
The Role of the IL-6 Cytokine Family in Epithelial–Mesenchymal Plasticity in Cancer Progression


Abstract
:1. Introduction
2. Role of IL-6 and OSM in Epithelial–Mesenchymal Plasticity
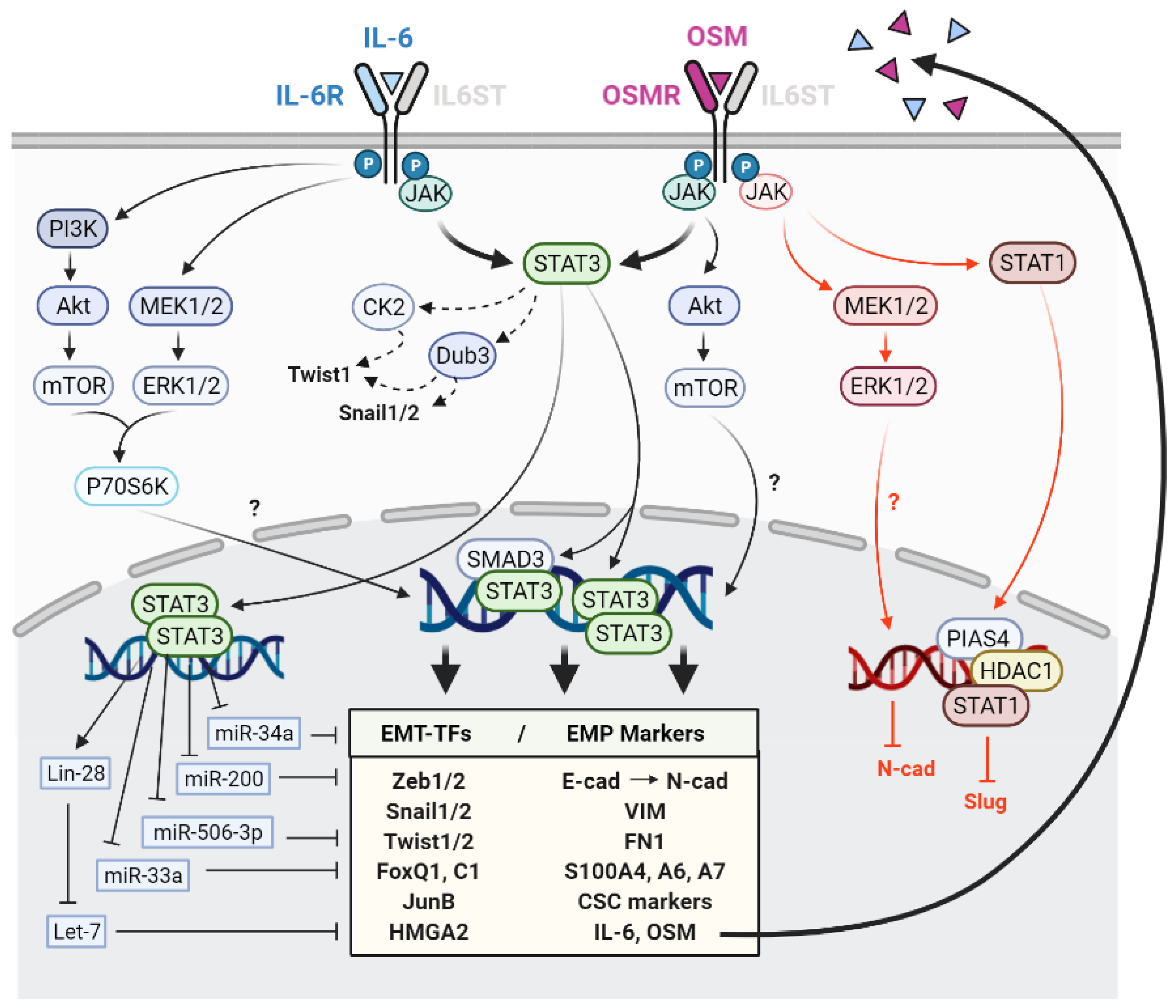
2.1. Signalling Pathways Mediating IL-6 and OSM-Induced EMP
2.2. Activation of Epithelial-Mesenchymal Transition Transcription Factors by IL-6 and OSM
2.3. EMP Regulation by IL-6 and OSM through Micro-RNAs (miRNAs)
2.4. Molecular and Functional Implications of IL-6 and OSM-Induced EMP in Cancer Cells
2.5. Association between OSM, IL-6 and EMP in Cancer Patient Samples
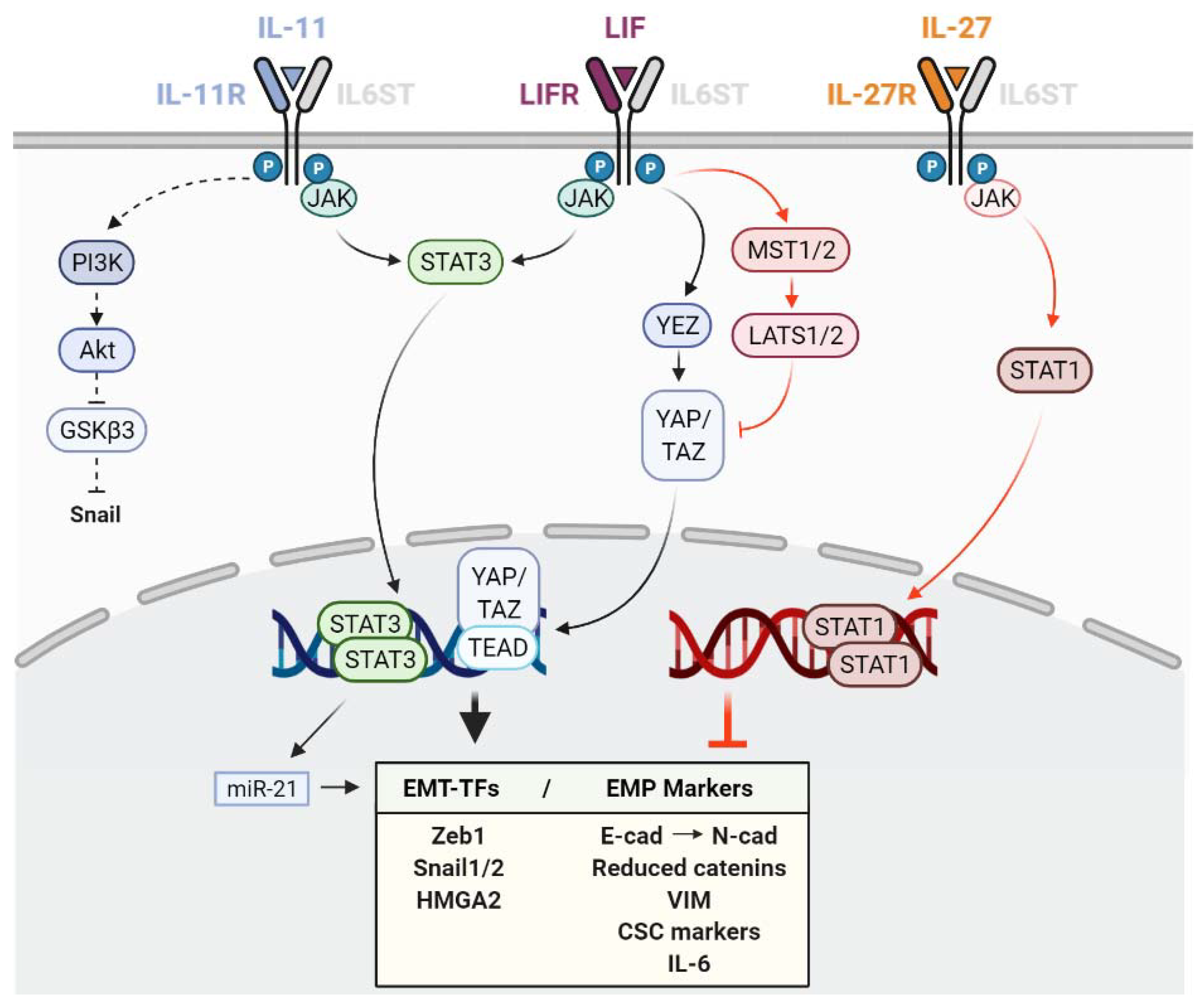
3. Role of LIF on Epithelial-Mesenchymal Plasticity
4. Role of IL-11 on Epithelial–Mesenchymal Plasticity
5. Role of IL-27 and IL-30 on Epithelial–Mesenchymal Plasticity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarin, D. The Fallacy of Epithelial Mesenchymal Transition in Neoplasia. Cancer Res. 2005, 65, 5996–6000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.W.; Newgreen, D.F. Carcinoma Invasion and Metastasis: A Role for Epithelial-Mesenchymal Transition? Cancer Res. 2005, 65, 5991–5995. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-Mesenchymal Transition Is Not Required for Lung Metastasis but Contributes to Chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; Lebleu, V.S.; Kalluri, R. Epithelial-to-Mesenchymal Transition Is Dispensable for Metastasis but Induces Chemoresistance in Pancreatic Cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a Role for EMT in Breast Cancer Metastasis. Nature 2017, 547, E1–E6. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Kang, Y. Context-Dependent EMT Programs in Cancer Metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around Epithelial–Mesenchymal Plasticity in Cancer Metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [Green Version]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.P.; Vanderhyden, B.C. Context Specificity of the EMT Transcriptional Response. Nat. Commun. 2020, 11, 2142. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci. 2020, 7, 71. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Onuchic, J.N.; Levine, H.; Ben-Jacob, E. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, H.; Melendez-Alvarez, J.R.; Chen, L.; Tian, X.J. A Plausible Accelerating Function of Intermediate States in Cancer Metastasis. PLoS Comput. Biol. 2020, 16, e1007682. [Google Scholar] [CrossRef]
- Sistigu, A.; Di Modugno, F.; Manic, G.; Nisticò, P. Deciphering the Loop of Epithelial-Mesenchymal Transition, Inflammatory Cytokines and Cancer Immunoediting. Cytokine Growth Factor Rev. 2017, 36, 67–77. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling Mechanisms of the Epithelial-Mesenchymal Transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirkan, B. The Roles of Epithelial-to-Mesenchymal Transition (EMT) and Mesenchymal-to-Epithelial Transition (MET) in Breast Cancer Bone Metastasis: Potential Targets for Prevention and Treatment. J. Clin. Med. 2013, 2, 264. [Google Scholar] [CrossRef] [Green Version]
- De Craene, B.; Berx, G. Regulatory Networks Defining EMT during Cancer Initiation and Progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and Inflammation: Inseparable Actors of Cancer Progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An Alliance towards Organ Fibrosis and Cancer Progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, C.; David, J.M.; Palena, C. Epithelial-Mesenchymal Transition and Inflammation at the Site of the Primary Tumor. Semin. Cancer Biol. 2017, 47, 177–184. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Piotrowski, I.; Kulcenty, K.; Suchorska, W. Interplay between Inflammation and Cancer. Rep. Pract. Oncol. Radiother. 2020, 25, 422–427. [Google Scholar] [CrossRef]
- Cytokines in the Balance. Nat. Immunol. 2019, 20, 1557. [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Il-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, 16295–16296. [Google Scholar] [CrossRef]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.A.; Jenkins, B.J. Recent Insights into Targeting the IL-6 Cytokine Family in Inflammatory Diseases and Cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a Keystone Cytokine in Health and Disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of Interleukin (IL)-6-Type Cytokine Signalling and Its Regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.A. The JAK/STAT Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011205. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The Molecular Basis of JAK/STAT Inhibition by SOCS1. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Carow, B.; Rottenberg, M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.; Dahmen, H.; Grimm, C.; Gendo, C.; Müller-Newen, G.; Heinrich, P.C.; Schaper, F. The Cytoplasmic Tyrosine Motifs in Full-Length Glycoprotein 130 Have Different Roles in IL-6 Signal Transduction. J. Immunol. 