
Mechanistic and Predictive QSAR Analysis of Diverse Molecules to Capture Salient and Hidden Pharmacophores for Anti-Thrombotic Activity

 and
and 
Abstract
:1. Introduction
- (1)
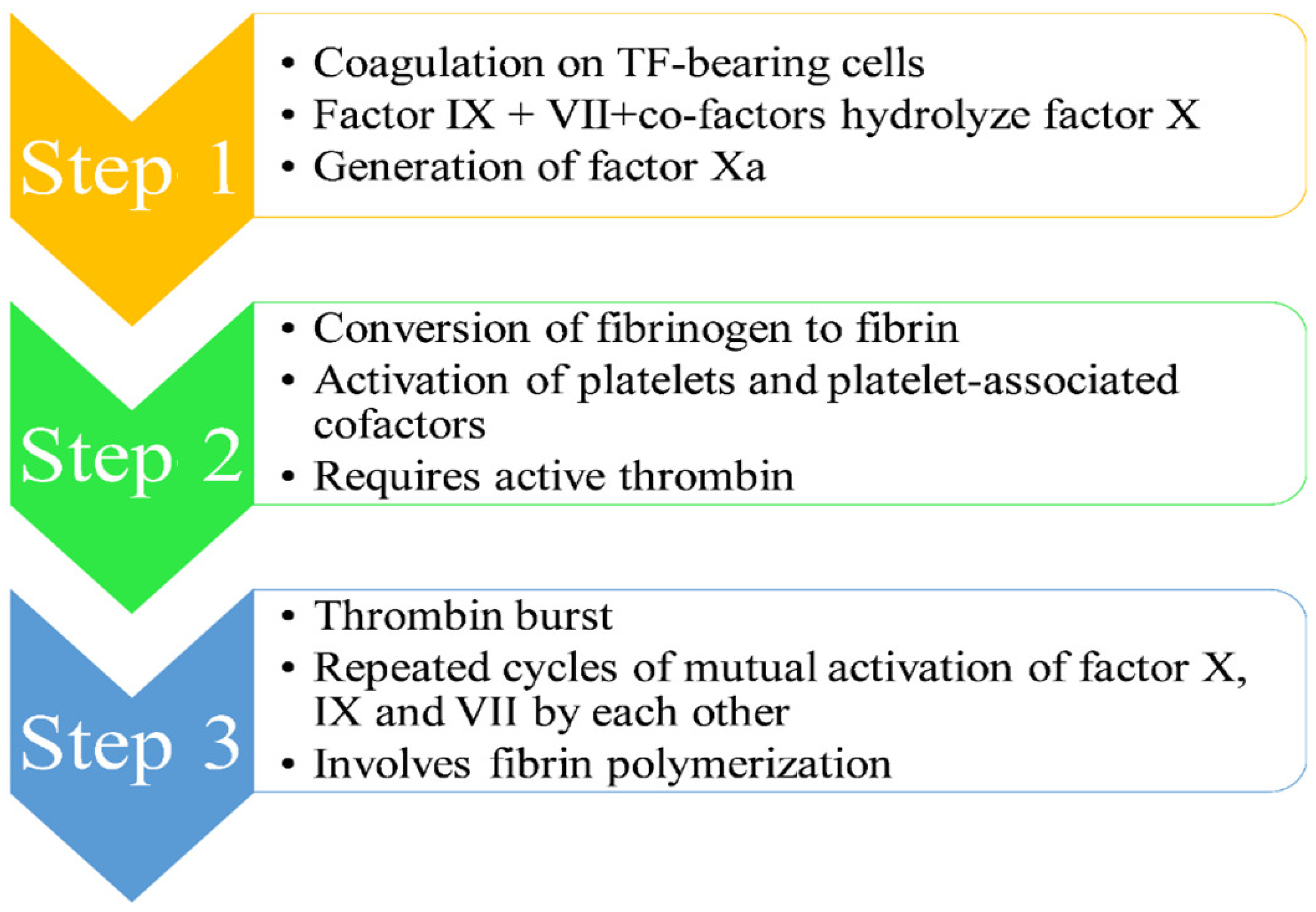
- Step 1 [1,8,9,10,11,13]: The mechanism of the coagulation cascade begins with coagulation on TF-bearing cells. Factor IX and VII, along with their respective co-factors, are responsible for the hydrolysis of factor X, leading to its conversion to its activated form Xa. The activated factor Xa is accountable for the dual breaking of prothrombin first at an arg-thr and then at an arg-ile bond, thereby generating active thrombin, which is a coagulation protease. A single factor X converts several prothrombin molecules, thus generating multiple thrombin molecules.
