
Effects of Urolithin A on Mitochondrial Parameters in a Cellular Model of Early Alzheimer Disease

Abstract
:1. Introduction
2. Results
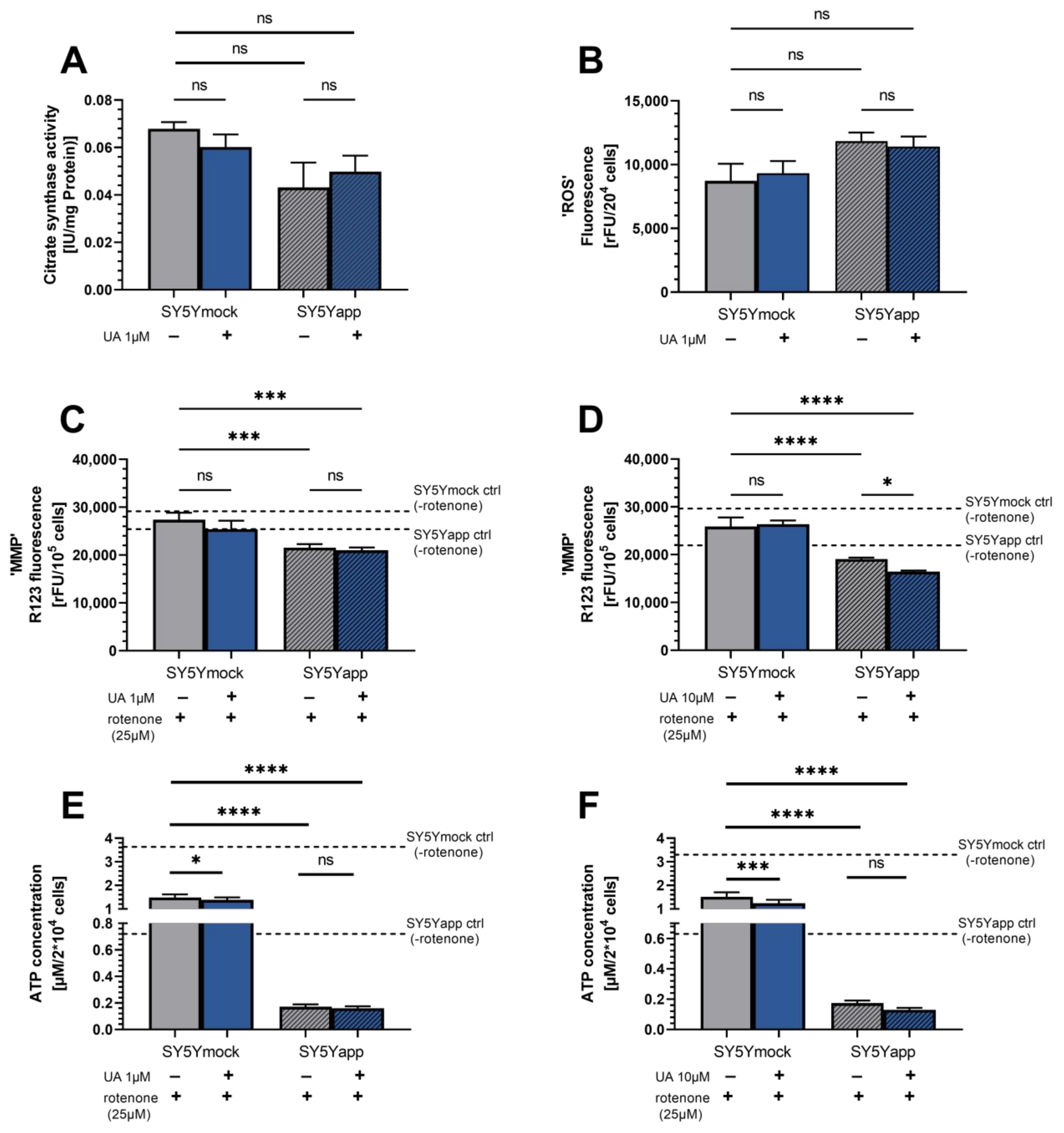
2.1. Urolithin A Does Not Affect Mitochondrial Function in SY5Y-APP695 Cells
2.2. Urolithin A Enhances Gene Expression for OXPHOS and Mitochondrial Biogenesis
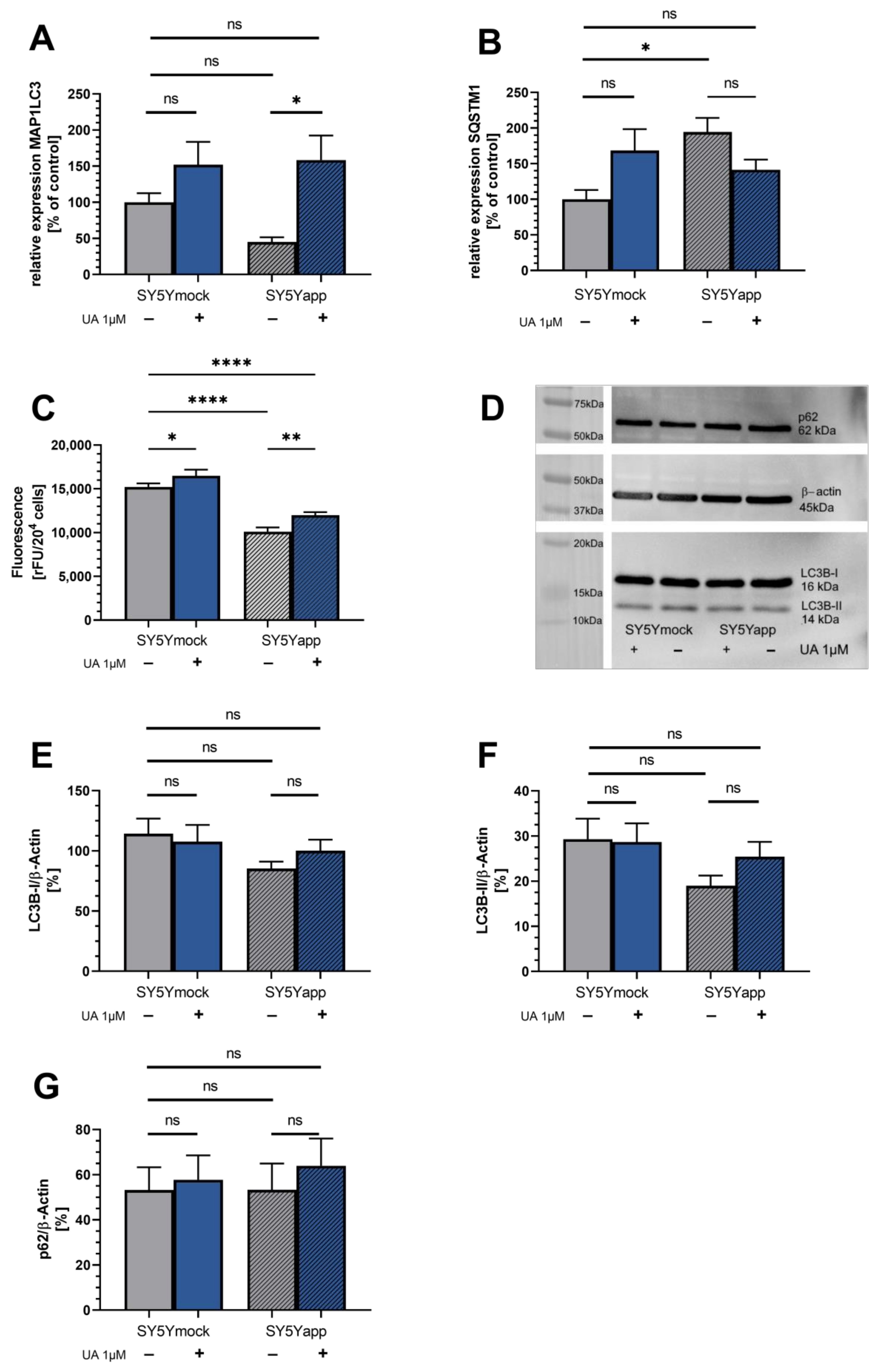
2.3. Urolithin Does Not Affect Autophagy in SY5Y-APP695 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cells
4.3. Autophagy
4.4. Western Blotting
4.5. High-Resolution Respirometry
4.6. Citrate Synthase Activity
4.7. Protein Content
4.8. ROS Determination
4.9. Mitochondrial Membrane Potential (MMP)
4.10. Adenosine Triphosphate Levels
4.11. Aβ1–40 Concentrations
4.12. Quantitative Real-Time PCR (qRT-PCR)
4.13. Data Handling and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albensi, B.C. Dysfunction of mitochondria: Implications for Alzheimer’s disease. Int. Rev. Neurobiol. 2019, 145, 13–27. [Google Scholar]
- Kukreja, L.; Kujoth, G.C.; Prolla, T.A.; van Leuven, F.; Vassar, R. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Navarro, A.; Boveris, A. The mitochondrial energy transduction system and the aging process. American journal of physiology. Cell Physiol. 2007, 292, C670–C686. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Ershov, N.I.; Maksimova, K.Y.; Muraleva, N.A.; Tyumentsev, M.A.; Kolosova, N.G. The Rat Prefrontal-Cortex Transcriptome: Effects of Aging and Sporadic Alzheimer’s disease-Like Pathology. J. Gerontol. Ser. A 2019, 74, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Friedland, K.; Eckert, A. Mitochondrial dysfunction: The missing link between aging and sporadic Alzheimer’s disease. Biogerontology 2016, 17, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Farmer, T.; Naslavsky, N.; Caplan, S. Tying trafficking to fusion and fission at the mighty mitochondria. Traffic 2018, 19, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.-M.; Jung, Y.-K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s disease. Cell. Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H. Alzheimer’s disease pathologic cascades: Who comes first, what drives what. Neurotox. Res. 2012, 22, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, L.; Faustino, C. Therapeutic Strategies Targeting Amyloid-β in Alzheimer’s disease. Curr. Alzheimer Res. 2019, 16, 418–452. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Rhein, V.; Baysang, G.; Rao, S.; Meier, F.; Bonert, A.; Müller-Spahn, F.; Eckert, A. Amyloid-beta leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell. Mol. Neurobiol. 