2000, 164, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Wendt, M.K.; Balanis, N.; Carlin, C.R.; Schiemann, W.P. STAT3 and Epithelial–Mesenchymal Transitions in Carcinomas. JAK-STAT 2014, 3, e28975. [Google Scholar] [CrossRef] [Green Version]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial–Mesenchymal Transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.H.; Chang, Y.W.; Hong, M.X.; Hsu, T.C.; Lee, K.C.; Lin, C.; Lee, J.L. STAT3 Phosphorylation at Ser727 and Tyr705 Differentially Regulates the EMT–MET Switch and Cancer Metastasis. Oncogene 2021, 40, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Junk, D.J.; Bryson, B.L.; Smigiel, J.M.; Parameswaran, N.; Bartel, C.A.; Jackson, M.W. Oncostatin M Promotes Cancer Cell Plasticity through Cooperative STAT3-SMAD3 Signaling. Oncogene 2017, 36, 4001–4013. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Jeong, J.; Seo, J.; Kim, H.S.; Kim, S.J.; Jin, W. Dysregulated JAK2 Expression by TrkC Promotes Metastasis Potential, and EMT Program of Metastatic Breast Cancer. Sci. Rep. 2016, 6, 33899. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 Promotes Head and Neck Tumor Metastasis by Inducing Epithelial-Mesenchymal Transition via the JAK-STAT3-SNAIL Signaling Pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Cheng, J.; Sun, G.; Wu, S.; Li, M.; Gao, Z.; Zhai, S.; Li, P.; Su, D.; Wang, X. P70S6K Promotes IL-6-Induced Epithelial-Mesenchymal Transition and Metastasis of Head and Neck Squamous Cell Carcinoma. Oncotarget 2016, 7, 36539–36550. [Google Scholar] [CrossRef]
- Yu, Z.; Li, Z.; Wang, C.; Pan, T.; Chang, X.; Wang, X.; Zhou, Q.; Wu, X.; Li, J.; Zhang, J.; et al. Oncostatin M Receptor, Positively Regulated by SP1, Promotes Gastric Cancer Growth and Metastasis upon Treatment with Oncostatin M. Gastric Cancer 2019, 22, 955–966. [Google Scholar] [CrossRef] [Green Version]
- West, N.R.; Murray, J.I.; Watson, P.H. Oncostatin-M Promotes Phenotypic Changes Associated with Mesenchymal and Stem Cell-like Differentiation in Breast Cancer. Oncogene 2014, 33, 1485–1494. [Google Scholar] [CrossRef]
- Bryson, B.L.; Junk, D.J.; Cipriano, R.; Jackson, M.W. STAT3-Mediated SMAD3 Activation Underlies Oncostatin M-Induced Senescence. Cell Cycle 2017, 16, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Bryson, B.L.; Tamagno, I.; Taylor, S.E.; Parameswaran, N.; Chernosky, N.M.; Balasubramaniam, N.; Jackson, M.W. Aberrant Induction of a Mesenchymal/Stem-Cell Program Engages Senescence in Normal Mammary Epithelial Cells. Mol. Cancer Res. 2020, 19. [Google Scholar] [CrossRef]
- Sterbova, S.; Karlsson, T.; Persson, E. Oncostatin M Induces Tumorigenic Properties in Non-Transformed Human Prostate Epithelial Cells, in Part through Activation of Signal Transducer and Activator of Transcription 3 (STAT3). Biochem. Biophys. Res. Commun. 2018, 498, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Colomiere, M.; Ward, A.C.; Riley, C.; Trenerry, M.K.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross Talk of Signals between EGFR and IL-6R through JAK2/STAT3 Mediate Epithelial-Mesenchymal Transition in Ovarian Carcinomas. Br. J. Cancer 2009, 100, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal Growth Factor Receptor Cooperates with Signal Transducer and Activator of Transcription 3 to Induce Epithelial-Mesenchymal Transition in Cancer Cells via up-Regulation of TWIST Gene Expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [Green Version]
- Rojas, A.; Liu, G.; Coleman, I.; Nelson, P.S.; Zhang, M.; Dash, R.; Fisher, P.B.; Plymate, S.R.; Wu, J.D. IL-6 Promotes Prostate Tumorigenesis and Progression through Autocrine Cross-Activation of IGF-IR. Oncogene 2011, 30, 2345–2355. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Lu, G.; Yao, Y.; Gu, W. An Autocrine IL-6/IGF-1R Loop Mediates Emt and Promotes Tumor Growth in Non-Small Cell Lung Cancer. Int. J. Biol. Sci. 2019, 15, 1882–1891. [Google Scholar] [CrossRef]
- Yao, C.; Su, L.; Shan, J.; Zhu, C.; Liu, L.; Liu, C.; Xu, Y.; Yang, Z.; Bian, X.; Shao, J.; et al. IGF/STAT3/NANOG/Slug Signaling Axis Simultaneously Controls Epithelial-Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer. Stem Cells 2016, 34, 820–831. [Google Scholar] [CrossRef]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.P. Senescence-Associated IL-6 and IL-8 Cytokines Induce a Self- and Cross-Reinforced Senescence/Inflammatory Milieu Strengthening Tumorigenic Capabilities in the MCF-7 Breast Cancer Cell Line. Cell Commun. Signal. 2017, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Hu, J.; Wu, X.; Liang, Z. PMA Treated THP-1-Derived-IL-6 Promotes EMT of SW48 through STAT3/ERK-Dependent Activation of Wnt/β-Catenin Signaling Pathway. Biomed. Pharmacother. 2018, 108, 618–624. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, D.; Kobayashi, S.; Wada, H.; Kawamoto, K.; Marubashi, S.; Eguchi, H.; Ishii, H.; Nagano, H.; Doki, Y.; Mori, M. Role of Crosstalk between Interleukin-6 and Transforming Growth Factor-Beta 1 in Epithelial-Mesenchymal Transition and Chemoresistance in Biliary Tract Cancer. Eur. J. Cancer 2013, 49, 1725–1740. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.R.; Parvani, J.G.; Tamagno, I.; Junk, D.J.; Bryson, B.L.; Cheon, H.J.; Stark, G.R.; Jackson, M.W. The Opposing Effects of Interferon-Beta and Oncostatin-M as Regulators of Cancer Stem Cell Plasticity in Triple-Negative Breast Cancer. Breast Cancer Res. 2019, 21, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Bai, J.; Long, Q.; Wei, Y.; Pan, J.; Li, X.; Tang, Q. TCF-3-Mediated Transcription of LncRNA HNF1A-AS1 Targeting Oncostatin M Expression Inhibits Epithelial-Mesenchymal Transition via TGFβ Signaling in Gastroenteropancreatic Neuroendocrine Neoplasms. Aging 2021, 13, 14065–14077. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, J.W.; Cho, Y.S.; Cho, C.H.; Kim, J.S.; Shin, H.W.; Chung, D.H.; Kim, S.J.; Chun, Y.S. Pathogenic Role of HIF-1α in Prostate Hyperplasia in the Presence of Chronic Inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An Epigenetic Switch Involving NF-ΚB, Lin28, Let-7 MicroRNA, and IL6 Links Inflammation to Cell Transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, H.; Bai, F.; Ding, L.; Huang, Y.; Lu, C.; Chen, S.; Li, C.; Yue, X.; Liang, X.; et al. IL-6 Promotes Metastasis of Non-Small-Cell Lung Cancer by up-Regulating TIM-4 via NF-ΚB. Cell Prolif. 2020, 53, e12776. [Google Scholar] [CrossRef]
- Li, W.; Sun, L.; Lei, J.; Wu, Z.; Ma, Q.; Wang, Z. Curcumin Inhibits Pancreatic Cancer Cell Invasion and EMT by Interfering with Tumor-Stromal Crosstalk under Hypoxic Conditions via the IL-6/ERK/NF-B Axis. Oncol. Rep. 2020, 44, 382–392. [Google Scholar] [CrossRef]
- Gao, X.; Liu, X.; Lu, Y.; Wang, Y.; Cao, W.; Liu, X.; Hu, H.; Wang, H. PIM1 Is Responsible for IL-6-Induced Breast Cancer Cell EMT and Stemness via c-Myc Activation. Breast Cancer 2019, 26, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Kan, C.E.; Cipriano, R.; Jackson, M.W. C-MYC Functions as a Molecular Switch to Alter the Response of Human Mammary Epithelial Cells to Oncostatin M. Cancer Res. 2011, 71, 6930–6939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, C.M.; Wang, M.L.; Chiou, S.H.; Chen, H.Y.; Wu, C.W. Oncostatin M Suppresses Metastasis of Lung Adenocarcinoma by Inhibiting SLUG Expression through Coordination of STATs and PIASs Signalings. Oncotarget 2016, 7, 60395–60406. [Google Scholar] [CrossRef] [Green Version]
- Sarközi, R.; Hauser, C.; Noppert, S.-J.; Kronbichler, A.; Pirklbauer, M.; Haller, V.M.; Grillari, J.; Grillari-Voglauer, R.; Mayer, G.; Schramek, H. Oncostatin M Is a Novel Inhibitor of TGF-Β1-Induced Matricellular Protein Expression. Am. J. Physiol. Physiol. 2011, 301, F1014–F1025. [Google Scholar] [CrossRef]
- Pollack, V.; Sarközi, R.; Banki, Z.; Feifel, E.; Wehn, S.; Gstraunthaler, G.; Stoiber, H.; Mayer, G.; Montesano, R.; Strutz, F.; et al. Oncostatin M-Induced Effects on EMT in Human Proximal Tubular Cells: Differential Role of ERK Signaling. Am. J. Physiol. Physiol. 2007, 293, F1714–F1726. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 Induces an Epithelial-Mesenchymal Transition Phenotype in Human Breast Cancer Cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [Green Version]
- Gyamfi, J.; Lee, Y.H.; Eom, M.; Choi, J. Interleukin-6/STAT3 Signalling Regulates Adipocyte Induced Epithelial-Mesenchymal Transition in Breast Cancer Cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Liang, S.; Ghosh, S.; Hornsby, P.J.; Li, R. Interleukin 6 Secreted from Adipose Stromal Cells Promotes Migration and Invasion of Breast Cancer Cells. Oncogene 2009, 28, 2745–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wang, Y.; Lin, Y.; Liu, Y.; Wang, Y.; Jia, J.; Singh, P.; Chi, Y.I.; Wang, C.; Dong, C.; et al. Dub3 Inhibition Suppresses Breast Cancer Invasion and Metastasis by Promoting Snail1 Degradation. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Yao, Q.; Liu, Y.; Du, S.; Liu, A.; Guo, Z.; Sun, A.; Ruan, J.; Chen, L.; Ye, C.; et al. IL-6-Induced Epithelial-Mesenchymal Transition Promotes the Generation of Breast Cancer Stem-like Cells Analogous to Mammosphere Cultures. Int. J. Oncol. 2012, 40, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Kim, G., II; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 Inflammatory Loop Mediates Trastuzumab Resistance in HER2+ Breast Cancer by Expanding the Cancer Stem Cell Population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotta, L.L.C.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 Signaling Pathway Is Required for Growth of CD44 +CD24- Stem Cell-like Breast Cancer Cells in Human Tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Zhao, Z.; Cheng, X.; Wang, Y.; Han, R.; Li, L.; Xiang, T.; He, L.; Long, H.; Zhu, B.; He, Y. Metformin Inhibits the IL-6-Induced Epithelial-Mesenchymal Transition and Lung Adenocarcinoma Growth and Metastasis. PLoS ONE 2014, 9, e95884. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.O.; Yang, X.; Duan, S.; Tsai, Y.; Strojny, L.R.; Keng, P.; Chen, Y. IL-6 Promotes Growth and Epithelial-Mesenchymal Transition of CD133+ Cells of Non-Small Cell Lung Cancer. Oncotarget 2016, 7, 6626–6638. [Google Scholar] [CrossRef] [Green Version]
- Che, D.; Zhang, S.; Jing, Z.; Shang, L.; Jin, S.; Liu, F.; Shen, J.; Li, Y.; Hu, J.; Meng, Q.; et al. Macrophages Induce EMT to Promote Invasion of Lung Cancer Cells through the IL-6-Mediated COX-2/PGE2/β-Catenin Signalling Pathway. Mol. Immunol. 2017, 90, 197–210. [Google Scholar] [CrossRef]
