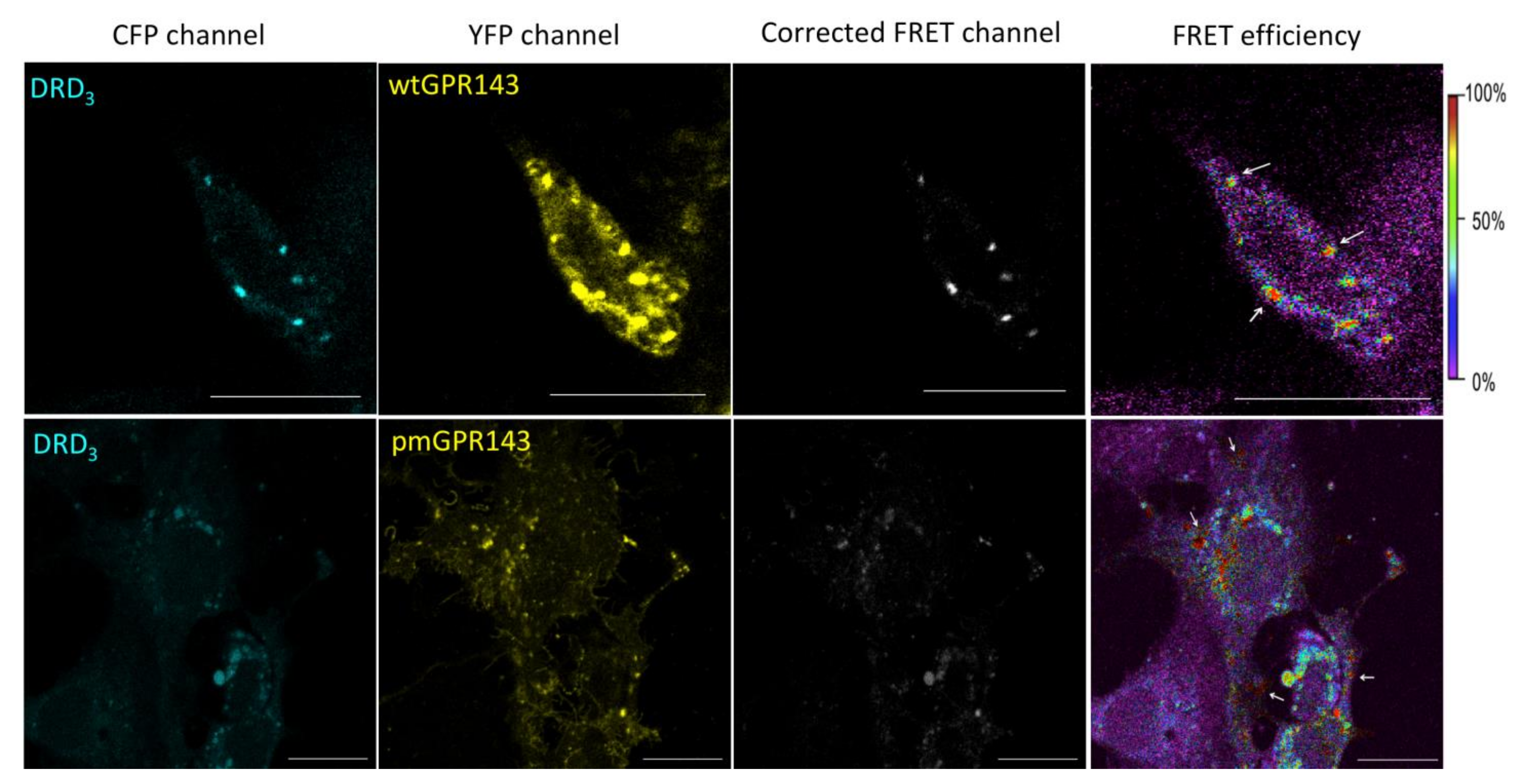
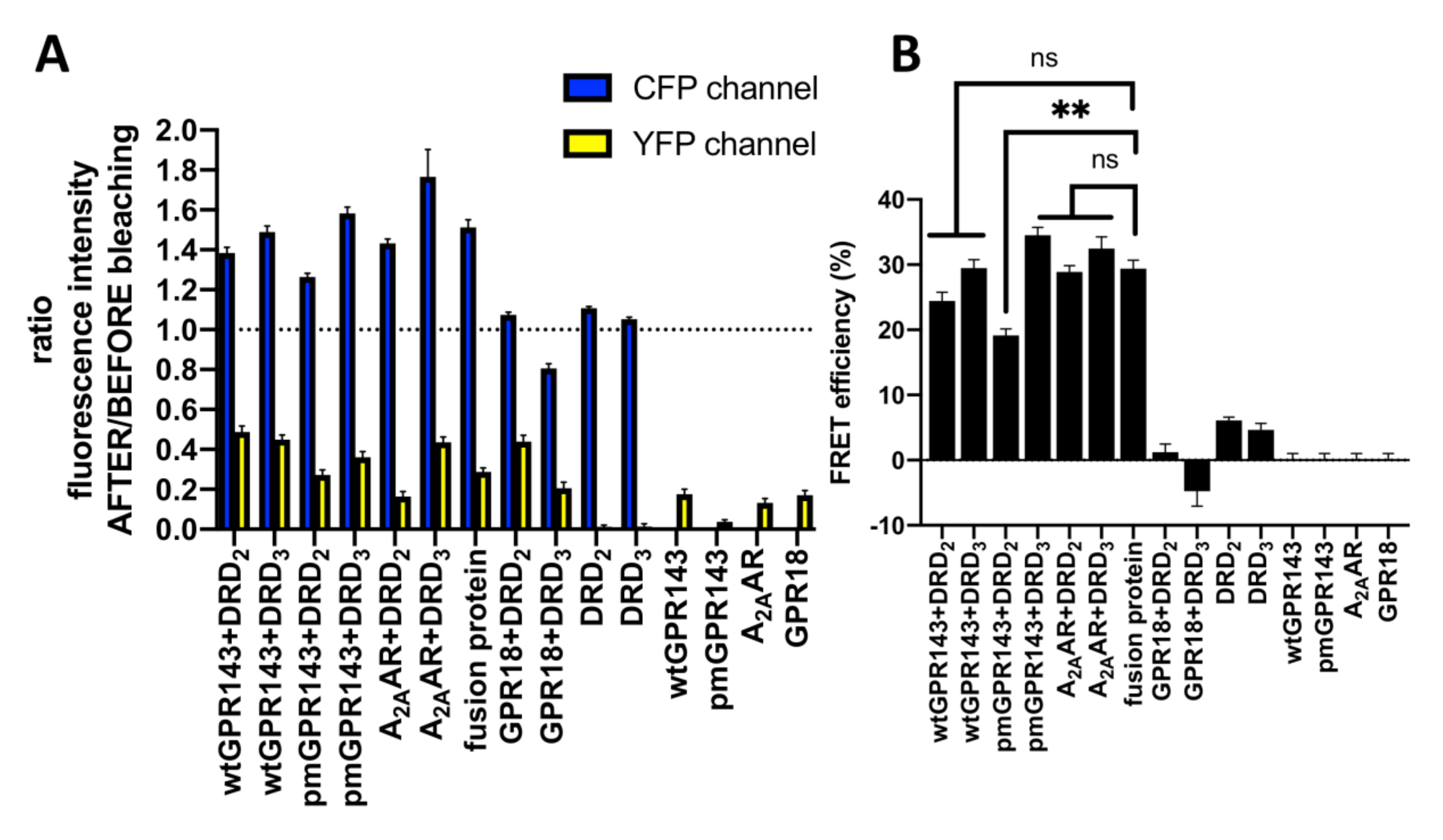
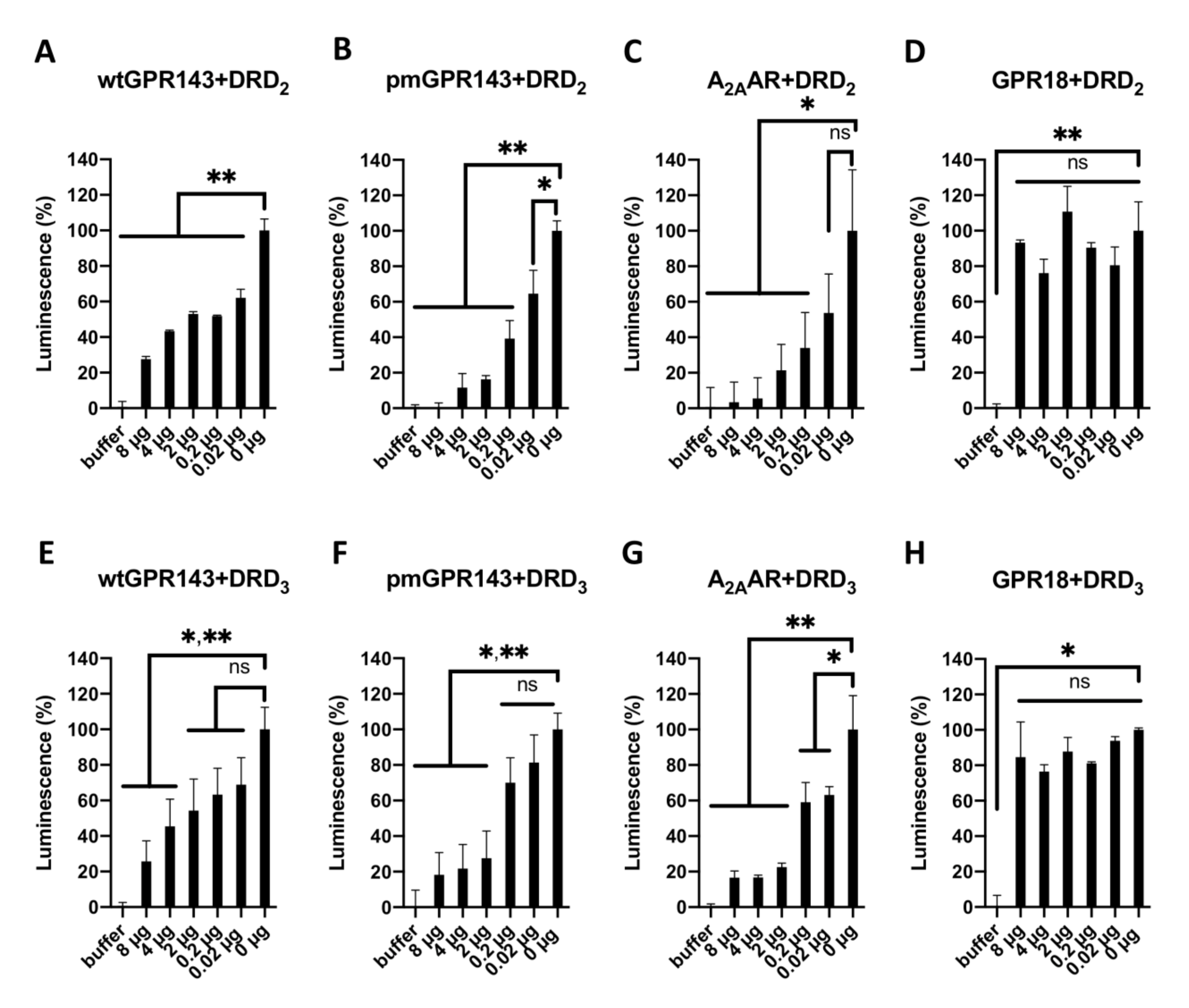
2.1. Fluorescence Resonance Energy Transfer
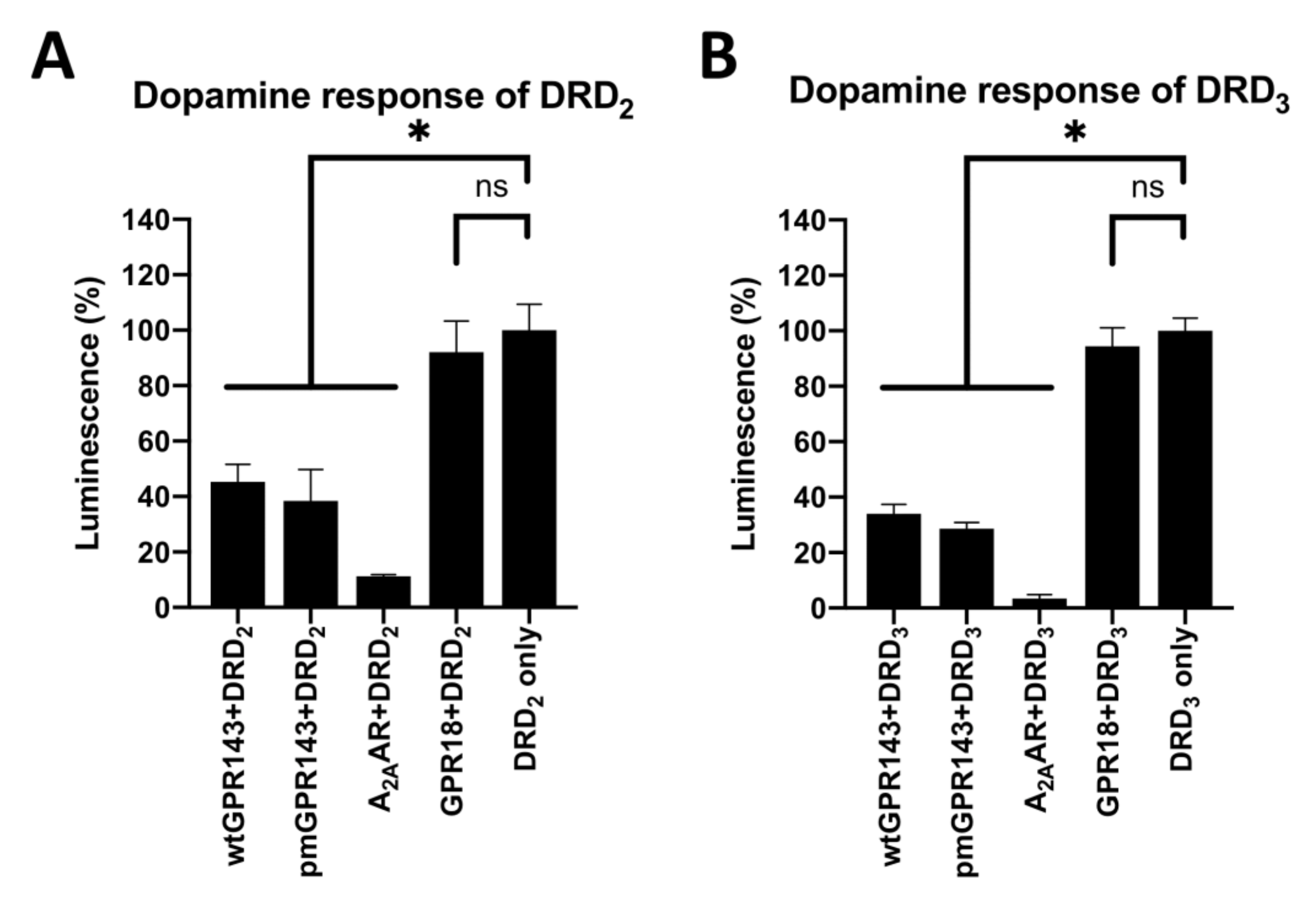
2.2. GPR143 Influences Dopaminergic Signaling in the β-Arrestin Recruitment Assay
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cell Culture and Transfection
4.3. β-. Arrestin Assay
4.4. Immunostaining
4.5. Fluorescence Resonance Energy Transfer (FRET)
4.6. Western Blot
4.7. Data Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Filippo, E.; Schiedel, A.C.; Manga, P. Interaction between G Protein-Coupled Receptor 143 and Tyrosinase: Implications for Understanding Ocular Albinism Type 1. J. Investig. Dermatol. 2017, 137, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Jabeen, A.; Ranganathan, S. Applications of machine learning in GPCR bioactive ligand discovery. Curr. Opin. Struct. Biol. 2019, 55, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Somvanshi, R.K.; Kumar, U. Pathophysiology of GPCR homo- and heterodimerization: Special emphasis on Somatostatin receptors. Pharmaceuticals 2012, 5, 417–446. [Google Scholar] [CrossRef] [Green Version]
- Ferré, S.; Casadó, V.; Devi, L.A.; Filizola, M.; Jockers, R.; Lohse, M.J.; Milligan, G.; Pin, J.P.; Guitart, X. G protein-coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharmacol. Rev. 2014, 66, 413–434. [Google Scholar] [CrossRef] [Green Version]
- Fuxe, K.; Borroto-Escuela, D.O.; Marcellino, D.; Romero-Fernandez, W.; Frankowska, M.; Guidolin, D.; Filip, M.; Ferraro, L.; Woods, A.S.; Tarakanov, A.; et al. GPCR Heteromers and their Allosteric Receptor-Receptor Interactions. Curr. Med. Chem. 2012, 19, 356–363. [Google Scholar] [CrossRef]
- Smith, L.N.; Bachus, S.E.; McDonald, C.G.; Smith, R.F. Role of the D3 dopamine receptor in nicotine sensitization. Behav. Brain Res. 2015, 289, 92–104. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Espinoza, S.; Gainetdinov, R.R. Dopamine receptors—IUPHAR review 13. Br. J. Pharmacol. 2015, 172, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cokan, K.B.; Mavri, M.; Rutland, C.S.; Glisic, S.; Sencanski, M.; Vreci, M.; Kubale, V. Critical Impact of Different Conserved Endoplasmic Retention Motifs and Dopamine Receptor Interacting Proteins (DRIPs) on Intracellular Localization and Trafficking of the D2 Dopamine Receptor (D2-R) Isoforms Kaja. Biomolecules 2020, 10, 1355. [Google Scholar] [CrossRef]
