
The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses

 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. α-Syn Aggregation and Cytotoxicity
3. α-Syn Cell-to-Cell Propagation
4. α-Syn Propagation in the Clinical Pathology of PD: Models and Hypotheses
4.1. ‘Dual-Hit’ Hypothesis
4.2. Selective Neuronal Vulnerability/Threshold Hypothesis
5. Future Prospects and Opportunities
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Parkinson’s Disease; Oxford University Press: Oxford, NY, USA, 2010; p. 116. [Google Scholar]
- Frank, C.; Pari, G.; Rossiter, J.P. Approach to diagnosis of Parkinson disease. Can. Fam. Physician Med. Fam. Can. 2006, 52, 862–868. [Google Scholar]
- Gray, F.O.; Duyckaerts, C.; De Girolami, U.; Escourolle, R.; Gray, F.O. Escourolle & Poirier’s Manual of Basic Neuropathology, 5th ed.; Oxford University Press: Oxford, NY, USA, 2014; p. xiii. 406p. [Google Scholar]
- Rajput, A.H.; Voll, A.; Rajput, M.L.; Robinson, C.A. Course in Parkinson disease subtypes: A 39-year clinicopathologic study. Neurology 2009, 73, 206–212. [Google Scholar] [CrossRef]
- Selikhova, M.; Williams, D.R.; Kempster, P.A.; Holton, J.L.; Revesz, T.; Lees, A.J. A clinico-pathological study of subtypes in Parkinson’s disease. Brain 2009, 132, 2947–2957. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. Is Braak staging valid for all types of Parkinson’s disease? J. Neural Transm. 2018, 126, 423–431. [Google Scholar] [CrossRef]
- Dickson, D.W.; Fujishiro, H.; Orr, C.; DelleDonne, A.; Josephs, K.A.; Frigerio, R.; Burnett, M.; Parisi, J.E.; Klos, K.J.; Ahlskog, J.E. Neuropathology of non-motor features of Parkinson disease. Park. Relat. Disord. 2009, 15, S1–S5. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 509. [Google Scholar] [CrossRef]
- Dickson, D.W. Neuropathology of Parkinson disease. Park. Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef]
- McCann, H.; Stevens, C.; Cartwright, H.; Halliday, G. α-Synucleinopathy phenotypes. Park. Relat. Disord. 2014, 20, S62–S67. [Google Scholar] [CrossRef] [Green Version]
- Scudamore, O.; Ciossek, T. Increased Oxidative Stress Exacerbates α-Synuclein Aggregation In Vivo. J. Neuropathol. Exp. Neurol. 2018, 77, 443–453. [Google Scholar] [CrossRef]
- Beal, M.F. Mitochondria, Oxidative Damage, and Inflammation in Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2006, 991, 120–131. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, Y.C.W.D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Schaeffer, W.J. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2012, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Sandmann-Keil, D.; Gai, W.P.; De Vos, R.A.I.; Steur, E.N.H.J.; Arai, K.; Braak, E. Parkinson’s disease: Affection of brain stem nuclei controlling premotor and motor neurons of the somatomotor system. Acta Neuropathol. 2000, 99, 489–495. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Lue, L.; Sue, L.I.; Bachalakuri, J.; Henry-Watson, J.; Sasse, J.; Boyer, S.; Shirohi, S.; Brooks, R.; et al. Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009, 117, 613–634. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, A.A.; Voorn, P.; Berendse, H.W.; Groenewegen, H.J.; Rozemuller, A.J.; van de Berg, W.; Bank, N.B. Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson’s disease. Mov. Disord. 2014, 29, 1244–1251. [Google Scholar] [CrossRef]
- Uchihara, T.; Giasson, B.I. Propagation of alpha-synuclein pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2015, 131, 49–73. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Lassen, L.B.; Reimer, L.; Ferreira, N.; Betzer, C.; Jensen, P.H. Protein Partners of α-Synuclein in Health and Disease. Brain Pathol. 2016, 26, 389–397. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and Dynamics of Micelle-bound Human α-Synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, J.M.; Giasson, B.I.; Lee, V.M.-Y.; Ischiropoulos, H. Chaperone-like activity of synucleins. FEBS Lett. 2000, 474, 116–119. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Murray, I.; Trojanowski, J.Q.; Lee, V.M.-Y. A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of α-Synuclein Is Essential for Filament Assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.B.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2003, 55, 164–173. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.; Südhof, T.C. Properties of native brain α-synuclein. Nat. Cell Biol. 2013, 498, E4–E6. [Google Scholar] [CrossRef]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Mapping Long-Range Interactions in α-Synuclein using Spin-Label NMR and Ensemble Molecular Dynamics Simulations. J. Am. Chem. Soc. 2005, 127, 476–477. [Google Scholar] [CrossRef]
- Bertoncini, C.W.; Jung, Y.-S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured -synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a Partially Folded Intermediate in α-Synuclein Fibril Formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.-P.; Weinstock, D.S.; Narayanan, C.; Levy, R.M.; Baum, J. Structural Reorganization of α-Synuclein at Low pH Observed by NMR and REMD Simulations. J. Mol. Biol. 2009, 391, 784–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtilerman, M.D.; Ding, A.T.T.; Lansbury, J.P.T. Molecular Crowding Accelerates Fibrillization of α-Synuclein: Could an Increase in the Cytoplasmic Protein Concentration Induce Parkinson’s Disease? Biochemistry 2002, 41, 3855–3860. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered Structural Transformations, Aggregation, and Fibrillation of Human α-Synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [Green Version]