- Son, H.K.; Park, I.; Kim, J.Y.; Kim, D.K.; Illeperuma, R.P.; Bae, J.Y.; Lee, D.Y.; Oh, E.S.; Jung, D.W.; Williams, D.R.; et al. A Distinct Role for Interleukin-6 as a Major Mediator of Cellular Adjustment to an Altered Culture Condition. J. Cell. Biochem. 2015, 116, 2552–2562. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-W.W.; Xie, T.-X.X.; Sano, D.; Myers, J.N. IL-6 Stabilizes Twist and Enhances Tumor Cell Motility in Head and Neck Cancer Cells through Activation of Casein Kinase 2. PLoS ONE 2011, 6, e19412. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ren, G.; Wang, T.; Chen, Y.; Gong, C.; Bai, Y.; Wang, B.; Qi, H.; Shen, J.; Zhu, L.; et al. Aberrantly Expressed Fra-1 by IL-6/STAT3 Transactivation Promotes Colorectal Cancer Aggressiveness through Epithelial-Mesenchymal Transition. Carcinogenesis 2015, 36, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between Cancer Cells and Tumor Associated Macrophages Is Required for Mesenchymal Circulating Tumor Cell-Mediated Colorectal Cancer Metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef]
- Rokavec, M.; Öner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/MiR-34a Feedback Loop Promotes EMT-Mediated Colorectal Cancer Invasion and Metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.S.; Chung, I.; Wong, W.F.; Masamune, A.; Sim, M.S.; Looi, C.Y. Paracrine IL-6 Signaling Mediates the Effects of Pancreatic Stellate Cells on Epithelial-Mesenchymal Transition via Stat3/Nrf2 Pathway in Pancreatic Cancer Cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 296–306. [Google Scholar] [CrossRef]
- Kesh, K.; Garrido, V.T.; Dosch, A.; Durden, B.; Gupta, V.K.; Sharma, N.S.; Lyle, M.; Nagathihalli, N.; Merchant, N.; Saluja, A.; et al. Stroma Secreted IL6 Selects for “Stem-like” Population and Alters Pancreatic Tumor Microenvironment by Reprogramming Metabolic Pathways. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Al-Ismaeel, Q.; Neal, C.P.; Al-Mahmoodi, H.; Almutairi, Z.; Al-Shamarti, I.; Straatman, K.; Jaunbocus, N.; Irvine, A.; Issa, E.; Moreman, C.; et al. ZEB1 and IL-6/11-STAT3 Signalling Cooperate to Define Invasive Potential of Pancreatic Cancer Cells via Differential Regulation of the Expression of S100 Proteins. Br. J. Cancer 2019, 121, 65–75. [Google Scholar] [CrossRef]
- Ebbing, E.A.; Van Der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-Derived Interleukin 6 Drives Epithelial-to-Mesenchymal Transition and Therapy Resistance in Esophageal Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-Associated Fibroblasts Induce Epithelial-Mesenchymal Transition of Bladder Cancer Cells through Paracrine IL-6 Signalling. BMC Cancer 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Gong, W.; Zuo, B.; Chu, B.; Tang, Z.; Zhang, Y.; Yang, Y.; Zhou, D.; Weng, M.; Qin, Y.; et al. The MicroRNA MiR-33a Suppresses IL-6-Induced Tumor Progression by Binding Twist in Gallbladder Cancer. Oncotarget 2016, 7, 78640–78652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Shen, J.; Fang, Z.; Qiao, L.; Feng, R.; Lin, X.; Li, S. Abnormally Expressed Junb Transactivated by Il-6/Stat3 Signaling Promotes Uveal Melanoma Aggressiveness via Epithelial–Mesenchymal Transition. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, J.W.; Liu, L.J.; Huang, J. Interleukin-6-Induced Epithelial-Mesenchymal Transition through Signal Transducer and Activator of Transcription 3 in Human Cervical Carcinoma. Int. J. Oncol. 2014, 45, 165–176. [Google Scholar] [CrossRef]
- Smigiel, J.M.; Parameswaran, N.; Jackson, M.W. Potent EMT and CSC Phenotypes Are Induced by Oncostatin-M in Pancreatic Cancer. Mol. Cancer Res. 2017, 15, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Kucia-Tran, J.A.; Tulkki, V.; Smith, S.; Scarpini, C.G.; Hughes, K.; Araujo, A.M.; Yan, K.Y.M.; Botthof, J.; Pérez-Gómez, E.; Quintanilla, M.; et al. Overexpression of the Oncostatin-M Receptor in Cervical Squamous Cell Carcinoma Is Associated with Epithelial-Mesenchymal Transition and Poor Overall Survival. Br. J. Cancer 2016, 115, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-Coordinated Lin-28-Let-7-HMGA2 and MiR-200-ZEB1 Circuits Initiate and Maintain Oncostatin M-Driven Epithelial-Mesenchymal Transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef]
- Lapeire, L.; Hendrix, A.; Lambein, K.; Van Bockstal, M.; Braems, G.; Van Den Broecke, R.; Limame, R.; Mestdagh, P.; Vesompele, J.; Vanhove, C.; et al. Cancer-Associated Adipose Tissue Promotes Breast Cancer Progression by Paracrine Oncostatin M and Jak/STAT3 Signaling. Cancer Res. 2014, 74, 6806–6819. [Google Scholar] [CrossRef] [Green Version]
- Parashar, D.; Geethadevi, A.; Aure, M.R.; Mishra, J.; George, J.; Chen, C.; Mishra, M.K.; Tahiri, A.; Zhao, W.; Nair, B.; et al. MiRNA551b-3p Activates an Oncostatin Signaling Module for the Progression of Triple-Negative Breast Cancer. Cell Rep. 2019, 29, 4389–4406.e10. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.C.; Hsu, T.; Chang, Y.S.; Chung, A.K.; Jiang, S.S.; OuYang, C.N.; Yuh, C.H.; Hsueh, C.; Liu, Y.P.; Tsang, N.M. Cytoplasmic LIF Reprograms Invasive Mode to Enhance NPC Dissemination through Modulating YAP1-FAK/PXN Signaling. Nat. Commun. 2018, 9, 333. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Zhao, Y.; Zhang, C.; Li, J.; Liu, Z.; Liu, J.; Hu, W. Leukemia Inhibitory Factor Promotes EMT through STAT3- Dependent MiR-21 Induction. Oncotarget 2016, 7, 3777–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Sun, Y.; Wei, Y.; Zhang, P.; Rezaeian, A.H.; Teruya-Feldstein, J.; Gupta, S.; Liang, H.; Lin, H.K.; Hung, M.C.; et al. LIFR Is a Breast Cancer Metastasis Suppressor Upstream of the Hippo-YAP Pathway and a Prognostic Marker. Nat. Med. 2012, 18, 1511–1517. [Google Scholar] [CrossRef] [Green Version]