2009, 29, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.I.; Lee, K.H.; Oh, J.Y.; Kim, J.S.; Han, H.J. Relationship Between β-Amyloid and Mitochondrial Dynamics. Cell. Mol. Neurobiol. 2017, 37, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Quntanilla, R.A.; Tapia-Monsalves, C. The Role of Mitochondrial Impairment in Alzheimer´s Disease Neurodegeneration: The Tau Connection. Curr. Neuropharmacol. 2020, 18, 1076–1091. [Google Scholar] [CrossRef] [PubMed]
- Poirier, Y.; Grimm, A.; Schmitt, K.; Eckert, A. Link between the unfolded protein response and dysregulation of mitochondrial bioenergetics in Alzheimer’s disease. Cell. Mol. Life Sci. 2019, 76, 1419–1431. [Google Scholar] [CrossRef] [Green Version]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Sasso, G.L.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Ma, H.; Liu, W.; Niesen, D.B.; Shah, N.; Crews, R.; Rose, K.N.; Vattem, D.A.; Seeram, N.P. Pomegranate’s Neuroprotective Effects against Alzheimer’s disease Are Mediated by Urolithins, Its Ellagitannin-Gut Microbial Derived Metabolites. ACS Chem. Neurosci. 2016, 7, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Gasperotti, M.; Passamonti, S.; Tramer, F.; Masuero, D.; Guella, G.; Mattivi, F.; Vrhovsek, U. Fate of microbial metabolites of dietary polyphenols in rats: Is the brain their target destination? ACS Chem. Neurosci. 2015, 6, 1341–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Shi, F.; Guo, Z.; Zhao, J.; Song, X.; Yang, H. Metabolite of ellagitannins, urolithin A induces autophagy and inhibits metastasis in human sw620 colorectal cancer cells. Mol. Carcinog. 2018, 57, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Y.; Halemahebai, G.; Tian, L.; Dong, H.; Aisker, G. Urolithin A, a pomegranate metabolite, protects pancreatic β cells from apoptosis by activating autophagy. J. Ethnopharmacol. 2020, 272, 113628. [Google Scholar] [CrossRef]
- Ahsan, A.; Zheng, Y.-R.; Wu, X.-L.; Tang, W.-D.; Liu, M.-R.; Ma, S.-J.; Jiang, L.; Hu, W.W.; Zhang, X.N.; Chen, Z. Urolithin A-activated autophagy but not mitophagy protects against ischemic neuronal injury by inhibiting ER stress in vitro and in vivo. CNS Neurosci. Ther. 2019, 25, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Cásedas, G.; Les, F.; Choya-Foces, C.; Hugo, M.; López, V. The Metabolite Urolithin-A Ameliorates Oxidative Stress in Neuro-2a Cells, Becoming a Potential Neuroprotective Agent. Antioxidants 2020, 9, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Sarrías, A.; Núñez-Sánchez, M.