- Munishkina, L.A.; Phelan, C.; Uversky, V.N.; Fink, A.L. Conformational Behavior and Aggregation of α-Synuclein in Organic Solvents: Modeling the Effects of Membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar] [CrossRef]
- Galvagnion, C.; Brown, J.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohlberg, J.A.; Li, J.; Uversky, A.V.N.; Fink, A.L. Heparin and Other Glycosaminoglycans Stimulate the Formation of Amyloid Fibrils from α-Synuclein in Vitro. Biochemistry 2002, 41, 1502–1511. [Google Scholar] [CrossRef]
- Goers, J.; Uversky, V.N.; Fink, A.L. Polycation-induced oligomerization and accelerated fibrillation of human alpha-synuclein in vitro. Protein Sci. 2003, 12, 702–707. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Pesticides directly accelerate the rate of α-synuclein fibril formation: A possible factor in Parkinson’s disease. FEBS Lett. 2001, 500, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Engelender, S.; Kaminsky, Z.; Guo, X.; Sharp, A.H.; Amaravi, R.K.; Kleiderlein, J.J.; Margolis, R.L.; Troncoso, J.C.; Lanahan, A.A.; Worley, P.F.; et al. Synphilin-1 associates with α-synuclein and promotes the formation of cytosolic inclusions. Nat. Genet. 1999, 22, 110–114. [Google Scholar] [CrossRef]
- Lindersson, E.; Lundvig, D.; Petersen, C.; Madsen, P.S.; Nyengaard, J.R.; Højrup, P.; Moos, T.; Otzen, D.; Gai, W.-P.; Blumbergs, P.C.; et al. p25α Stimulates α-Synuclein Aggregation and Is Co-localized with Aggregated α-Synuclein in α-Synucleinopathies. J. Biol. Chem. 2005, 280, 5703–5715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.; Gram, H.; Sorrentino, Z.A.; Gregersen, E.; Schmidt, S.I.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M.; et al. Multiple system atrophy-associated oligodendroglial protein p25α stimulates formation of novel α-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 142, 87–115. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of α-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 Is Enhanced in Synucleinopathies, Inhibits α-Synuclein Oligomerization, and Influences Synuclein-Membrane Interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamashita, H.; Takahashi, T.; Nakamura, S. Activated Fyn Phosphorylates α-Synuclein at Tyrosine Residue 125. Biochem. Biophys. Res. Commun. 2001, 280, 1085–1092. [Google Scholar] [CrossRef]
- Kofoed, R.H.; Zheng, J.; Ferreira, N.; Lykke-Andersen, S.; Salvi, M.; Betzer, C.; Reimer, L.; Jensen, T.H.; Fog, K.; Jensen, P.H. Polo-like kinase 2 modulates α-synuclein protein levels by regulating its mRNA production. Neurobiol. Dis. 2017, 106, 49–62. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated α-Synuclein Is Ubiquitinated in α-Synucleinopathy Lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef] [Green Version]
- Tofaris, G.; Razzaq, A.; Ghetti, B.; Lilley, K.S.; Spillantini, M.G. Ubiquitination of α-Synuclein in Lewy Bodies Is a Pathological Event Not Associated with Impairment of Proteasome Function. J. Biol. Chem. 2003, 278, 44405–44411. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.-Y. Oxidative Damage Linked to Neurodegeneration by Selective alpha -Synuclein Nitration in Synucleinopathy Lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the Role of Site-Specific Nitration of α-Synuclein in the Pathogenesis of Parkinson’s Disease via Protein Semisynthesis and Mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Elghani, F.A.; Bandopadhyay, R.; Engelender, S. SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.-H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits α-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowther, R.; Jakes, R.; Spillantini, M.G.; Goedert, M. Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett. 1998, 436, 309–312. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.-H.; Kim, Y.-J.; Kim, Y.-G.; Joh, T.H.; Beal, M.F.; Kim, Y.-S. Role of Matrix Metalloproteinase 3-mediated α-Synuclein Cleavage in Dopaminergic Cell Death. J. Biol. Chem. 2011, 286, 14168–14177. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Janowska, M.K.; Moriarty, G.M.; Baum, J. Mechanistic Insight into the Relationship between N-Terminal Acetylation of α-Synuclein and Fibril Formation Rates by NMR and Fluorescence. PLoS ONE 2013, 8, e75018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikiy, I.; Eliezer, D. N-terminal Acetylation Stabilizes N-terminal Helicity in Lipid- and Micelle-bound α-Synuclein and Increases Its Affinity for Physiological Membranes. J. Biol. Chem. 2014, 289, 3652–3665. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Fournier, M.; Lashuel, H.A. Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 2010, 183, 115–145. [Google Scholar] [CrossRef]
- Oueslati, A. Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade? J. Park. Dis. 2016, 6, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Burgold, S.; Filser, S.; Dorostkar, M.M.; Schmidt, B.; Herms, J. In vivo imaging reveals sigmoidal growth kinetic of beta-amyloid plaques. Acta. Neuropathol. Com. 2014, 2, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, G.; Udgaonkar, J.B. Understanding the Kinetic Roles of the Inducer Heparin and of Rod-like Protofibrils during Amyloid Fibril Formation by Tau Protein. J. Biol. Chem. 2011, 286, 38948–38959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, M.J.; Magalhaes, J.; Ferreira, N.; Almeida, M. Transthyretin Deposition in Familial Amyloidotic Polyneuropathy. Curr. Med. Chem. 2012, 19, 2304–2311. [Google Scholar] [CrossRef]