- Gulluoglu, S.; Sahin, M.; Tuysuz, E.C.; Yaltirik, C.K.; Kuskucu, A.; Ozkan, F.; Sahin, F.; Ture, U.; Bayrak, O.F. Leukemia Inhibitory Factor Promotes Aggressiveness of Chordoma. Oncol. Res. 2017, 25, 1177–1188. [Google Scholar] [CrossRef]
- Bian, S.; Yang, Y.; Liang, W.; Zhang, K.; Chen, L.; Zhang, Z. Leukemia Inhibitory Factor Promotes Gastric Cancer Cell Proliferation, Migration, and Invasion via the LIFR–Hippo–YAP Pathway. Ann. N. Y. Acad. Sci. 2021, 1484, 74–89. [Google Scholar] [CrossRef]
- Seeneevassen, L.; Giraud, J.; Molina-Castro, S.; Sifré, E.; Tiffon, C.; Beauvoit, C.; Staedel, C.; Mégraud, F.; Lehours, P.; Martin, O.C.B.; et al. Leukaemia Inhibitory Factor (LIF) Inhibits Cancer Stem Cells Tumorigenic Properties through Hippo Kinases Activation in Gastric Cancer. Cancers 2020, 12, 2011. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Hu, Z.; Jiang, Y.; Sun, R.; Chen, X.; Chu, H.; Zeng, M.; Sun, C. Interleukin-11 Promotes Epithelial-Mesenchymal Transition in Anaplastic Thyroid Carcinoma Cells through PI3K/Akt/GSK3β Signaling Pathway Activation. Oncotarget 2016, 7, 59652–59663. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Liu, Y.; Liu, R.; Qi, J.; Hou, Y.; Chang, J.; Ren, L. Upregulation of IL-11, an IL-6 Family Cytokine, Promotes Tumor Progression and Correlates with Poor Prognosis in Non-Small Cell Lung Cancer. Cell. Physiol. Biochem. 2018, 45, 2213–2224. [Google Scholar] [CrossRef]
- Peng, N.; Lu, M.; Kang, M.; Liu, X.; Li, B.; Dong, C. Recombinant Human IL-11 Promotes Lung Adenocarcinoma A549 Cell Growth and EMT through Activating STAT3/HIF-1α/EMT Signaling Pathway. Anticancer Agents Med. Chem. 2020, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bockhorn, J.; Dalton, R.; Nwachukwu, C.; Huang, S.; Prat, A.; Yee, K.; Chang, Y.F.; Huo, D.; Wen, Y.; Swanson, K.E.; et al. MicroRNA-30c Inhibits Human Breast Tumour Chemotherapy Resistance by Regulating TWF1 and IL-11. Nat. Commun. 2013, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, Y.; Xu, X.; Cao, H.; Sahengbieke, S.; Sheng, H.; Huang, Q.; Lai, M. Transcriptional Activation of FN1 and IL11 by HMGA2 Promotes the Malignant Behavior of Colorectal Cancer. Carcinogenesis 2016, 37, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Airoldi, I.; Tupone, M.G.; Esposito, S.; Russo, M.V.; Barbarito, G.; Cipollone, G.; Di Carlo, E. Interleukin-27 Re-Educates Intratumoral Myeloid Cells and down-Regulates Stemness Genes in Non-Small Cell Lung Cancer. Oncotarget 2015, 6, 3694–3708. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Tai, W.; Lei, W.; Wang, Y.; Li, Z.; Zhang, T. IL-27 Inhibits the TGF-Β1-Induced Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells. BMC Cell Biol. 2016, 17, 7. [Google Scholar] [CrossRef] [Green Version]
- Kachroo, P.; Lee, M.H.; Zhang, L.; Baratelli, F.; Lee, G.; Srivastava, M.K.; Wang, G.; Walser, T.C.; Krysan, K.; Sharma, S.; et al. IL-27 Inhibits Epithelial-Mesenchymal Transition and Angiogenic Factor Production in a STAT1-Dominant Pathway in Human Non-Small Cell Lung Cancer. J. Exp. Clin. Cancer Res. 2013, 32, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Wang, Y.; Shi, Q.; Yu, Q.; Liu, C.; Feng, J.; Deng, J.; Mark Evers, B.; Zhou, B.P.; Wu, Y. Stabilization of the Transcription Factors Slug and Twist by the Deubiquitinase Dub3 Is a Key Requirement for Tumor Metastasis. Oncotarget 2017, 8, 75127–75140. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.G.; Patel, V.; Roue, N.C.; Fok, S.Y.; Soon, L.L.; Halliday, G.M.; Gutkind, J.S. Snail Up-Regulates Proinflammatory Mediators and Inhibits Differentiation in Oral Keratinocytes. Cancer Res. 2008, 68, 4525–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Carmona, M.; Bourcy, M.; Lesage, J.; Leroi, N.; Syne, L.; Blacher, S.; Hubert, P.; Erpicum, C.; Foidart, J.M.; Delvenne, P.; et al. Soluble Factors Regulated by Epithelial-Mesenchymal Transition Mediate Tumour Angiogenesis and Myeloid Cell Recruitment. J. Pathol. 2015, 236, 491–504. [Google Scholar] [CrossRef]