- (2)
- (3)
- Step 3 [1,8,9,10,11,13]: The third step involves “thrombin burst”, which occurs due to continuous generation of thrombin on the platelet surface, thereby leading to repeated cycles of mutual activation of factor X, IX and VII by each other. This thrombin burst through fibrin polymerization is vital for a thrombus formation.
2. Results
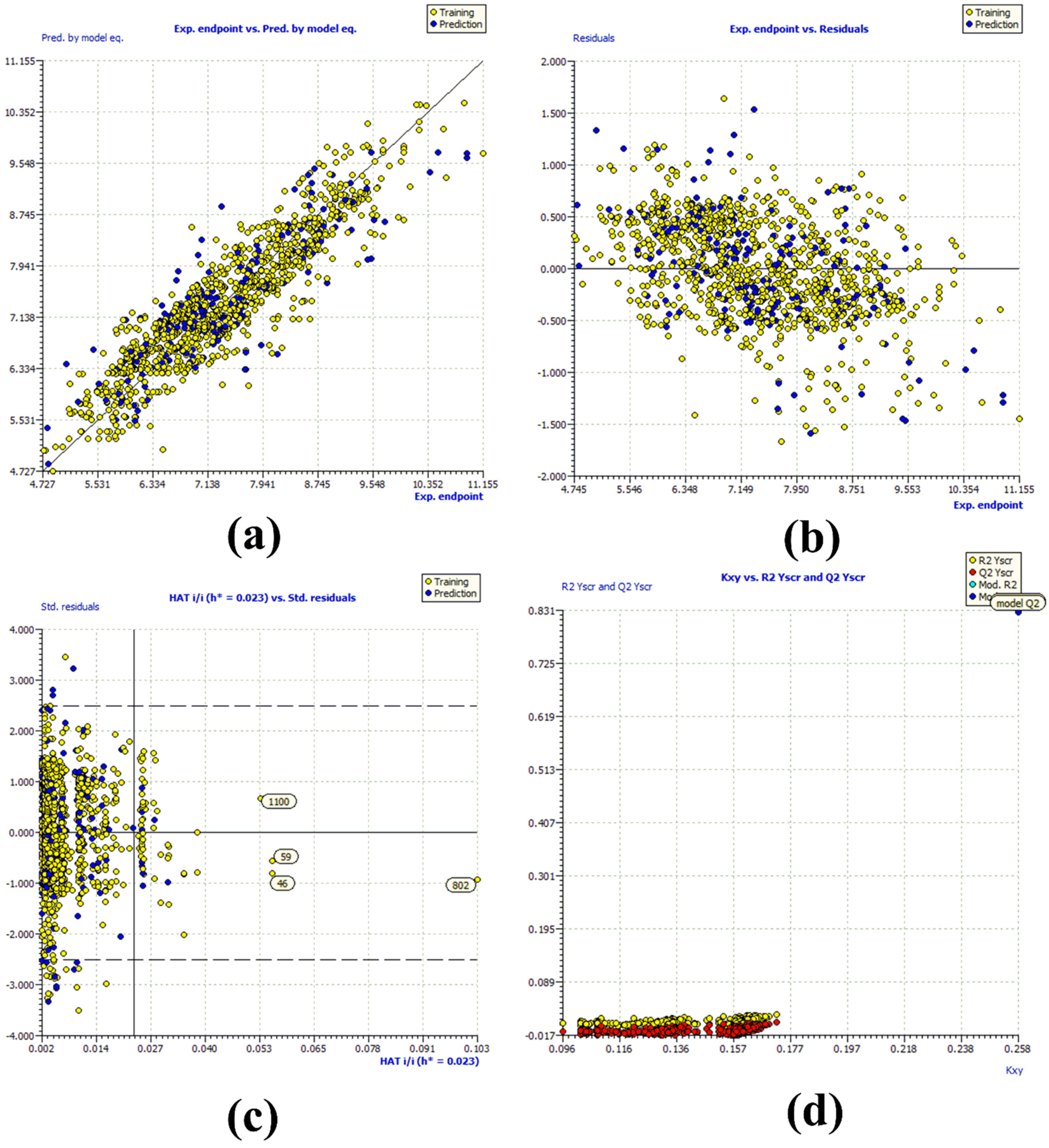
R2tr = 0.831, R2adj. = 0.83, RMSEtr = 0.476, CCCtr = 0.908, s = 0.478, F = 731.048, R2cv (Q2loo) = 0.829, RMSEcv = 0.479, CCCcv = 0.907, Q2LMO = 0.828, R2Yscr = 0.007, RMSEex = 0.526, R2ex = 0.783, Q2 − F1 = 0.782, Q2 − F2 = 0.782, Q2 − F3 = 0.794, CCCex = 0.874, R2 − ExPy = 0.783, R′o2 = 0.704, k′ = 0.996, 1 − (R2/R′o2) = 0.101, Ro2 = 0.782, k = 0.999, 1 − (R2 − ExPy/Ro2) = 0.001
3. Discussion
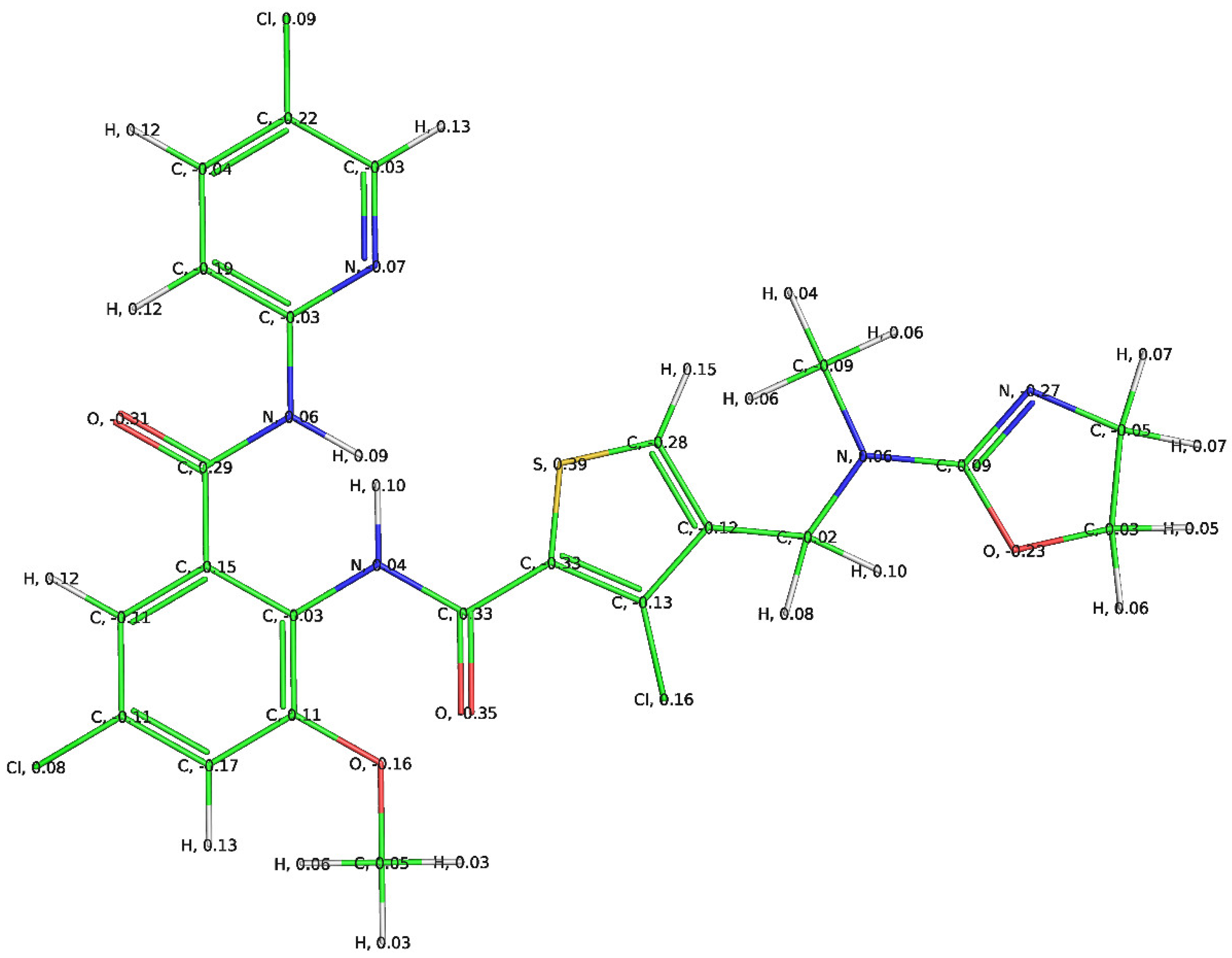
3.1. Mechanistic Interpretation of QSAR Model
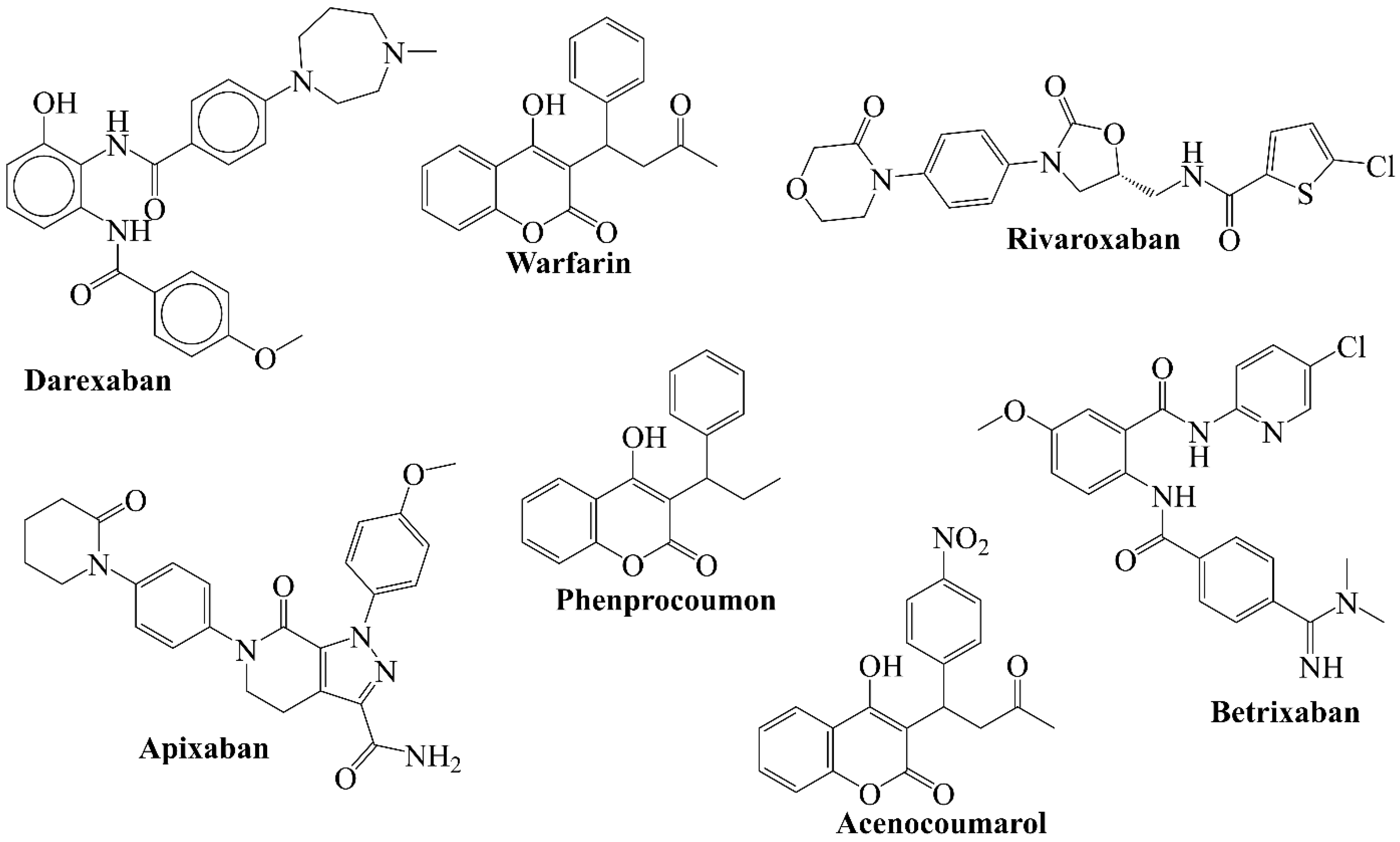
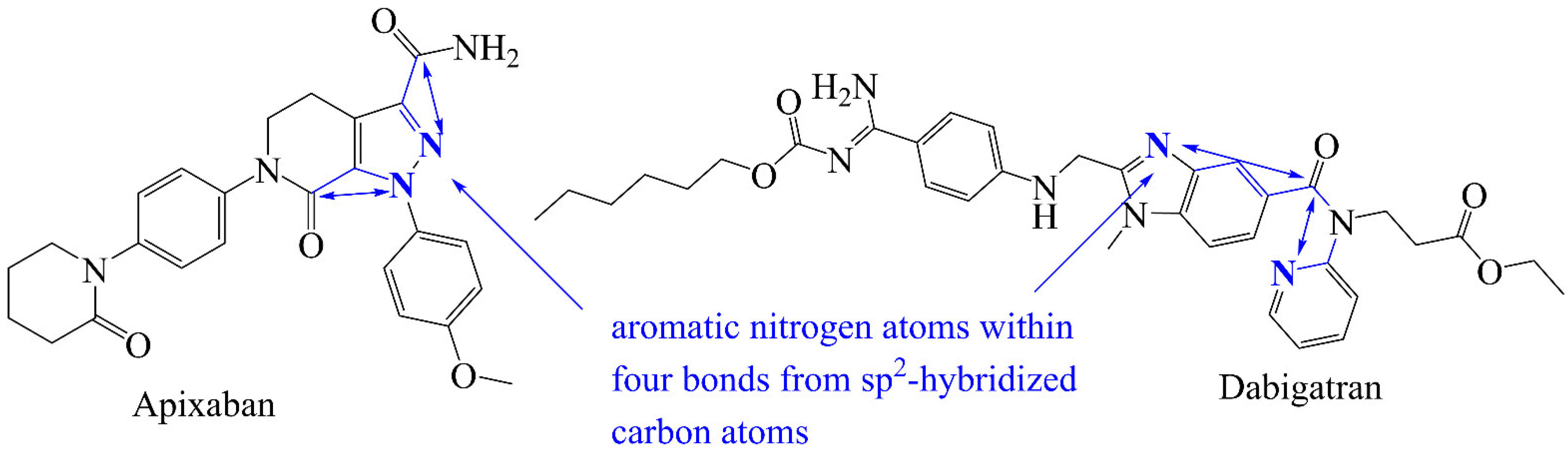
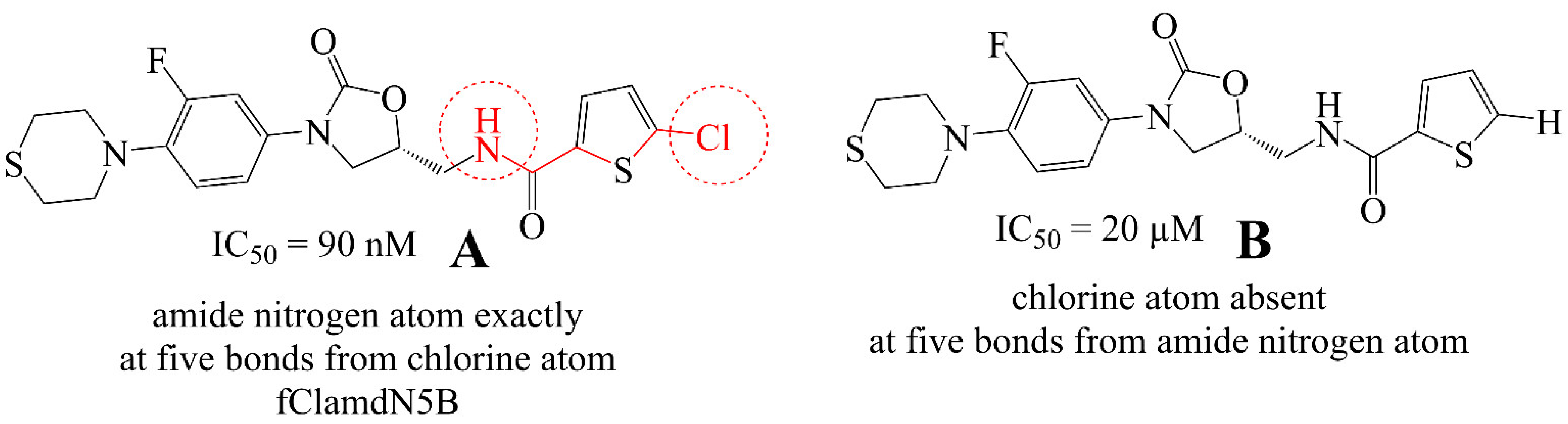
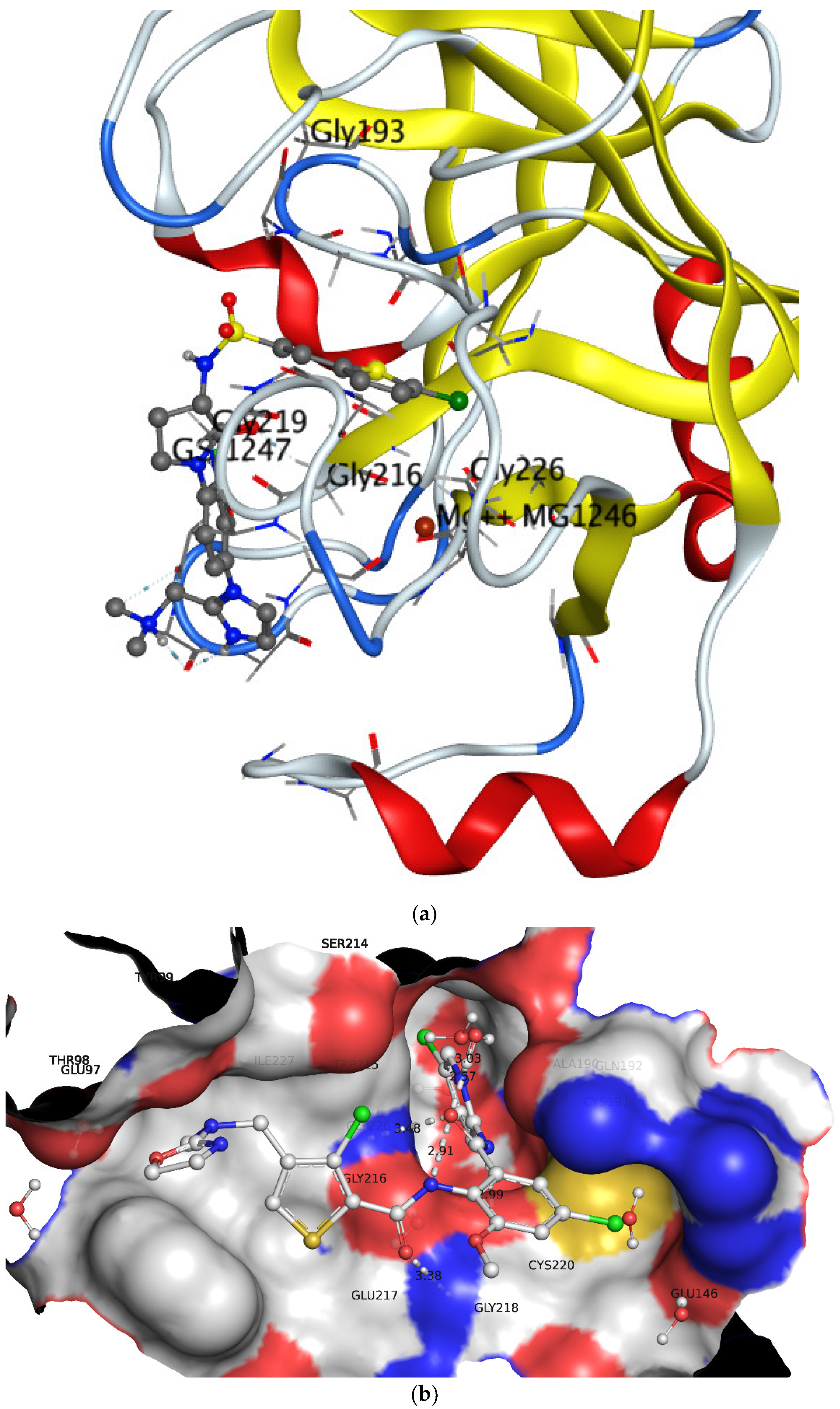
3.2. Comparison of QSAR Results with Reported Crystal Structures
4. Materials and Methods
4.1. Data Collection & Curation
4.2. Calculation of Molecular Descriptors and Objective Feature Selection (OFS)
4.3. Splitting the Data Set into Training and External Sets and Subjective Feature Selection (SFS)