- Sant’Anna, R.; Gallego, P.; Robinson, L.Z.; Pereira-Henriques, A.; Ferreira, N.; Pinheiro, F.; Esperante, S.; Pallares, I.; Huertas, O.; Almeida, M.; et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat. Commun. 2016, 7, 10787. [Google Scholar] [CrossRef]
- Ferreira, N.; Saraiva, M.J.; Almeida, M. Natural polyphenols inhibit different steps of the process of transthyretin (TTR) amyloid fibril formation. FEBS Lett. 2011, 585, 2424–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Uncovering the Neuroprotective Mechanisms of Curcumin on Transthyretin Amyloidosis. Int. J. Mol. Sci. 2019, 20, 1287. [Google Scholar] [CrossRef] [Green Version]
- Wood, S.J.; Wypych, J.; Steavenson, S.; Louis, J.-C.; Citron, M.; Biere, A.L. α-Synuclein Fibrillogenesis Is Nucleation-dependent—Implications for the pathogenesis of Parkinson’s disease. J. Biol. Chem. 1999, 274, 19509–19512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Dong, C.; Hoffmann, M.; Garen, C.R.; Cortez, L.M.; Petersen, N.O.; Woodside, M.T. Early stages of aggregation of engineered α-synuclein monomers and oligomers in solution. Sci. Rep. 2019, 9, 1734. [Google Scholar] [CrossRef]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-synuclein oligomers and fibrils: A spectrum of species, a spectrum of toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, N.; Otzen, D.E. Oligomers of α-synuclein: Picking the culprit in the line-up. Essays Biochem. 2014, 56, 137–148. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olanow, C.W.; Perl, D.P.; DeMartino, G.N.; McNaught, K.S.P. Lewy-body formation is an aggresome-related process: A hypothesis. Lancet Neurol. 2004, 3, 496–503. [Google Scholar] [CrossRef]
- Lindström, V.; Fagerqvist, T.; Nordström, E.; Eriksson, F.; Lord, A.; Tucker, S.; Andersson, J.; Johannesson, M.; Schell, H.; Kahle, P.J.; et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, K.E.; Kragh, C.L.; Mann, D.M.A.; Salem, S.A.; Al-Shami, R.; Allsop, D.; Hassan, A.H.; Jensen, P.H.; El-Agnaf, O.M.A. Detection of elevated levels of soluble α-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain 2008, 132, 1093–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.; Selkoe, D.J. The Formation of Highly Soluble Oligomers of α-Synuclein Is Regulated by Fatty Acids and Enhanced in Parkinson’s Disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Groveman, B.R.; Orrù, C.D.; Hughson, A.G.; Raymond, L.D.; Zanusso, G.; Ghetti, B.; Campbell, K.; Safar, J.; Galasko, D.; Caughey, B. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol. Commun. 2018, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Hall, S.; Öhrfelt, A.; Zetterberg, H.; Blennow, K.; Minthon, L.; Nägga, K.; Londos, E.; Varghese, S.; Majbour, N.; et al. Levels of cerebrospinal fluid α-synuclein oligomers are increased in Parkinson’s disease with dementia and dementia with Lewy bodies compared to Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Reitboeck, P.G.; Anichtchik, O.; Bellucci, A.; Iovino, M.; Ballini, C.; Fineberg, E.; Ghetti, B.; Della Corte, L.; Spano, P.; Tofaris, G.; et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 2010, 133, 2032–2044. [Google Scholar] [CrossRef] [PubMed]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef] [Green Version]
- Bridi, J.; Hirth, F. Mechanisms of α-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, K.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous Induction of Cerebral -Amyloidogenesis Is Governed by Agent and Host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Riquelme, A.I.R.; Lau, H.H.C.; Stuart, E.; Goczi, A.N.; Wang, Z.; Schmitt-Ulms, G.; Watts, J.C. Prion-like propagation of β-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol. Commun. 2018, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M.-Y. Seeding of Normal Tau by Pathological Tau Conformers Drives Pathogenesis of Alzheimer-like Tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef] [Green Version]
- Ren, P.-H.; Lauckner, J.E.; Kachirskaia, I.; Heuser, J.E.; Melki, R.; Kopito, R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 2009, 11, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Pearce, M.P.; Kopito, R.R. Prion-Like Characteristics of Polyglutamine-Containing Proteins. Cold Spring Harb. Perspect. Med. 2017, 8, a024257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.; McGarvey, N.H.; Ayers, J.; Notterpek, L.; Borchelt, D.R.; et al. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10732–10737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.; Gonçalves, N.P.; Jan, A.; Jensen, N.M.; van der Laan, A.; Mohseni, S.; Vægter, C.B.; Jensen, P.H. Trans-synaptic spreading of alphaα-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 1–17. [Google Scholar] [CrossRef]
- Berge, N.V.D.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpicelli-Daley, L.A.; Luk, K.; Patel, T.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfarrash, S.; Jensen, N.M.; Ferreira, N.; Betzer, C.; Thevathasan, J.V.; Diekmann, R.; Adel, M.; Omar, N.M.; Boraie, M.Z.; Gad, S.; et al. Organotypic slice culture model demonstrates inter-neuronal spreading of alpha-synuclein aggregates. Acta Neuropathol. Commun. 2019, 7, 1–16. [Google Scholar] [CrossRef]
- Berge, N.V.D.; Ferreira, N.; Mikkelsen, T.W.; Alstrup, A.K.O.; Tamgüney, G.; Karlsson, P.; Terkelsen, A.J.; Nyengaard, J.R.; Jensen, P.H.; Borghammer, P. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain 2021. [Google Scholar] [CrossRef]