- Katsura, A.; Tamura, Y.; Hokari, S.; Harada, M.; Morikawa, M.; Sakurai, T.; Takahashi, K.; Mizutani, A.; Nishida, J.; Yokoyama, Y.; et al. ZEB1-Regulated Inflammatory Phenotype in Breast Cancer Cells. Mol. Oncol. 2017, 11, 1241–1262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ma, L. MicroRNA Control of Epithelial-Mesenchymal Transition and Metastasis. Cancer Metastasis Rev. 2012, 31, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The MiR-200 Family and MiR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The MiR-200 Family Determines the Epithelial Phenotype of Cancer Cells by Targeting the E-Cadherin Repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A P53/MiRNA-34 Axis Regulates Snail1-Dependent Cancer Cell Epithelial-Mesenchymal Transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Siemens, H.; Jackstadt, R.; Hünten, S.; Kaller, M.; Menssen, A.; Götz, U.; Hermeking, H. MiR-34 and SNAIL Form a Double-Negative Feedback Loop to Regulate Epithelial-Mesenchymal Transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. P53 Regulates Epithelial-Mesenchymal Transition and Stem Cell Properties through Modulating MiRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Lin, C.P.; Ho, J.J.; He, X.; Okada, N.; Bu, P.; Zhong, Y.; Kim, S.Y.; Bennett, M.J.; Chen, C.; et al. MiR-34 MiRNAs Provide a Barrier for Somatic Cell Reprogramming. Nat. Cell Biol. 2011, 13, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.S.; Tseng, H.Y.; Chen, Y.A.; Shen, P.C.; Al Haq, A.T.; Chen, L.M.; Tung, Y.C.; Hsu, H.L. MCT-1/MiR-34a/IL-6/IL-6R Signaling Axis Promotes EMT Progression, Cancer Stemness and M2 Macrophage Polarization in Triple-Negative Breast Cancer. Mol. Cancer 2019, 18, 42. [Google Scholar] [CrossRef] [Green Version]
- Biddle, A.; Mackenzie, I.C. Cancer Stem Cells and EMT in Carcinoma. Cancer Metastasis Rev. 2012, 31, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, Cancer Stem Cells and Drug Resistance: An Emerging Axis of Evil in the War on Cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- May, C.D.; Sphyris, N.; Evans, K.W.; Werden, S.J.; Guo, W.; Mani, S.A. Epithelial-Mesenchymal Transition and Cancer Stem Cells: A Dangerously Dynamic Duo in Breast Cancer Progression. Breast Cancer Res. 2011, 13, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, V.L.; Lim, C.L. Epithelial-Mesenchymal Plasticity—Engaging Stemness in an Interplay of Phenotypes. Stem Cell Investig. 2019, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.B. Interleukin-6 Induces Increased Motility, Cell-Cell and Cell-Substrate Dyshesion and Epithelial-to-Mesenchymal Transformation in Breast Cancer Cells. Oncogene 2010, 29, 2599–2600. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Liu, Q.; Liao, Q.; Wu, Q.; Sun, B.; Yang, Z.; Hu, X.; Tan, M.; Li, L. Interleukin-6/Signal Transducer and Activator of Transcription 3 Promotes Prostate Cancer Resistance to Androgen Deprivation Therapy via Regulating Pituitary Tumor Transforming Gene 1 Expression. Cancer Sci. 2018, 109, 678–687. [Google Scholar] [CrossRef]
- Giladi, N.D.; Ziv-Av, A.; Lee, H.K.; Finniss, S.; Cazacu, S.; Xiang, C.; Ben-Asher, H.W.; de Carvalho, A.; Mikkelsen, T.; Poisson, L.; et al. RTVP-1 Promotes Mesenchymal Transformation of Glioma via a STAT-3/IL-6-Dependent Positive Feedback Loop. Oncotarget 2015, 6, 22680–22697. [Google Scholar] [CrossRef]
- Bharti, R.; Dey, G.; Mandal, M. Cancer Development, Chemoresistance, Epithelial to Mesenchymal Transition and Stem Cells: A Snapshot of IL-6 Mediated Involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-Renewal of CD133hi Cells by IL6/Notch3 Signalling Regulates Endocrine Resistance in Metastatic Breast Cancer. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Han, R.; Xiao, H.; Lin, C.; Wang, Y.; Liu, H.; Li, K.; Chen, H.; Sun, F.; Yang, Z.; et al. Metformin Sensitizes EGFR-TKI-Resistant Human Lung Cancer Cells in Vitro and in Vivo through Inhibition of IL-6 Signaling and EMT Reversal. Clin. Cancer Res. 2014, 20, 2714–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Fenoglio, S.; Gao, D.C.; Camiolo, M.; Stiles, B.; Lindsted, T.; Schlederer, M.; Johns, C.; Altorki, N.; Mittal, V.; et al. TGF-β IL-6 Axis Mediates Selective and Adaptive Mechanisms of Resistance to Molecular Targeted Therapy in Lung Cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 15535–15540. [Google Scholar] [CrossRef] [Green Version]
- Fofaria, N.M.; Srivastava, S.K. Critical role of STAT3 in melanoma metastasis through anoikis resistance. Oncotarget 2014, 5, 7051–7064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehner, C.; Miller, E.; Hockla, A.; Coban, M.; Weroha, S.J.; Radisky, D.C.; Radisky, E.S. Targeting an autocrine IL-6-SPINK1 signaling axis to suppress metastatic spread in ovarian clear cell carcinoma. Oncogene 2020, 39, 6606–6618. [Google Scholar] [CrossRef]
- Fofaria, N.M.; Srivastava, S.K. STAT3 induces anoikis resistance, promotes cell invasion and metastatic potential in pancreatic cancer cells. Carcinogenesis 2015, 36, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Chen, H.; Duan, C.; Liu, D.; Qian, L.; Yang, Z.; Guo, L.; Song, L.; Yu, M.; Hu, M.; et al. Deficiency of Erbin induces resistance of cervical cancer cells to anoikis in a STAT3-dependent manner. Oncogenesis 2013, 2, e52. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [Green Version]
- Covert, H.; Mellor, L.F.; Wolf, C.L.; Ankenbrandt, N.; Emathinger, J.M.; Tawara, K.; Oxford, J.T.; Jorcyk, C.L. OSM-Induced CD44 Contributes to Breast Cancer Metastatic Potential through Cell Detachment but Not Epithelial-Mesenchymal Transition. Cancer Manag. Res. 2019, 11, 7721–7737. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Ichikawa, T.; Kurozumi, K.; Otani, Y.; Fujimura, A.; Fujii, K.; Tomita, Y.; Hattori, Y.; Uneda, A.; Tsuboi, N.; et al. Annexin A2-STAT3-Oncostatin M Receptor Axis Drives Phenotypic and Mesenchymal Changes in Glioblastoma. Acta Neuropathol. Commun. 2020, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Chanoch-Myers, R.; Mathewson, N.D.; Myskiw, C.; Atta, L.; Bussema, L.; Eichhorn, S.W.; Greenwald, A.C.; Kinker, G.S.; Rodman, C.; et al. Interactions between Cancer Cells and Immune Cells Drive Transitions to Mesenchymal-like States in Glioblastoma. Cancer Cell 2021, 39, 779.e11–792.e11. [Google Scholar] [CrossRef]