- Farran, B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol. Res. 2017, 117, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Wouters, E.; Marin, A.R.; Dalton, J.A.R.; Giraldo, J.; Stove, C. Distinct Dopamine D2 Receptor Antagonsits Differntially Impact D2 Receptor Oligomerization. Int. J. Mol. Sci. 2019, 20, 1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borroto-Escuela, D.O.; Rodriguez, D.; Romero-Fernandez, W.; Kapla, J.; Jaiteh, M.; Ranganathan, A.; Lazarova, T.; Fuxe, K.; Carlsson, J. Mapping the interface of a GPCR Dimer: A structural model of the A2A Adenosine and D2 dopamine receptor heteromer. Front. Pharmacol. 2018, 9, 829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuxe, K.; Marcellino, D.; Genedani, S.; Agnati, L. Adenosine A2A receptors, dopamine D2 receptors and their interactions in Parkinson’s disease. Mov. Disord. 2007, 22, 1990–2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wan, Q.; Pristupa, Z.B.; Yu, X.M.; Wang, Y.T.; Niznik, H.B. Direct protein-protein coupling enables cross-talk between dopamine D5 and γ-aminobutyric acid A receptors. Nature 2000, 403, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, A.C.; Köse, M.; Barreto, C.; Bueschbell, B.; Morra, G.; Sensoy, O.; Moreira, I.S. Prediction and targeting of interaction interfaces in G-protein coupled receptor oligomers. Curr. Top. Med. Chem. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Canals, M.; Marcellino, D.; Fanelli, F.; Ciruela, F.; De Benedetti, P.; Goldberg, S.R.; Neve, K.; Fuxe, K.; Agnati, L.F.; Woods, A.S.; et al. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: Qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J. Biol. Chem. 2003, 278, 46741–46749. [Google Scholar] [CrossRef] [Green Version]
- Pinna, A.; Bonaventura, J.; Farré, D.; Sánchez, M.; Simola, N.; Mallol, J.; Lluís, C.; Costa, G.; Baqi, Y.; Müller, C.E.; et al. L-DOPA disrupts adenosine A2A–cannabinoid CB1–dopamine D2 receptor heteromer cross-talk in the striatum of hemiparkinsonian rats: Biochemical and behavioral studies Annalisa. Exp. Neurol. J. 2014, 253, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Fuxe, K. Oligomeric Receptor Complexes and Their Allosteric Receptor-Receptor Interactions in the Plasma Membrane Represent a New Biological Principle for Integration of Signals in the CNS. Front. Mol. Neurosci. 2019, 12, 230. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.C.; Gatti McArthur, S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. [Google Scholar] [CrossRef] [PubMed]