- Song, Y.J.C.; Halliday, G.M.; Holton, J.L.; Lashley, T.; O’Sullivan, S.S.; McCann, H.; Lees, A.J.; Ozawa, T.; Williams, D.R.; Lockhart, P.; et al. Degeneration in Different Parkinsonian Syndromes Relates to Astrocyte Type and Astrocyte Protein Expression. J. Neuropathol. Exp. Neurol. 2009, 68, 1073–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, Z.A.; Giasson, B.I.; Chakrabarty, P. α-Synuclein and astrocytes: Tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol. 2019, 138, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.L. Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000, 10, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Mikolaenko, I.; Pletnikova, O.; Kawas, C.H.; O’Brien, R.; Resnick, S.M.; Crain, B.; Troncoso, J.C. Alpha-Synuclein Lesions in Normal Aging, Parkinson Disease, and Alzheimer Disease: Evidence from the Baltimore Longitudinal Study of Aging (BLSA). J. Neuropathol. Exp. Neurol. 2005, 64, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R.; Jicha, G.A.; Liu, H.; Schmitt, F.A. Lewy Body Pathology in Normal Elderly Subjects. J. Neuropathol. Exp. Neurol. 2009, 68, 816–822. [Google Scholar] [CrossRef]
- Uemura, N.; Uemura, M.; Luk, K.; Lee, V.M.-Y.; Trojanowski, J.Q. Cell-to-Cell Transmission of Tau and α-Synuclein. Trends Mol. Med. 2020, 26, 936–952. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M.-Y. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.B.; Jagannath, S.; Lashuel, H.A. Reverse engineering Lewy bodies: How far have we come and how far can we go? Nat. Rev. Neurosci. 2021, 22, 256. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, T.; Zhang, B. Exosomes in Parkinson’s Disease. Neurosci. Bull. 2016, 33, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, C.-Q.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Stuendl, A.; Kunadt, M.; Kruse, N.; Bartels, C.; Moebius, W.; Danzer, K.M.; Mollenhauer, B.; Schneider, A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 2015, 139, 481–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain 2020, 143, 1476–1497. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, G.; Han, C.; Ma, K.; Guo, X.; Wan, F.; Kou, L.; Yin, S.; Liu, L.; Huang, J.; et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019, 10, 174. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.-Q.; Jia, C.; Lim, Y.-J.; Feng, G.; Xu, E.; Long, H.; Kimura, Y.; Tao, Y.; Zhao, C.; et al. Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.T.; Karuppagounder, S.; Kam, T.-I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.-H.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef] [Green Version]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buée, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gath, J.; Bousset, L.; Habenstein, B.; Melki, R.; Böckmann, A.; Meier, B.H. Unlike Twins: An NMR Comparison of Two α-Synuclein Polymorphs Featuring Different Toxicity. PLoS ONE 2014, 9, e90659. [Google Scholar] [CrossRef]
- Gath, J.; Bousset, L.; Habenstein, B.; Melki, R.; Meier, B.H.; Böckmann, A. Yet another polymorph of α-synuclein: Solid-state sequential assignments. Biomol. NMR Assign. 2013, 8, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gath, J.; Habenstein, B.; Bousset, L.; Melki, R.; Meier, B.H.; Böckmann, A. Solid-state NMR sequential assignments of α-synuclein. Biomol. NMR Assign. 2011, 6, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [Green Version]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Haute, C.V.D.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nat. Cell Biol. 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.; et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nat. Cell Biol. 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nat. Cell Biol. 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Surguchov, A. Analysis of Protein Conformational Strains—A Key for New Diagnostic Methods of Human Diseases. Int. J. Mol. Sci. 2020, 21, 2801. [Google Scholar] [CrossRef]
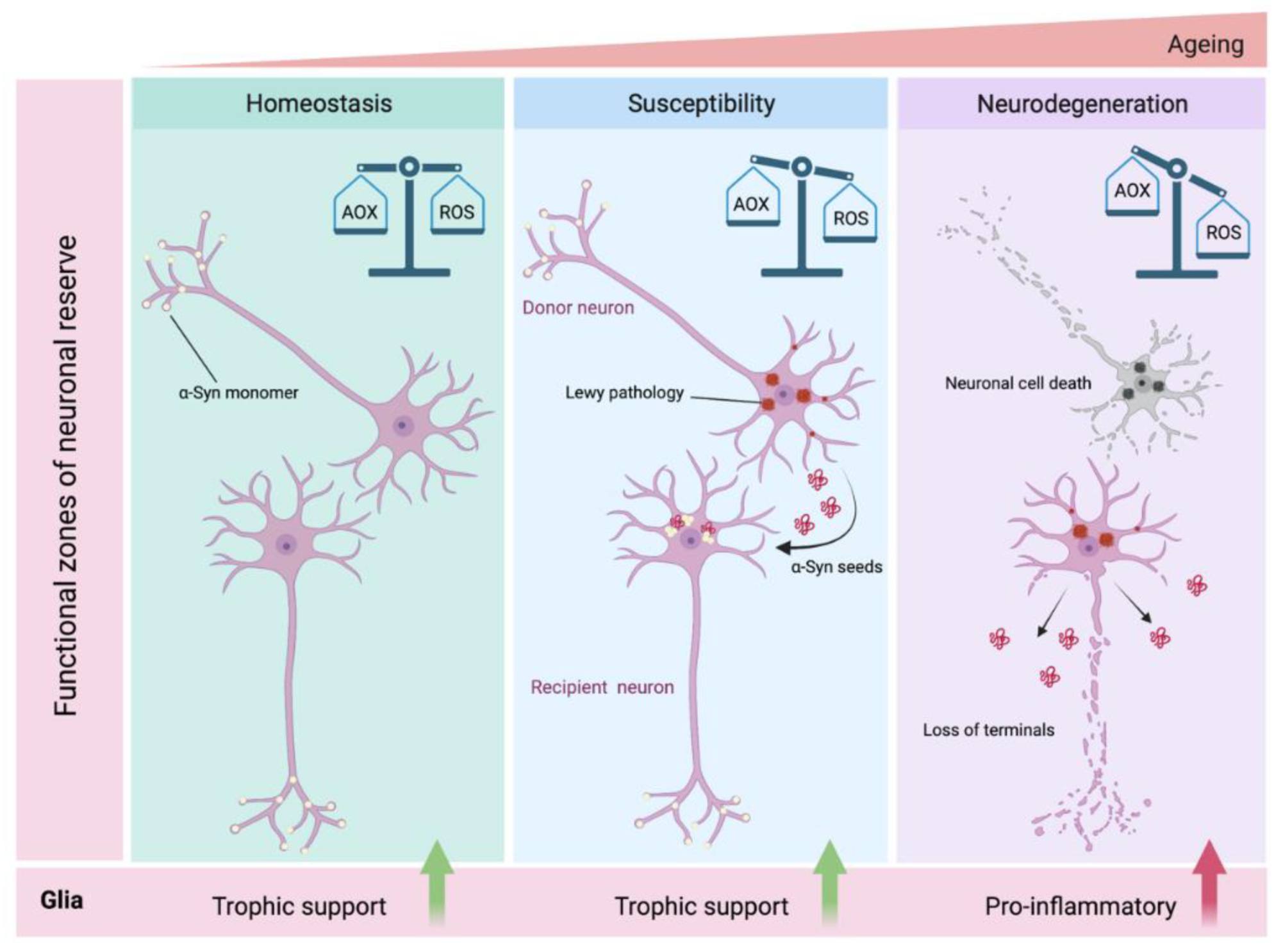
- Del Tredici, K.; Braak, H. Review: Sporadic Parkinson’s disease: Development and distribution of α-synuclein pathology. Neuropathol. Appl. Neurobiol. 2015, 42, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S.; Isacson, O. The Threshold Theory for Parkinson’s Disease. Trends Neurosci. 2017, 40, 4–14. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448. [Google Scholar] [CrossRef]
- Halliday, G.; McRitchie, D.; Cartwright, H.; Pamphlett, R.; Hely, M.; Morris, J. Midbrain neuropathology in idiopathic Parkinson’s disease and diffuse Lewy body disease. J. Clin. Neurosci. 1996, 3, 52–60. [Google Scholar] [CrossRef]
- Cheng, H.-C.; Ulane, C.M.; Burke, R. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Jellinger, K.A. A critical evaluation of current staging of α-synuclein pathology in Lewy body disorders. Biochim. Biophys. Acta 2009, 1792, 730–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaitzakis, M.E.; Graeber, M.B.; Gentleman, S.M.; Pearce, R.K.B. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: A critical analysis of α-synuclein staging. Neuropathol. Appl. Neurobiol. 2008, 34, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Doherty, K.M.; Silveira-Moriyama, L.; Parkkinen, L.; Healy, D.G.; Farrell, M.; Mencacci, N.E.; Ahmed, Z.; Brett, F.M.; Hardy, J.; Quinn, N.; et al. Parkin Disease: A clinicopathologic entity? JAMA Neurol. 2013, 70, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; Iii, C.L.W.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Krogh, K.; Ostergaard, K.; Sabroe, S.; Laurberg, S. Clinical aspects of bowel symptoms in Parkinson?s disease. Acta Neurol. Scand. 2007, 117, 60–64. [Google Scholar] [CrossRef]