- Tawara, K.; Scott, H.; Emathinger, J.; Wolf, C.; LaJoie, D.; Hedeen, D.; Bond, L.; Montgomery, P.; Jorcyk, C. HIGH Expression of OSM and IL-6 Are Associated with Decreased Breast Cancer Survival: Synergistic Induction of IL-6 Secretion by OSM and IL-1β. Oncotarget 2019, 10, 2068–2085. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Gong, W.; Zhang, Y.; Yang, Y.; Zhou, D.; Weng, M.; Qin, Y.; Jiang, A.; Ma, F.; Quan, Z. Expression of Interleukin-6 Is Associated with Epithelial-Mesenchymal Transition and Survival Rates in Gallbladder Cancer. Mol. Med. Rep. 2015, 11, 3539–3546. [Google Scholar] [CrossRef] [Green Version]
- Bottai, G.; Diao, L.; Baggerly, K.A.; Paladini, L.; Győrffy, B.; Raschioni, C.; Pusztai, L.; Calin, G.A.; Santarpia, L. Integrated MicroRNA–MRNA Profiling Identifies Oncostatin M as a Marker of Mesenchymal-like ER-Negative/HER2-Negative Breast Cancer. Int. J. Mol. Sci. 2017, 18, 194. [Google Scholar] [CrossRef] [Green Version]
- Onishi, K.; Zandstra, P.W. LIF Signaling in Stem Cells and Development. Development 2015, 142, 2230–2236. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, M.; Wang, Z.; Yu, X.; Song, Y.; Wang, C.; Xu, Y.; Wei, F.; Zhao, Y.; Xu, Y. Long Non-Coding RNA-CTD-2108O9.1 Represses Breast Cancer Metastasis by Influencing Leukemia Inhibitory Factor Receptor. Cancer Sci. 2018, 109, 1764–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massagué, J. Breast Cancer Bone Metastasis Mediated by the Smad Tumor Suppressor Pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar] [CrossRef] [Green Version]
- Deckers, M.; Van Dinther, M.; Buijs, J.; Que, I.; Löwik, C.; Van Der Pluijm, G.; Ten Dijke, P. The Tumor Suppressor Smad4 Is Required for Transforming Growth Factor β-Induced Epithelial to Mesenchymal Transition and Bone Metastasis of Breast Cancer Cells. Cancer Res. 2006, 66, 2202–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, T.; Chiba, Y.; Furusawa, J.I.; Xu, M.; Tsunoda, R.; Higuchi, K.; Mizoguchi, I. Potential Clinical Application of Interleukin-27 as an Antitumor Agent. Cancer Sci. 2015, 106, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.; Shi, W.; Li, P.; Wu, Y.; Li, Y.; Bao, C. The Mechanism of and the Association between Interleukin-27 and Chemotherapeutic Drug Sensitivity in Lung Cancer. Oncol. Lett. 2021, 21. [Google Scholar] [CrossRef]
- Di Carlo, E. Decoding the Role of Interleukin-30 in the Crosstalk between Cancer and Myeloid Cells. Cells 2020, 9, 615. [Google Scholar] [CrossRef] [Green Version]
- Di Carlo, E. Interleukin-30: A Novel Microenvironmental Hallmark of Prostate Cancer Progression. Oncoimmunology 2014, 3, e27618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airoldi, I.; Cocco, C.; Sorrentino, C.; Angelucci, D.; Di Meo, S.; Manzoli, L.; Esposito, S.; Ribatti, D.; Bertolotto, M.; Iezzi, L.; et al. Interleukin-30 Promotes Breast Cancer Growth and Progression. Cancer Res. 2016, 76, 6218–6229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, S.; Airoldi, I.; Sorrentino, C.; Zorzoli, A.; Esposito, S.; Di Carlo, E. Interleukin-30 Expression in Prostate Cancer and Its Draining Lymph Nodes Correlates with Advanced Grade and Stage. Clin. Cancer Res. 2014, 20, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Ciummo, S.L.; Cipollone, G.; Caputo, S.; Bellone, M.; Di Carlo, E. Interleukin-30/Il27p28 Shapes Prostate Cancer Stem-like Cell Behavior and Is Critical for Tumor Onset and Metastasization. Cancer Res. 2018, 78, 2654–2668. [Google Scholar] [CrossRef] [Green Version]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in Tumorigenesis. JAK-STAT 2012, 1, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Xu, Y.; Li, Y.; Xu, W.; Luo, F.; Wang, B.; Pang, Y.; Xiang, Q.; Zhou, J.; Wang, X.; et al. NF-ΚB-Mediated Inflammation Leading to EMT via MiR-200c Is Involved in Cell Transformation Induced by Cigarette Smoke Extract. Toxicol. Sci. 2013, 135, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, J.; Parvani, J.G.; Tamagno, I.; Polak, K.; Jackson, M.W. Breaking the Oncostatin M Feed-Forward Loop to Suppress Metastasis and Therapy Failure. J. Pathol. 2018, 245, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Cancer Type | Main Pathway | EMT-TFs | EMT Markers | References |
|---|---|---|---|---|
| Breast | STAT3 | Snail Twist1 Zeb1 | Mesenchymal-like morphology Decreased E-cadherin Increased N-cadherin, vimentin, CSC markers (CD24low/CD44high) | Walter 2009 [74]; Sullivan 2009 [72]; Gyamfi 2018 [73]; Y Wu 2017 [75]; Xie 2012 [76]; Korkaya 2012 [77]; Marotta 2011 [78] |
| Lung | STAT3 | Snail Slug Twist1 | Mesenchymal-like morphology Decreased E-cadherin Increased vimentin, nuclear β-catenin, CSC markers (CD133) | Zhao 2014 [79]; Shintani 2016 [59]; Lee 2016 [80]; Liu 2020 [65]; Che 2017 [81] |