- Hillefors, M.; Hedlund, P.B.; Von Euler, G. Effects of adenosine A(2A) receptor stimulation in vivo on dopamine D3 receptor agonist binding in the rat brain. Biochem. Pharmacol. 1999, 58, 1961–1964. [Google Scholar] [CrossRef]
- Beaulieu, J.-M.; Gainetdinov, R.R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [Green Version]
- Borroto-Escuela, D.O.; Ferraro, L.; Narvaez, M.; Tanganelli, S.; Beggiato, S.; Liu, F.; Rivera, A.; Fuxe, K. Multiple Adenosine-Dopamine (A2A-D2 Like) Heteroreceptor Complexes in the Brain and Their Role in Schizophrenia. Cells 2020, 9, 1077. [Google Scholar] [CrossRef]
- De Filippo, E.; Manga, P.; Schiedel, A.C. Identification of novel G Protein-Coupled Receptor 143 ligands as pharmacologic tools for investigating X-linked ocular albinism. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3118–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwinn, G.; Schwarck, H.; McIntosch, C.; Milstrey, H.-R.; Willms, B.; Köbberling, J. Effect of the dopamine receptor blocking agent pimozide on the growth hormone response to arginine and exercise and on the spontaneous growth hormone fluctuations. J. Clin. Endocrinol. Metab. 1976, 43, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.B.; Patel, S.; Marwood, R.; Emms, F.; Seabrook, G.R.; Knowles, M.R.; McAllister, G. Expression and pharmacological characterization of the human D3 dopamine receptor. J. Pharmacol. Exp. Ther. 1994, 268, 417–426. [Google Scholar]
- Sokoloff, P.; Andrieux, M.; Besançon, R.; Pilon, C.; Martres, M.-P.; Giros, B.; Schwartz, J.-C. Pharmacology of human dopamine D3 receptor expressed in a mammalian cell line: Comparison with D2 receptor. Eur. J. Pharmacol. Mol. Pharmacol. 1992, 225, 331–337. [Google Scholar] [CrossRef]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Masukawa, D.; Nakamura, F.; Koga, M.; Kamiya, M.; Chen, S.; Yamashita, N.; Arai, N.; Goshima, Y. Localization of ocular albinism-1 gene product GPR143 in the rat central nervous system. Neurosci. Res. 2014, 88, 49–57. [Google Scholar] [CrossRef]
- Fukuda, N.; Naito, S.; Masukawa, D.; Kaneda, M.; Miyamoto, H.; Abe, T.; Yamashita, Y.; Endo, I.; Nakamura, F.; Goshima, Y. Expression of ocular albinism 1 (OA1), 3, 4- dihydroxy- L-phenylalanine (DOPA) receptor, in both neuronal and non-neuronal organs. Brain Res. 2015, 1602, 62–74. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Miyamoto, H.; Nakamura, F.; Masukawa, D.; Yamamoto, T.; Muraoka, H.; Kamiya, M.; Yamashita, N.; Suzuki, T.; Matsuzaki, S.; et al. The protein Ocular albinism 1 is the orphan GPCR GPR143 and mediates depressor and bradycardic responses to DOPA in the nucleus tractus solitarii. Br. J. Pharmacol. 