Á.; Tomás-Barberán, F.A.; Espín, J.C. Neuroprotective Effects of Bioavailable Polyphenol-Derived Metabolites against Oxidative Stress-Induced Cytotoxicity in Human Neuroblastoma SH-SY5Y Cells. J. Agric. Food Chem. 2017, 65, 752–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DaSilva, N.A.; Nahar, P.P.; Ma, H.; Eid, A.; Wei, Z.; Meschwitz, S.; Zawia, N.H.; Slitt, A.L.; Seeram, N.P. Pomegranate ellagitannin-gut microbial-derived metabolites, urolithins, inhibit neuroinflammation in vitro. Nutr. Neurosci. 2019, 22, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Yoon, J.H.; Cho, J.H.; Lee, S.J.; et al. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2021, 28, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Toney, A.M.; Fan, R.; Xian, Y.; Chaidez, V.; Ramer-Tait, A.E.; Chung, S. Urolithin A, a Gut Metabolite, Improves Insulin Sensitivity Through Augmentation of Mitochondrial Function and Biogenesis. Obesity 2019, 27, 612–620. [Google Scholar] [CrossRef]
- Ghosh, N.; Das, A.; Biswas, N.; Gnyawali, S.; Singh, K.; Gorain, M.; Polcyn, C.; Khanna, S.; Roy, S.; Sen, C.K. Urolithin A augments angiogenic pathways in skeletal muscle by bolstering NAD+ and SIRT1. Sci. Rep. 2020, 10, 20184. [Google Scholar] [CrossRef]
- Stockburger, C.; Gold, V.A.M.; Pallas, T.; Kolesova, N.; Miano, D.; Leuner, K.; Müller, W.E. A cell model for the initial phase of sporadic Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Grewal, R.; Reutzel, M.; Dilberger, B.; Hein, H.; Zotzel, J.; Marx, S.; Tretzel, J.; Sarafeddinov, A.; Fuchs, C.; Eckert, G.P. Purified oleocanthal and ligstroside protect against mitochondrial dysfunction in models of early Alzheimer’s disease and brain ageing. Exp. Neurol. 2020, 328, 113248. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; Donnell, M.O.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef]
- Pakrashi, S.; Chakraborty, J.; Bandyopadhyay, J. Neuroprotective Role of Quercetin on Rotenone-Induced Toxicity in SH-SY5Y Cell Line Through Modulation of Apoptotic and Autophagic Pathways. Neurochem. Res. 2020, 45, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Yuyun, X.; Jinjun, Q.; Minfang, X.; Jing, Q.; Juan, X.; Rui, M.; Li, Z.; Jing, G. Effects of Low Concentrations of Rotenone upon Mitohormesis in SH-SY5Y Cells. Dose-Response 2013, 11, 270–280. [Google Scholar] [CrossRef]