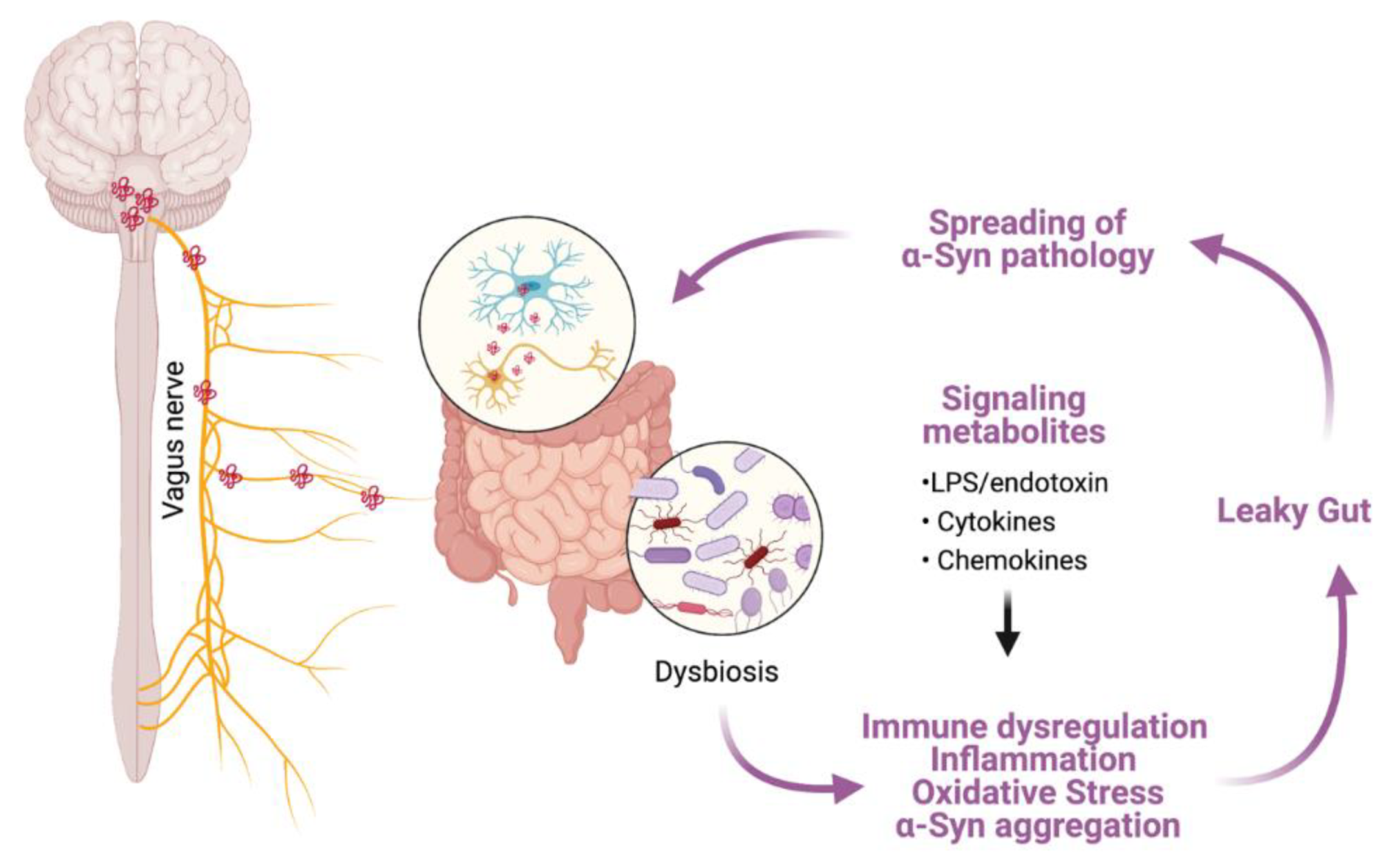
- Breen, D.P.; Halliday, G.M.; Lang, A.E. Gut–brain axis and the spread of α-synuclein pathology: Vagal highway or dead end? Mov. Disord. 2019, 34, 307–316. [Google Scholar] [CrossRef]
- Menozzi, E.; Macnaughtan, J.; Schapira, A.H.V. The gut-brain axis and Parkinson disease: Clinical and pathogenetic relevance. Ann. Med. 2021, 53, 611–625. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Danielsen, E.H.; Hamilton-Dutoit, S.; Borghammer, P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol. 2016, 79, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, F.S.; Stefanova, N.; Gelpi, E.; Seppi, K.; Navarro-Otano, J.; Offner, F.; Vilas, D.; Valldeoriola, F.; Pont-Sunyer, C.; Aldecoa, I.; et al. Enteric nervous system α-synuclein immunoreactivity in idiopathic REM sleep behavior disorder. Neurology 2015, 85, 1761–1768. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Fang, F.; Pedersen, N.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Fedorova, T.D.; Seidelin, L.B.; Knudsen, K.; Schacht, A.C.; Geday, J.; Pavese, N.; Brooks, D.; Borghammer, P. Decreased intestinal acetylcholinesterase in early Parkinson disease: An (11)C-donepezil PET study. Neurology 2017, 88, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.; Fedorova, T.D.; Hansen, A.K.; Sommerauer, M.; Otto, M.; Svendsen, K.B.; Nahimi, A.; Stokholm, M.G.; Pavese, N.; Beier, C.P.; et al. In-vivo staging of pathology in REM sleep behaviour disorder: A multimodality imaging case-control study. Lancet Neurol. 2018, 17, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Bjorklund, T.; Wang, Z.-Y.; Roybon, L.; Melki, R.; Li, J.-Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol. Neurodegener. 2018, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Boertien, J.M.; Pereira, P.A.; Aho, V.T.; Scheperjans, F. Increasing Comparability and Utility of Gut Microbiome Studies in Parkinson’s Disease: A Systematic Review. J. Park. Dis. 2019, 9, S297–S312. [Google Scholar] [CrossRef] [Green Version]
- Pierantozzi, M.; Pietroiusti, A.; Brusa, L.; Galati, S.; Stefani, A.; Lunardi, G.; Fedele, E.; Sancesario, G.; Bernardi, G.; Bergamaschi, A.; et al. Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology 2006, 66, 1824–1829. [Google Scholar] [CrossRef]
- Schwiertz, A.; Spiegel, J.; Dillmann, U.; Grundmann, D.; Bürmann, J.; Fassbender, K.; Schäfer, K.-H.; Unger, M. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Park. Relat. Disord. 2018, 50, 104–107. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased Intestinal Permeability Correlates with Sigmoid Mucosa alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [Green Version]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti–Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients with Inflammatory Bowel Disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Brudek, T. Inflammatory Bowel Diseases and Parkinson’s Disease. J. Park. Dis. 2019, 9, S331–S344. [Google Scholar] [CrossRef] [Green Version]
- Harapan, B.N.; Frydrychowicz, C.; Classen, J.; Wittekind, C.; Gradistanac, T.; Rumpf, J.-J.; Mueller, W. No enhanced (p-) α-synuclein deposition in gastrointestinal tissue of Parkinson’s disease patients. Park. Relat. Disord. 2020, 80, 82–88. [Google Scholar] [CrossRef]
- Barrenschee, M.; Zorenkov, D.; Böttner, M.; Lange, C.; Cossais, F.; Scharf, A.B.; Deuschl, G.; Schneider, S.A.; Ellrichmann, M.; Fritscher-Ravens, A.; et al. Distinct pattern of enteric phospho-alpha-synuclein aggregates and gene expression profiles in patients with Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Adler, C.H.; Connor, D.J.; Ms, J.G.H.; Sabbagh, M.N.; Caviness, J.N.; Shill, H.A.; Noble, B.; Beach, T.G. Incidental Lewy body disease: Clinical comparison to a control cohort. Mov. Disord. 2010, 25, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2016, 140, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Kingsbury, A.E.; Bandopadhyay, R.; Moriyama, L.S.; Ayling, H.; Kallis, C.; Sterlacci, W.; Maeir, H.; Poewe, W.; Lees, A.J. Brain stem pathology in Parkinson’s disease: An evaluation of the Braak staging model. Mov. Disord. 2010, 25, 2508–2515. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef] [Green Version]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [Green Version]
- Koprich, J.B.; Kalia, L.V.; Brotchie, J. Animal models of α-synucleinopathy for Parkinson disease drug development. Nat. Rev. Neurosci. 2017, 18, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Riffe, C.J.; Sorrentino, Z.A.; Diamond, J.; Fagerli, E.; Brooks, M.; Galaleldeen, A.; Hart, P.J.; Giasson, B.I. Localized Induction of Wild-Type and Mutant Alpha-Synuclein Aggregation Reveals Propagation along Neuroanatomical Tracts. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Van der Perren, A.; Haute, C.V.D.; Baekelandt, V. Viral Vector-Based Models of Parkinson’s Disease. Curr. Top. Behav. Neurosci. 2014, 22, 271–301. [Google Scholar] [CrossRef]
- Watts, J.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560. [Google Scholar] [CrossRef] [Green Version]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.-O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2013, 75, 351–362. [Google Scholar] [CrossRef]