2014, 171, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiaffino, M.V.; Tacchetti, C. The ocular albinism type 1 (OA1) protein and the evidence for an intracellular signal transduction system involved in melanosome biogenesis. Pigment Cell Res. 2005, 18, 227–233. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, F.E.; Hambrick, G.W.; Green, W.R.; Iliff, J.; Stone, D.L. X-Linked Ocular Albinism—An Oculocutaneous Macromelanosomal Disorder. Arch. Ophthalmol. 1976, 94, 1883–1892. [Google Scholar] [CrossRef] [PubMed]
- Samaraweera, P.; Shen, B.; Newton, J.; Barsh, G.S.; Orlow, S.J. The mouse ocular albinism 1 gene product is an endolysosomal protein. Exp. Eye Res. 2001, 72, 319–329. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, D.B. Albinism, or the NOACH syndrome. Clin. Genet. 1987, 31, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Charles, S.J.; Green, J.S.; Grant, J.W.; Yates, J.R.W.; Moore, A.T. Clinical features of affected males with X linked ocular albinism. Br. J. Ophthalmol. 1993, 77, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creel, D.J.; Summers, C.G.; King, R.A. Visual anomalies associated with albinism. Ophthalmic Genet. 1990, 11, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.; Bonetti, C.; Surace, E.M.; Marigo, V.; Raposo, G. The ocular albinism type 1 (OA1) G-protein-coupled receptor functions with MART-1 at early stages of melanogenesis to control melanosome identity and composition. Hum. Mol. Genet. 2009, 18, 4530–4545. [Google Scholar] [CrossRef] [Green Version]
- Palmisano, I.; Bagnato, P.; Palmigiano, A.; Innamorati, G.; Rotondo, G.; Altimare, D.; Venturi, C.; Sviderskaya, E.V.; Piccirillo, R.; Coppola, M.; et al. The ocular albinism type 1 protein, an intracellular G protein-coupled receptor, regulates melanosome transport in pigment cells. Hum. Mol. Genet. 2008, 17, 3487–3501. [Google Scholar] [CrossRef]
- Young, A.; Jiang, M.; Wang, Y.; Ahmedli, N.B.; Ramirez, J.; Reese, B.E.; Birnbaumer, L.; Farber, D.B. Specific interaction of Gαi3 with the Oa1 G-protein coupled receptor controls the size and density of melanosomes in retinal pigment epithelium. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Masukawa, D.; Koga, M.; Sezaki, A.; Nakao, Y.; Kamikubo, Y.; Hashimoto, T.; Okuyama-Oki, Y.; Aladeokin, A.C.; Nakamura, F.; Yokoyama, U.; et al. L-DOPA sensitizes vasomotor tone by modulating the vascular alpha1-adrenergic receptor. JCI Insight 2017, 2, e90903. [Google Scholar] [CrossRef] [Green Version]