- Friedland-Leuner, K.; Stockburger, C.; Denzer, I.; Eckert, G.P.; Müller, W.E. Mitochondrial dysfunction: Cause and consequence of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2014, 127, 183–210. [Google Scholar] [PubMed]
- Stockburger, C.; Eckert, S.; Eckert, G.P.; Friedland, K.; Müller, W.E. Mitochondrial Function, Dynamics, and Permeability Transition: A Complex Love Triangle as A Possible Target for the Treatment of Brain Aging and Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 64, S455–S467. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nature reviews. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar]
- Deng, Y.-N.; Shi, J.; Liu, J.; Qu, Q.-M. Celastrol protects human neuroblastoma SH-SY5Y cells from rotenone-induced injury through induction of autophagy. Neurochem. Int. 2013, 63, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Lee, S.; Kim, J.H. Neuroprotective effects of urolithin A on H2O2-induced oxidative stress-mediated apoptosis in SK-N-MC cells. Nutr. Res. Pract. 2020, 14, 3–11. [Google Scholar] [CrossRef]
- Liu, C.-F.; Li, X.-L.; Zhang, Z.-L.; Qiu, L.; Ding, S.-X.; Xue, J.-X.; Zhao, G.P.; Li, J. Antiaging Effects of Urolithin A on Replicative Senescent Human Skin Fibroblasts. Rejuvenation Res. 2019, 22, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M.; Tokudome, S.; Shimizu, N.; Yoshikawa, N.; Ogawa, C.; Shirakawa, K.; Endo, J.; Katayama, T.; Yuasa, S.; Ieda, M.; et al. Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator-activated receptor gamma coactivator 1alpha. J. Biol. Chem. 2007, 282, 25970–25980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Nucleus-encoded regulators of mitochondrial function: Integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim. Biophys. Acta 2012, 1819, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential Expression of Oxidative Phosphorylation Genes in Patients with Alzheimer’s disease: Implications for Early Mitochondrial Dysfunction and Oxidative Damage. NeuroMol. Med. 2004, 5, 147–162. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2002, 1576, 1–14. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.-Y. Thyroid hormone receptor and ERRα coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal. 2018, 11, eaam5855. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-F.; Drumea, K.; Mott, S.; Wang, J.; Rosmarin, A.G. GABP transcription factor (nuclear respiratory factor 2) is required for mitochondrial biogenesis. Mol. Cell. Biol. 2014, 34, 3194–3201. [Google Scholar] [CrossRef] [Green Version]