4.4. Building Regression Model and Its Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SMILES | Simplified molecular-input line-entry system |
| GA | Genetic algorithm |
| MLR | Multiple linear regression |
| QSAR | Quantitative structure−activity relationship |
| WHO | World Health Organization |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| OLS | Ordinary least square |
| QSARINS | QSAR Insubria |
| OECD | Organisation for Economic Co-operation and Development |
References
- Satoh, K.; Satoh, T.; Yaoita, N.; Shimokawa, H. Recent Advances in the Understanding of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e159–e165. [Google Scholar] [CrossRef]
- Tripathi, N.; Tripathi, N.; Goshisht, M.K. COVID-19: Inflammatory responses, structure-based drug design and potential therapeutics. Mol. Divers. 2021, 1–17. [Google Scholar] [CrossRef]
- Kirby, T. New variant of SARS-CoV-2 in UK causes surge of COVID-19. Lancet Respir. Med. 2021, 9, e20–e21. [Google Scholar] [CrossRef]
- Chilamakuri, R.; Agarwal, S. COVID-19: Characteristics and Therapeutics. Cells 2021, 10, 206. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.A.; Urquiza, J.; Ramirez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef] [PubMed]
- Livingston, J.R.; Sutherland, M.R.; Friedman, H.M.; Pryzdial, E.L.G. Herpes simplex virus type 1-encoded glycoprotein C contributes to direct coagulation Factor X–virus binding. Biochem. J. 2005, 393, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Zalpour, A.; Kroll, M.H.; Afshar-Kharghan, V.; Yusuf, S.W.; Escalante, C. Role of factor xa inhibitors in cancer-associated thrombosis: Any new data? Adv. Hematol. 2011, 2011, 196135. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, H.E.; McBane, R.D., II; Wysokinski, W.E.; Tafur, A.J.; Loprinzi, C.L.; Murad, M.H.; Riaz, I.B. Direct Oral Factor Xa Inhibitors for the Treatment of Acute Cancer-Associated Venous Thromboembolism: A Systematic Review and Network Meta-analysis. Mayo Clin. Proc. 2019, 94, 2444–2454. [Google Scholar] [CrossRef]
- Mackman, N.; Bergmeier, W.; Stouffer, G.A.; Weitz, J.I. Therapeutic strategies for thrombosis: New targets and approaches. Nat. Rev. Drug Discov. 2020, 19, 333–352. [Google Scholar] [CrossRef]
- Nar, H. The role of structural information in the discovery of direct thrombin and factor Xa inhibitors. Trends Pharm. Sci 2012, 33, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Turpie, A.G.G. Oral, Direct Factor Xa Inhibitors in Development for the Prevention and Treatment of Thromboembolic Diseases. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1238–1247. [Google Scholar] [CrossRef]