- Innamorati, G.; Piccirillo, R.; Bagnato, P.; Palmisano, I.; Schiaffino, M.V. The melanosomal/lysosomal protein OA1 has properties of a G protein-coupled receptor. Pigment Cell Res. 2006, 19, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Sone, M.; Orlow, S.J. The Ocular Albinism Type 1 Gene Product, OA1, Spans Intracellular Membranes 7 Times. Exp. Eye Res. 2007, 85, 806–816. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Rosenberg, B.; Orlow, S.J. Intracellular distribution and late endosomal effects of the ocular albinism type 1 gene product: Consequences of disease-causing mutations and implications for melanosome biogenesis. Traffic 2001, 2, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Staleva, L.; Orlow, S.J. Ocular albinism 1 protein: Trafficking and function when expressed in Saccharomyces cerevisiae. Exp. Eye Res. 2006, 82, 311–318. [Google Scholar] [CrossRef]
- Neumann, A.; Engel, V.; Mahardhika, A.B.; Schoeder, C.T.; Namasivayam, V.; Kiec-Kononowicz, K.; Müller, C.E. Computational Investigations on the Binding Mode of Ligands for the Cannabinoid-Activated G Protein-Coupled Receptor GPR18 Alexander. Biomolecules 2020, 10, 686. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Di, W.L.; Kelsell, D.P.; Zicha, D. Quantitative fluorescence resonance energy transfer (FRET) measurement with acceptor photobleaching and spectral unmixing. J. Microsc. 2004, 215, 162–173. [Google Scholar] [CrossRef]
- Wilson, M.C.; Meredith, D.; Halestrap, A.P. Fluorescence resonance energy transfer studies on the interaction between the lactate transporter MCT1 and CD147 provide information on the topology and stoichiometry of the complex in situ. J. Biol. Chem. 2002, 277, 3666–3672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A guide to fluorescent protein FRET pairs. Sensors 2016, 16, 1488. [Google Scholar] [CrossRef]
- Mott, A.M.; Nunes, E.J.; Collins, L.E.; Port, R.G.; Sink, K.S.; Hockemeyer, J.; Müller, C.E.; Salamone, J.D. The adenosine A2A antagonist MSX-3 reverses the effects of the dopamine antagonist haloperidol on effort-related decision making in a T-maze cost/benefit procedure. Psychopharmacology 2009, 204, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Romero-Fernandez, W.; Zhou, Z.; Beggiato, S.; Wydra, K.; Filip, M.; Tanganelli, S.; Borroto-Escuela, D.O.; Ferraro, L.; Fuxe, K. Acute cocaine treatment enhances the antagonistic allosteric adenosine A2A-dopamine D2 receptor–receptor interactions in rat dorsal striatum without increasing significantly extracellular dopamine levels. Pharmacol. Rep. 2020, 72, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Torvinen, M.; Marcellino, D.; Canals, M.; Agnati, L.; Lluis, C.; Franco, R.; Fuxe, K. Adenosine A2A receptor and dopamine D3 receptor interactions: Evidence of functional A2A/D3 heteromeric complexes. Mol. Pharmacol. 