- Schaser, A.J.; Stackhouse, T.L.; Weston, L.J.; Kerstein, P.C.; Osterberg, V.R.; López, C.S.; Dickson, D.W.; Luk, K.C.; Meshul, C.K.; Woltjer, R.L.; et al. Trans-synaptic and retrograde axonal spread of Lewy pathology following pre-formed fibril injection in an in vivo A53T alpha-synuclein mouse model of synucleinopathy. Acta Neuropathol. Commun. 2020, 8, 1–23. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Xia, Y.; Funk, C.; Riffe, C.J.; Rutherford, N.J.; Diaz, C.C.; Sacino, A.N.; Price, N.; Golde, T.E.; Giasson, B.I.; et al. Motor neuron loss and neuroinflammation in a model of α-synuclein-induced neurodegeneration. Neurobiol. Dis. 2018, 120, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Giguère, N.; Nanni, S.B.; Trudeau, L.-E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv. Anat. Embryol. Cell Biol. 2009, 201, 1–119. [Google Scholar] [PubMed]
- Matsuda, W.; Furuta, T.; Nakamura, K.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single Nigrostriatal Dopaminergic Neurons Form Widely Spread and Highly Dense Axonal Arborizations in the Neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Cabello, C.R.; Thune, J.J.; Pakkenberg, H.; Pakkenberg, B. Ageing of substantia nigra in humans: Cell loss may be compensated by hypertrophy. Neuropathol. Appl. Neurobiol. 2002, 28, 283–291. [Google Scholar] [CrossRef]
- Chan, C.S.; Gertler, T.S.; Surmeier, D.J. A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson’s disease. Mov. Disord. 2010, 25, S63–S70. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Bergamasco, B.; Lopiano, L. Is neuromelanin changed in Parkinson’s disease? Investigations by magnetic spectroscopies. J. Neural Transm. 2006, 113, 769–774. [Google Scholar] [CrossRef]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
- Delaidelli, A.; Richner, M.; Jiang, L.; van der Laan, A.; Christiansen, I.B.J.; Ferreira, N.; Nyengaard, J.R.; Vægter, C.B.; Jensen, P.H.; Mackenzie, I.R.; et al. α-Synuclein pathology in Parkinson disease activates homeostatic NRF2 anti-oxidant response. Acta Neuropathol. Commun. 2021, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.; Jordan-Sciutto, K.L. Expression of Nrf2 in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Tanji, K.; Maruyama, A.; Odagiri, S.; Mori, F.; Itoh, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Keap1 Is Localized in Neuronal and Glial Cytoplasmic Inclusions in Various Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2013, 72, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipperab, H.M.; Liberman, A.; Stopa, E. Neural Heme Oxygenase-1 Expression in Idiopathic Parkinson’s Disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; García-Yagüe, J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansorge, O.; Daniel, S.E.; Pearce, R.K. Neuronal loss and plasticity in the supraoptic nucleus in Parkinson’s disease. Neurology 1997, 49, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Greffard, S.; Verny, M.; Bonnet, A.-M.; Seilhean, D.; Hauw, J.-J.; Duyckaerts, C. A stable proportion of Lewy body bearing neurons in the substantia nigra suggests a model in which the Lewy body causes neuronal death. Neurobiol. Aging 2010, 31, 99–103. [Google Scholar] [CrossRef]
- Porritt, M.J.; Kingsbury, A.E.; Hughes, A.J.; Howells, D.W. Striatal dopaminergic neurons are lost with Parkinson’s disease progression. Mov. Disord. 2006, 21, 2208–2211. [Google Scholar] [CrossRef]
- Burke, R.E.; Dauer, W.; Vonsattel, J.P.G. A critical evaluation of the Braak staging scheme for Parkinson’s disease. Ann. Neurol. 2008, 64, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G.; Milenkovic, I.J.; Preusser, M.; Budka, H. Nigral burden of α-synuclein correlates with striatal dopamine deficit. Mov. Disord. 2008, 23, 1608–1612. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sánchez-Padilla, J.; Chan, C.S.; Surmeier, D.J. Robust Pacemaking in Substantia Nigra Dopaminergic Neurons. J. Neurosci. 2009, 29, 11011–11019. [Google Scholar] [CrossRef]
- Nedergaard, S.; Flatman, J.A.; Engberg, I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J. Physiol. 1993, 466, 727–747. [Google Scholar]
- Puopolo, M.; Raviola, E.; Bean, B.P. Roles of Subthreshold Calcium Current and Sodium Current in Spontaneous Firing of Mouse Midbrain Dopamine Neurons. J. Neurosci. 2007, 27, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nat. Cell Biol. 2007, 447, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, N.B.; Bond, A.; Calabresi, P.; Stratta, F.; Stefani, A.; Bernardi, G. Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br. J. Pharmacol. 1994, 113, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Foehring, R.C.; Zhang, X.F.; Lee, J.; Callaway, J.C. Endogenous Calcium Buffering Capacity of Substantia Nigral Dopamine Neurons. J. Neurophysiol. 2009, 102, 2326–2333. [Google Scholar] [CrossRef] [Green Version]
- Khaliq, Z.M.; Bean, B.P. Pacemaking in Dopaminergic Ventral Tegmental Area Neurons: Depolarizing Drive from Background and Voltage-Dependent Sodium Conductances. J. Neurosci. 2010, 30, 7401–7413. [Google Scholar] [CrossRef] [PubMed]
- Philippart, F.; Destreel, G.; Merino-Sepúlveda, P.; Henny, P.; Engel, D.; Seutin, V. Differential Somatic Ca2+ Channel Profile in Midbrain Dopaminergic Neurons. J. Neurosci. 2016, 36, 7234–7245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta 2009, 1787, 1334–1341. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, J.; Guzman, J.N.; Estep, C.M.; Ilijic, E.; Kondapalli, J.; Sanchez-Padilla, J.; Surmeier, D.J. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson’s disease. Nat. Neurosci. 2012, 15, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nat. Cell Biol. 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, C.; Paxinos, G.; Puelles, L. The Mouse Nervous System, 1st ed.; Elsevier Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2012; p. xvii. 795p. [Google Scholar]