- Patel, N.R.; Patel, D.V.; Murumkar, P.R.; Yadav, M.R. Contemporary developments in the discovery of selective factor Xa inhibitors: A review. Eur. J. Med. Chem. 2016, 121, 671–698. [Google Scholar] [CrossRef]
- Santana-Romo, F.; Lagos, C.F.; Duarte, Y.; Castillo, F.; Moglie, Y.; Maestro, M.A.; Charbe, N.; Zacconi, F.C. Innovative Three-Step Microwave-Promoted Synthesis of N-Propargyltetrahydroquinoline and 1,2,3-Triazole Derivatives as a Potential Factor Xa (FXa) Inhibitors: Drug Design, Synthesis, and Biological Evaluation. Molecules 2020, 25, 491. [Google Scholar] [CrossRef] [Green Version]
- Gramatica, P. Principles of QSAR Modeling. Int. J. Quant. Struct. Prop. Relatsh. 2020, 5, 61–97. [Google Scholar] [CrossRef]
- Fujita, T.; Winkler, D.A. Understanding the Roles of the “Two QSARs”. J. Chem. Inf. Model. 2016, 56, 269–274. [Google Scholar] [CrossRef]
- Masand, V.H.; Mahajan, D.T.; Ben Hadda, T.; Jawarkar, R.D.; Alafeefy, A.M.; Rastija, V.; Ali, M.A. Does tautomerism influence the outcome of QSAR modeling? Med. Chem. Res. 2014, 23, 1742–1757. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [Green Version]
- Masand, V.H.; Patil, M.K.; El-Sayed, N.N.E.; Zaki, M.E.A.; Almarhoon, Z.; Al-Hussain, S.A. Balanced QSAR analysis to identify the structural requirements of ABBV-075 (Mivebresib) analogues as bromodomain and extraterminal domain (BET) family bromodomain inhibitor. J. Mol. Struct. 2021, 1229, 129597. [Google Scholar] [CrossRef]