| Head and neck | STAT3 | Snail Twist1 | Decreased E-cadherin Increased vimentin | Son 2015 [82]; Yadav 2011 [45]; Su 2011 [83] |
| Colon | STAT3 | Snail Slug Zeb1 TCF3 Fra-1 FoxQ1 | Mesenchymal-like morphology Decreased E-cadherin, ZO-1 Increased N-cadherin, vimentin, fibronectin, β-catenin, cell scattering | Liu 2015 [84]; Wei 2019 [85]; Rokavec 2014 [86]; Gao 2018 [58] |
| Pancreas | STAT3 | Snail Slug Zeb2 Twist2 Nrf2 | Mesenchymal-like morphology Decreased E-cadherin Increased N-cadherin, vimentin, fibronectin, collagen I, CSC markers (CD133), S100A4, S100A6, MMP9 | YS Wu 2017 [87]; Kesh 2020 [88]; Al-Ismaeel 2019 [89]; Li 2020 [66] |
| Oesophagus | STAT3 | Zeb1 Slug | Mesenchymal-like morphology Decreased E-cadherin, CK19, EPCAM Increased N-cadherin, vimentin, CSC markers (CD24low/CD44high, CD133) | Ebbing 2019 [90] |
| Bladder | Unknown | Snail Twist1 Zeb1 | Decreased E-cadherin, β-catenin Increased N-cadherin, vimentin | Goulet 2019 [91] |
| Gallbladder | Unknown | Twist1 | Increased CSC markers (CD44, CD133) | Zhang 2016 [92] |
| Uveal melanoma | STAT3 | JunB | Decreased E-cadherin, TJP2, TJP2 Increased fibronectin and fibronectin receptors, ICAM1 | Gong 2018 [93] |
| Cervix | STAT3 | Unknown | Mesenchymal-like morphology Decreased E-cadherin Increased vimentin, cell scattering | Miao 2014 [94] |
| Cancer Type | Main Pathway | EMT-TFs | EMT Markers | References |
|---|---|---|---|---|
| Pancreas | STAT3 | Snail Zeb1 | Mesenchymal-like morphology Decreased E-cadherin CSC markers (CD24low/CD44high) | Smigiel 2017 [95] |
| Cervix | STAT3 | Snail Zeb2 | Mesenchymal-like morphology Reduced E-cadherin, cell cohesion Increased vimentin, fibronectin, MMP9, MMP10 | Kucia-Tran 2016 [96] |
| Breast | STAT3 and PI3K/AKT | Snail Slug Zeb1 FoxC1 | Mesenchymal-like morphology Decreased E-cadherin, claudin-1, β-catenin Increased N-cadherin, vimentin, fibronectin, MMPs, S100A7, CSC markers (CD24low/CD44 high) | Doherty 2019 [61]; Guo 2013 [97]; Junk 2017 [43]; West 2014 [48]; Lapeire 2014 [98]; Bryson 2017 [49]; Parashar 2019 [99] |
| Prostate | STAT3 | Snail | Mesenchymal-like morphology, Decreased E-cadherin Increased vimentin | Sterbova 2018 [51] |
| Stomach | STAT3 | Decreased E-cadherin Increased N-cadherin | Yu 2019 [47] | |
| Lung | STAT1 | Slug | Increased E-cadherin | Pan 2016 [69] |
| Kidney | STAT3 | Decreased E-cadherin, claudin-2 Increased vimentin, collagen I, S100A4, cell scattering | Pollack 2007 [71] | |
| ERK1/2 | Decreased N-cadherin | Pollack 2007 [71] | ||
| STAT1 | FoxC2 | Decreased secretion of matricellular proteins (SPARC, thrombospondin, CTGF, TNC) | Sarközi 2011 [70] |
| Cytokine | Cancer Type | Main Pathway | EMT-TFs | EMT Markers | References |
|---|---|---|---|---|---|
| LIF | Head and Neck | Hippo pathway (YAP/FAK/PXN) | Mesenchymal-like morphology Decreased E-cadherin Increased N-cadherin, vimentin, IQAP1 | Liu 2018 [100] | |
| LIF | Colon | STAT3 | Mesenchymal-like morphology Decreased E-cadherin Increased N-cadherin, vimentin | Yue 2016 [101] | |
| LIF | Breast | STAT3 | Mesenchymal-like morphology Decreased E-cadherin Increased N-cadherin, vimentin | Yue 2016 [101] | |
| STAT3/ Hippo (MST/LATS/YAP) | Decreased YAP | Chen 2012 [102] | |||
| LIF | Chondroma | Zeb2 | Decreased E-cadherin, CK19, Increased CSC markers (CD15, CD133) | Gulluoglu 2017 [103] | |
| IL-11 | Stomach | Hippo pathway (Hippo/ YAP) | Decreased E-cadherin Increased YAP, MMP7 | Bian 2020 [104] | |
| STAT3/ Hippo (MST/LATS/YAP) | Decreased YAP and stemness | Seeneevassen 2020 [105] | |||
| IL-11 | Thyroid | PI3K/AKT | Snail | Decreased E-cadherin, ZO-1 Increased vimentin | Zhong 2016 [106] |
| IL-11 | Lung | STAT3 | Snail Slug Twist1 | Decreased E-cadherin, ZO-1, claudin-1 Increased N-cadherin, vimentin | Zhao 2018 [107] Peng 2020 [108] |
| IL-11 | Breast | Mesenchymal-like morphology F-actin organization, increased focal adhesions | Bockhorn 2013 [109] | ||
| IL-11 | Colon | STAT3 | HMGA2 | Increased vimentin | Wu 2016 [110] |
| IL-11 | Pancreas | STAT3 | Mesenchymal-like morphology Increased S100A4, S100A6 | Ai-Ismaeel 2019 [89] | |
| IL-27 | Lung | STAT1 | Snail Slug Zeb1 | Increased E-cadherin, β-catenin, γ-catenin Reduced N-cadherin, vimentin, CSC markers (SHH, OCT4A, SOX2, SOX9, NOTCH1, KLF4, nestin) | Airoldi 2015 [111]; Dong 2016 [112]; Kachroo 2013 [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abaurrea, A.; Araujo, A.M.; Caffarel, M.M. The Role of the IL-6 Cytokine Family in Epithelial–Mesenchymal Plasticity in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 8334. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158334
Abaurrea A, Araujo AM, Caffarel MM. The Role of the IL-6 Cytokine Family in Epithelial–Mesenchymal Plasticity in Cancer Progression. International Journal of Molecular Sciences. 2021; 22(15):8334. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158334
Chicago/Turabian StyleAbaurrea, Andrea, Angela M. Araujo, and Maria M. Caffarel. 2021. "The Role of the IL-6 Cytokine Family in Epithelial–Mesenchymal Plasticity in Cancer Progression" International Journal of Molecular Sciences 22, no. 15: 8334. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158334