2005, 67, 400–407. [Google Scholar] [CrossRef]
- Gines, S.; Hillion, J.; Torvinen, M.; Le Crom, S.; Casado, V.; Canela, E.I.; Rondin, S.; Lew, J.Y.; Watson, S.; Zoli, M.; et al. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc. Natl. Acad. Sci. USA 2000, 97, 8606–8611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, F.J.S.; Xue, S.; Pei, L.; Vukusic, B.; Chéry, N.; Wang, Y.; Wang, Y.T.; Niznik, H.B.; Yu, X.-M.; Liu, F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell 2002, 111, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Niewiarowska-Sendo, A.; Polit, A.; Piwowar, M.; Tworzydło, M.; Kozik, A.; Guevara-Lora, I. Bradykinin B2 and dopamine D2 receptors form a functional dimer. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1855–1866. [Google Scholar] [CrossRef]
- Kearn, C.S.; Blake-Palmer, K.; Daniel, E.; Mackie, K.; Glass, M. Concurrent Stimulation of Cannabinoid CB1 and Dopamine D2 Receptors Enhances Heterodimer Formation: A Mechanism for Receptor Cross-Talk? Mol. Pharmacol. 2005, 67, 1697–1704. [Google Scholar] [CrossRef] [Green Version]
- Cabello, N.; Gandía, J.; Bertarelli, D.C.G.; Watanabe, M.; Franco, R.; Ferré, S.; Luján, R.; Ciruela, F. Metabotropic Glutamate Type 5, Dopamine D2 and Adenosine A2A Receptors form Higher-Order Oligomers in Living Cells. J. Neurochem. 2014, 109, 1497–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalrymple, M.B.; Pfleger, K.D.G.; Eidne, K.A. G protein-coupled receptor dimers: Functional consequences, disease states and drug targets. Pharmacol. Ther. 2008, 118, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.M.; Kim, S.Y.; Lee, W.J.; Hwang, J.S.; Chang, S.E. Dopamine D4 receptor antagonist inhibits melanogenesis through transcriptional downregulation of MITF via ERK signalling. Exp. Dermatol. 2016, 25, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Incerti, B.; Cortese, K.; Pizzigoni, A.; Surace, E.M.; Varani, S.; Coppola, M.; Jeffery, G.; Seeliger, M.; Jaissle, G.; Bennett, D.C.; et al. Oa1 knock-out: New insights on the pathogenesis of ocular albinism type 1. Hum. Mol. Genet. 2002, 9, 2781–2788. [Google Scholar] [CrossRef] [Green Version]