- Matter, H.; Will, D.W.; Nazare, M.; Schreuder, H.; Laux, V.; Wehner, V. Structural requirements for factor Xa inhibition by 3-oxybenzamides with neutral P1 substituents: Combining X-ray crystallography, 3D-QSAR, and tailored scoring functions. J. Med. Chem. 2005, 48, 3290–3312. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Arnaiz, D.O.; Chou, Y.L.; Griedel, B.D.; Karanjawala, R.; Lee, W.; Morrissey, M.M.; Sacchi, K.L.; Sakata, S.T.; Shaw, K.J.; et al. Thiophene-anthranilamides as highly potent and orally available factor Xa inhibitors. J. Med. Chem. 2007, 50, 2967–2980. [Google Scholar] [CrossRef]
- Consonni, V.; Todeschini, R.; Ballabio, D.; Grisoni, F. On the Misleading Use of Q2F3 for QSAR Model Comparison. Mol. Inform. 2019, 38, e1800029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krstajic, D.; Buturovic, L.J.; Leahy, D.E.; Thomas, S. Cross-validation pitfalls when selecting and assessing regression and classification models. J. Cheminform. 2014, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramatica, P. External Evaluation of QSAR Models, in Addition to Cross-Validation Verification of Predictive Capability on Totally New Chemicals. Mol. Inform. 2014, 33, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P. On the development and validation of QSAR models. Methods Mol. Biol. 2013, 930, 499–526. [Google Scholar]