- Del Tredici, K.; Braak, H. Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson’s disease-related dementia. J. Neurol. Neurosurg. Psychiatry 2012, 84, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Suk, J.-E.; Patrick, C.; Bae, E.-J.; Cho, J.-H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.-J. Direct Transfer of α-Synuclein from Neuron to Astroglia Causes Inflammatory Responses in Synucleinopathies*. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.-L.; Long, C.-X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Beach, T.G.; Sue, L.I.; Walker, U.G.; Lue, L.F.; Connor, N.J.; Caviness, J.N.; Sabbagh, M.N.; Adler, C.H. Marked microglial reaction in normal aging human substantia nigra: Correlation with extraneuronal neuromelanin pigment deposits. Acta Neuropathol. 2007, 114, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Knott, C.; Stern, G.; Kingsbury, A.; Welcher, A.; Wilkin, G. Elevated glial brain-derived neurotrophic factor in Parkinson’s diseased nigra. Park. Relat. Disord. 2002, 8, 329–341. [Google Scholar] [CrossRef]
- Riederer, P.; Gerlach, M.; Müller, T.; Reichmann, H. Relating mode of action to clinical practice: Dopaminergic agents in Parkinson’s disease. Park. Relat. Disord. 2007, 13, 466–479. [Google Scholar] [CrossRef]
- Seidel, K.; Mahlke, J.; Siswanto, S.; Krueger, R.; Heinsen, H.; Auburger, G.; Bouzrou, M.; Grinberg, L.; Wicht, H.; Korf, H.; et al. The Brainstem Pathologies of Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathol. 2014, 25, 121–135. [Google Scholar] [CrossRef]
- Muntane, G.; Dalfo, E.; Martinez, A.; Ferrer, I. Phosphorylation of tau and α-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related α-synucleinopathies. Neuroscience 2008, 152, 913–923. [Google Scholar] [CrossRef]
- Obi, K.; Akiyama, H.; Kondo, H.; Shimomura, Y.; Hasegawa, M.; Iwatsubo, T.; Mizuno, Y.; Mochizuki, H. Relationship of phosphorylated α-synuclein and tau accumulation to A β deposition in the cerebral cortex of dementia with Lewy bodies. Exp. Neurol. 2008, 210, 409–420. [Google Scholar] [CrossRef]
- Wills, J.; Jones, J.; Haggerty, T.; Duka, V.; Joyce, J.N.; Sidhu, A. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp. Neurol. 2010, 225, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coakeley, S.; Strafella, A.P. Imaging tau pathology in Parkinsonisms. NPJ Park. Dis. 2017, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelsen, T.M.; Woldbye, D.P. Gene Therapy for Parkinson’s Disease, An Update. J. Park. Dis. 2018, 8, 195–215. [Google Scholar] [CrossRef] [Green Version]
- Coune, P.G.; Schneider, B.L.; Aebischer, P. Parkinson’s Disease: Gene Therapies. Cold Spring Harb. Perspect. Med. 2012, 2, a009431. [Google Scholar] [CrossRef]
- Parmar, M.; Grealish, S.; Henchcliffe, C. The future of stem cell therapies for Parkinson disease. Nat. Rev. Neurosci. 2020, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Dominey, T.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2020. J. Park. Dis. 2020, 10, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Fayyad, M.; Salim, S.; Majbour, N.; Erskine, D.; Stoops, E.; Mollenhauer, B.; El-Agnaf, O.M.A. Parkinson’s disease biomarkers based on α-synuclein. J. Neurochem. 2019, 150, 626–636. [Google Scholar] [CrossRef]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, P.; Hotter, A.; Hussl, A.; Esterhammer, R.; Schocke, M.; Seppi, K. Significance of MRI in Diagnosis and Differential Diagnosis of Parkinson’s Disease. Neurodegener. Dis. 2010, 7, 300–318. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Martinelli, P.; Manners, D.; Scaglione, C.; Tonon, C.; Cortelli, P.; Malucelli, E.; Capellari, S.; Testa, C.; Parchi, P.; et al. Diffusion-weighted brain imaging study of patients with clinical diagnosis of corticobasal degeneration, progressive supranuclear palsy and Parkinson’s disease. Brain 2008, 131, 2690–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weise, D.; Lorenz, R.; Schliesser, M.; Schirbel, A.; Reiners, K.; Classen, J. Substantia nigra echogenicity: A structural correlate of functional impairment of the dopaminergic striatal projection in Parkinson’s disease. Mov. Disord. 2009, 24, 1669–1675. [Google Scholar] [CrossRef]
- Rascol, O.; Schelosky, L. 123 I-metaiodobenzylguanidine scintigraphy in Parkinson’s disease and related disorders. Mov. Disord. 2009, 24, S732–S741. [Google Scholar] [CrossRef]
- Adams, J.L.; Lizarraga, K.J.; Waddell, E.M.; Myers, T.L.; Jensen-Roberts, S.; Modica, J.S.; Schneider, R.B. Digital Technology in Movement Disorders: Updates, Applications, and Challenges. Curr. Neurol. Neurosci. Rep. 2021, 21, 1–11. [Google Scholar] [CrossRef]
- Luis-Martínez, R.; Monje, M.H.G.; Antonini, A.; Sánchez-Ferro, Á.; Mestre, T.A. Technology-Enabled Care: Integrating Multidisciplinary Care in Parkinson’s Disease Through Digital Technology. Front. Neurol. 2020, 11, 575975. [Google Scholar] [CrossRef]
- Rockenstein, E.; Mallory, M.; Hashimoto, M.; Song, D.; Shults, C.W.; Lang, I.; Masliah, E. Differential neuropathological alterations in transgenic mice expressing α-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res. 2002, 68, 568–578. [Google Scholar] [CrossRef]