- Cortese, K.; Giordano, F.; Surace, E.M.; Venturi, C.; Ballabio, A.; Tacchetti, C.; Marigo, V. The ocular albinism type 1 (OA1) gene controls melanosome maturation and size. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4358–4364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, T.; Jolly, R.; Martin-Martin, B.; Seabra, M.C.; Piccirillo, R.; Schiaffino, M.V.; Futter, C.E. Expression of OA1 limits the fusion of a subset of MVBs with lysosomes—A mechanism potentially involved in the initial biogenesis of melanosomes. J. Cell Sci. 2014, 126, 5143–5152. [Google Scholar] [CrossRef] [Green Version]
- Carr, R.; Benovic, J.L. From biased signalling to polypharmacology: Unlocking unique intracellular signalling using pepducins. Biochem. Soc. Trans. 2016, 44, 555–561. [Google Scholar] [CrossRef]
- Falletta, P.; Bagnato, P.; Bono, M.; Monticone, M.; Schiaffino, M.V.; Bennett, D.C.; Goding, C.R.; Tacchetti, C.; Valetti, C. Melanosome-autonomous regulation of size and number: The OA1 receptor sustains PMEL expression. Pigment Cell Melanoma Res. 2014, 27, 565–579. [Google Scholar] [CrossRef]
- Vetrini, F.; Auricchio, A.; Du, J.; Angeletti, B.; Fisher, D.E.; Ballabio, A.; Marigo, V. The Microphthalmia Transcription Factor (Mitf) Controls Expression of the Ocular Albinism Type 1 Gene: Link between Melanin Synthesis and Melanosome Biogenesis. Mol. Cell. Biol. 2004, 24, 6550–6559. [Google Scholar] [CrossRef] [Green Version]
- Masukawa, D.; Nishizawa, D.; Kanai, K.; Kitamura, S.; Kasahara, Y.; Hashimoto, T.; Takahagi, R.; Hasegawa, J.; Nakayama, K.; Sato, N.; et al. Genetic associations of single nucleotide polymorphisms in the L-DOPA receptor (GPR143) gene with severity of nicotine dependence in Japanese individuals, and attenuation of nicotine reinforcement in Gpr143 gene-deficient mice. J. Pharmacol. Sci. 2020, 144, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, R.; Palmisano, I.; Innamorati, G.; Bagnato, P.; Altimare, D.; Schiaffino, M.V. An unconventional dileucine-based motif and a novel cytosolic motif are required for the lysosomal and melanosomal targeting of OA1. J. Cell Sci. 2006, 119, 2003–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, V.M.; Decatur, C.L.; Stamer, W.D.; Lynch, R.M.; McKay, B.S. L-DOPA is an endogenous ligand for OA1. PLoS Biol. 2008, 6, 1861–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levoye, A.; Dam, J.; Ayoub, M.A.; Guillaume, J.L.; Jockers, R. Do orphan G-protein-coupled receptors have ligand-independent functions? New insights from receptor heterodimers. EMBO Rep. 2006, 7, 1094–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levoye, A.; Dam, J.; Ayoub, M.A.; Guillaume, J.L.; Couturier, C.; Delagrange, P.; Jockers, R. The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J. 2006, 25, 3012–3023. [Google Scholar] [CrossRef]