- Huang, J.; Fan, X. Why QSAR fails: An empirical evaluation using conventional computational approach. Mol. Pharm. 2011, 8, 600–608. [Google Scholar] [CrossRef]
- Zaki, M.E.A.; Al-Hussain, S.A.; Masand, V.H.; Akasapu, S.; Lewaa, I. QSAR and Pharmacophore Modeling of Nitrogen Heterocycles as Potent Human N-Myristoyltransferase (Hs-NMT) Inhibitors. Molecules 2021, 26, 1834. [Google Scholar] [CrossRef]
- Masand, V.H.; El-Sayed, N.N.E.; Bambole, M.U.; Patil, V.R.; Thakur, S.D. Multiple quantitative structure-activity relationships (QSARs) analysis for orally active trypanocidal N-myristoyltransferase inhibitors. J. Mol. Struct. 2019, 1175, 481–487. [Google Scholar] [CrossRef]
- Masand, V.H.; Mahajan, D.T.; Nazeruddin, G.M.; Hadda, T.B.; Rastija, V.; Alfeefy, A.M. Effect of information leakage and method of splitting (rational and random) on external predictive ability and behavior of different statistical parameters of QSAR model. Med. Chem. Res. 2015, 24, 1241–1264. [Google Scholar] [CrossRef]
- Kar, S.; Roy, K.; Leszczynski, J. Applicability Domain: A Step toward Confident Predictions and Decidability for QSAR Modeling. In Computational Toxicology; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1800, pp. 141–169. [Google Scholar] [CrossRef]