- Bruni, F.; Polosa, P.L.; Gadaleta, M.N.; Cantatore, P.; Roberti, M. Nuclear respiratory factor 2 induces the expression of many but not all human proteins acting in mitochondrial DNA transcription and replication. J. Biol. Chem. 2010, 285, 3939–3948. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; McDonald, C.; Petrenko, N.B.; Leblanc, M.; Wang, T.; Giguere, V.; Evans, R.M.; Patel, V.V.; Pei, L. Estrogen-related receptor α (ERRα) and ERRγ are essential coordinators of cardiac metabolism and function. Mol. Cell. Biol. 2015, 35, 1281–1298. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Liu, Y.; Liu, X.; Zhu, L.; Cui, Y.; Cui, A.; Qiao, A.; Kong, X.; Liu, Y.; Chen, Q.; et al. PGC-1 beta-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERR alpha. Mitochondrion 2010, 10, 516–527. [Google Scholar] [CrossRef]
- Rouschop, K.M.A.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, B.; Goikuria, H.; Vega, R.; Rodríguez-Antigüedad, A.; López Medina, A.; Freijo, M.D.M.; Vandenbroeck, K.; Alloza, I. Autophagic marker MAP1LC3B expression levels are associated with carotid atherosclerosis symptomatology. PLoS ONE 2014, 9, e115176. [Google Scholar] [CrossRef]
- González-Rodríguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Iacono, O.L.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugan, S.; Amaravadi, R.K. Methods for Studying Autophagy Within the Tumor Microenvironment. Adv. Exp. Med. Biol. 2016, 899, 145–166. [Google Scholar] [PubMed] [Green Version]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; van Laar, T.; Huang, H.; Zhang, L. APP and APLP1 are degraded through autophagy in response to proteasome inhibition in neuronal cells. Protein Cell 2011, 2, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Jung, E.S.; Shin, H.J.; Byun, J.; Mook-Jung, I. Aβ-induced formation of autophagosomes is mediated by RAGE-CaMKKβ-AMPK signaling. Neurobiol. Aging 2012, 33, 1006.e11-23. [Google Scholar] [CrossRef]
- Esselun, C.; Bruns, B.; Hagl, S.; Grewal, R.; Eckert, G.P. Differential Effects of Silibinin A on Mitochondrial Function in Neuronal PC12 and HepG2 Liver Cells. Oxid. Med. Cell. Longev. 2019, 2019, 1652609. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial pathways and respiratory control. An introduction to OXPHOS analysis. Bioenerg. Commun. 2020, 2, 2. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SY5Y-APP695 | |||
|---|---|---|---|
| Ctrl | 1 µM UA | p | |
| Aβ1–40 [pg/mgProtein] | 53.97 ± 3.53 | 53.84 ± 2.15 | 0.98; ns |
| Primer | Sequence | Size [bp] | Conc [µM] | Annealing Temp. (Time) (Cycle No.) |
|---|---|---|---|---|