- Chen, T.; Hu, Y.; Lin, X.; Huang, X.; Liu, B.; Leung, P.; Chan, S.O.; Guo, D.; Jin, G. Dopamine signaling regulates the projection patterns in the mouse chiasm. Brain Res. 2015, 1625, 324–336. [Google Scholar] [CrossRef]
- Chagraoui, A.; Boulain, M.; Juvin, L.; Anouar, Y.; Barrière, G.; De Deurwaerdère, P. L-DOPA in parkinson’s disease: Looking at the “false” neurotransmitters and their meaning. Int. J. Mol. Sci. 2020, 21, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchese, A.; Paing, M.M.; Temple, B.R.S.; Trejo, J. G Protein–Coupled Receptor Sorting to Endosomes and Lysosomes. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 601–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanyaloglu, A.C. Advances in Membrane Trafficking and Endosomal Signaling of G Protein-Coupled Receptors. Int. Rev. Cell Mol. Biol. 2018, 339, 93–131. [Google Scholar] [CrossRef]
- Jensen, D.D.; Lieu, T.M.; Halls, M.L.; Veldhuis, N.A.; Imlach, W.L.; Mai, Q.N.; Poole, D.P.; Quach, T.; Aurelio, L.; Conner, J.; et al. Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, M.V.; D’Addio, M.; Alloni, A.; Baschirotto, C.; Valetti, C.; Cortese, K.; Puri, C.; Bassi, M.T.; Colla, C.; De Luca, M.; et al. Ocular albinism: Evidence for a defect in an intracellular signal transduction system. Nat. Genet. 1999, 23, 108–112. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bueschbell, B.; Manga, P.; Penner, E.; Schiedel, A.C. Evidence for Protein–Protein Interaction between Dopamine Receptors and the G Protein-Coupled Receptor 143. Int. J. Mol. Sci. 2021, 22, 8328. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158328
Bueschbell B, Manga P, Penner E, Schiedel AC. Evidence for Protein–Protein Interaction between Dopamine Receptors and the G Protein-Coupled Receptor 143. International Journal of Molecular Sciences. 2021; 22(15):8328. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158328
Chicago/Turabian StyleBueschbell, Beatriz, Prashiela Manga, Erika Penner, and Anke C. Schiedel. 2021. "Evidence for Protein–Protein Interaction between Dopamine Receptors and the G Protein-Coupled Receptor 143" International Journal of Molecular Sciences 22, no. 15: 8328. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158328