- Chesselet, M.-F.; Richter, F.; Zhu, C.; Magen, I.; Watson, M.B.; Subramaniam, S.R. A Progressive Mouse Model of Parkinson’s Disease: The Thy1-aSyn (“Line 61”) Mice. Neurotherapeutics 2012, 9, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Henrich, M.; Geibl, F.F.; Lee, B.; Chiu, W.-H.; Koprich, J.B.; Brotchie, J.M.; Timmermann, L.; Decher, N.; Matschke, L.A.; Oertel, W.H. A53T-α-synuclein overexpression in murine locus coeruleus induces Parkinson’s disease-like pathology in neurons and glia. Acta Neuropathol. Commun. 2018, 6, 39. [Google Scholar] [CrossRef]
- Butkovich, L.M.; Houser, M.C.; Chalermpalanupap, T.; Porter-Stransky, K.A.; Iannitelli, A.F.; Boles, J.; Lloyd, G.M.; Coomes, A.S.; Eidson, L.N.; Rodrigues, M.E.D.S.; et al. Transgenic Mice Expressing Human α-Synuclein in Noradrenergic Neurons Develop Locus Ceruleus Pathology and Nonmotor Features of Parkinson’s Disease. J. Neurosci. 2020, 40, 7559–7576. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Neuronal α-Synucleinopathy with Severe Movement Disorder in Mice Expressing A53T Human α-Synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Richner, M.; van der Laan, A.; Christiansen, I.B.J.; Vægter, C.B.; Nyengaard, J.R.; Halliday, G.M.; Weiss, J.; Giasson, B.I.; Mackenzie, I.R.; et al. Prodromal neuroinvasion of pathological α-synuclein in brainstem reticular nuclei and white matter lesions in a model of α-synucleinopathy. Brain Commun. 2021, 3, fcab104. [Google Scholar] [CrossRef]
- Lalonde, R.; Strazielle, C. Brain regions and genes affecting limb-clasping responses. Brain Res. Rev. 2011, 67, 252–259. [Google Scholar] [CrossRef]
- Linnman, C.; Moulton, E.; Barmettler, G.; Becerra, L.; Borsook, D. Neuroimaging of the periaqueductal gray: State of the field. NeuroImage 2012, 60, 505–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson-Poe, A.; Pocius, E.; Herschbach, M.; Morgan, M.M. The periaqueductal gray contributes to bidirectional enhancement of antinociception between morphine and cannabinoids. Pharmacol. Biochem. Behav. 2013, 103, 444–449. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Park. Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, M.C.; Growdon, W.; Gomez-Isla, T.; Newell, K.; George, J.M.; Clayton, D.F.; Hyman, B.T. Nigral and Cortical Lewy Bodies and Dystrophic Nigral Neurites in Parkinsonʼs Disease and Cortical Lewy Body Disease Contain α-synuclein Immunoreactivity. J. Neuropathol. Exp. Neurol. 1998, 57, 334–337. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy bodies. Nat. Cell Biol. 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.L.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments α-synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef]
- Tang, F.-L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.-M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.-C. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef]
- Ron, I.; Rapaport, D.; Horowitz, M. Interaction between parkin and mutant glucocerebrosidase variants: A possible link between Parkinson disease and Gaucher disease. Hum. Mol. Genet. 2010, 19, 3771–3781. [Google Scholar] [CrossRef] [Green Version]
- Doi, D.; Magotani, H.; Kikuchi, T.; Ikeda, M.; Hiramatsu, S.; Yoshida, K.; Amano, N.; Nomura, M.; Umekage, M.; Morizane, A.; et al. Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brauer, R.; Wei, L.; Ma, T.; Athauda, D.; Girges, C.; Vijiaratnam, N.; Auld, G.; Whittlesea, C.; Wong, I.; Foltynie, T. Diabetes medications and risk of Parkinson’s disease: A cohort study of patients with diabetes. Brain 2020, 143, 3067–3076. [Google Scholar] [CrossRef]
- De Pablo-Fernandez, E.; Goldacre, R.; Pakpoor, J.; Noyce, A.J.; Warner, T.T. Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology 2018, 91, e139–e142. [Google Scholar] [CrossRef]
- Henderson, M.X.; Sengupta, M.; Trojanowski, J.Q.; Lee, V.M.Y. Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 2019, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.; Fairfoul, G.; Tolosa, E.S.; Martí, M.J.; Green, A.; Barcelona, L.S.G. α-synuclein RT-QuIC in cerebrospinal fluid of LRRK 2-linked Parkinson’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Aasly, J.O.; Johansen, K.K.; Brã¸nstad, G.; Warã¸, B.J.; Majbour, N.; Evarghese, S.; Ealzahmi, F.; Paleologou, K.E.; Amer, D.A.M.; Eal-Hayani, A.; et al. Elevated levels of cerebrospinal fluid α-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front. Aging Neurosci. 2014, 6, 248. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.T.; Jagannath, S.; Francois, C.; Vanderstichele, H.; Stoops, E.; Lashuel, H.A. How specific are the conformation-specific α-synuclein antibodies? Characterization and validation of 16 α-synuclein conformation-specific antibodies using well-characterized preparations of α-synuclein monomers, fibrils and oligomers with distinct structures and morphology. Neurobiol. Dis. 2020, 146, 105086. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158338
Jan A, Gonçalves NP, Vaegter CB, Jensen PH, Ferreira N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. International Journal of Molecular Sciences. 2021; 22(15):8338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158338
Chicago/Turabian StyleJan, Asad, Nádia Pereira Gonçalves, Christian Bjerggaard Vaegter, Poul Henning Jensen, and Nelson Ferreira. 2021. "The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses" International Journal of Molecular Sciences 22, no. 15: 8338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158338