| ACTβ | 5′-GGACTTCGAGCAAGAGATGG-3′ 5′-AGCACTGTGTTGGCGTACAG-3′ | 234 | 0.2 | 58 °C (30 s), (45×) |
| PGK1 | 5′-CTGTGGGGGTATTTGAATGG-3′ 5′-CTTCCAGGAGCTCCAAACTG-3′ | 198 | 0.2 | 58 °C (30 s), (45×) |
| GAPDH | 5′-GAGTCAACGGATTTGGTCGT-3′ 5′-TTGATTTTGGAGGGATCTCG-3′ | 238 | 0.2 | 58 °C (30 s), (45×) |
| CS | 5′-GGGTCTGATGAAGTTTGTGG-3′ 5′-GATTAGGGAAGAAGGGACCA-3′ | 221 | 0.4 | 58 °C (30 s), (45×) |
| NDUVF1 (CI) | 5′-GCAGAAGAAGGCCATACGA-3′ 5′-CTCGCTTTATTGTCCAGCAT-3′ | 206 | 0.2 | 58 °C (30 s), (45×) |
| COX5A (CIV) | 5′-GCATGCAGACGGTTAAATGA-3′ 5′-AGTTCCTCCGGAGTGGAGAT-3′ | 152 | 0.2 | 58 °C (45 s), (45×) |
| ATP5D (CV) | 5′-CAACCAGATGTCCTTCACCT-3′ 5′-AACAACTGCACCGAAGAGTC-3′ | 240 | 0.2 | 58 °C (45 s), (45×) |
| MAP1LC3 | 5′-AGCAGCATCCAACCAAAATC-3′ 5′-CTGTGTCCGTTCACCAACAG-3′ | 187 | 0.1 | 58 °C (45 s), (45×) |
| SQSTM1 | 5′-CACCTCTCTGAGGGCTTCTC-3′ 5′AGTTTCCTGGTGGACCCATT-3′ | 97 | 0.2 | 58 °C (45 s), (45×) |
| SIRT1 | 5′-TGTGGTAGAGCTTGCATTGA-3′ 5′GCCTGTTGCTCTCCTCATTA-3′ | 153 | 02. | 58 °C (45 s), (45×) |
| CREB1 | 5′-TGGAGTTGTTATGGCATCCT-3′ 5′-ATTTTCAAGCACTGCCACTC-3′ | 169 | 0.2 | 58 °C (45 s), (45×) |
| PGC1α | 5′-CATCCCTCTGTCATCCTC-3′ 5′-GCAGACCTAGATTCAAACTC-3′ | 146 | 0.2 | 60 °C (30 s), (45×) |
| ESRRα | 5′-CCAATTCAGACTCTGTGC-3′ 5′-CTTCATACTCCAGCAGGG-3′ | 87 | 0.45 | 58 °C (30 s), (45×) |
| ESRRγ | 5′-GTGATGTGTACCATACTGTG-3′ 5′-TTAGCAGTCAAAAGTGGAAG-3′ | 198 | 0.45 | 58 °C (30 s), (45×) |
| NRF1 | 5′-GTAACCCTGATGGCACTGTC-3′ 5′-TCTGGATGGTCATCTCACT-3′ | 183 | 0.2 | 58 °C (45 s), (45×) |
| GABPα (NRF2) | 5′-CAACCTGTGCAAATTATTCC-3′ 5′-GGTGAGCTTCTATCTTCTCC-3′ | 131 | 0.45 | 58 °C (30 s), (45×) |
| TFAM | 5′-TCCCCCTTCAGTTTTGTGTA-3′ 5′-ATCAGGAAGTTCCCTCCAAC-3′ | 189 | 0.4 | 58 °C (30 s), (45×) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esselun, C.; Theyssen, E.; Eckert, G.P. Effects of Urolithin A on Mitochondrial Parameters in a Cellular Model of Early Alzheimer Disease. Int. J. Mol. Sci. 2021, 22, 8333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158333
Esselun C, Theyssen E, Eckert GP. Effects of Urolithin A on Mitochondrial Parameters in a Cellular Model of Early Alzheimer Disease. International Journal of Molecular Sciences. 2021; 22(15):8333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158333
Chicago/Turabian StyleEsselun, Carsten, Ellen Theyssen, and Gunter P. Eckert. 2021. "Effects of Urolithin A on Mitochondrial Parameters in a Cellular Model of Early Alzheimer Disease" International Journal of Molecular Sciences 22, no. 15: 8333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158333