- Schreuder, H.; Matter, H. Serine Proteinases from the Blood Coagulation Cascade. In Structural Biology in Drug Discovery; Structural Biology in Drug Discovery: Methods, Techniques, and Practices; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2020; pp. 395–422. [Google Scholar] [CrossRef]
- Zhou, Y.; Yao, Z.; Zhu, L.; Tang, Y.; Chen, J.; Wu, J. Safety of Dabigatran as an Anticoagulant: A Systematic Review and Meta-Analysis. Front. Pharm. 2021, 12, 626063. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Roy, P.P.; Kovarich, S.; Yap, C.W.; Papa, E. QSAR Modeling is not Push a Button and Find a Correlation: A Case Study of Toxicity of (Benzo-)triazoles on Algae. Mol. Inform. 2012, 31, 817–835. [Google Scholar] [CrossRef] [PubMed]
- Dearden, J.C.; Cronin, M.T.; Kaiser, K.L. How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR). SAR QSAR Environ. Res. 2009, 20, 241–266. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Ballabio, D.; Todeschini, R. Comments on the definition of the Q2 parameter for QSAR validation. J. Chem. Inf. Model. 2009, 49, 1669–1678. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Masand, V.H.; Rastija, V. PyDescriptor: A new PyMOL plugin for calculating thousands of easily understandable molecular descriptors. Chemom. Intell. Lab. Syst. 2017, 169, 12–18. [Google Scholar] [CrossRef]
- Martin, T.M.; Harten, P.; Young, D.M.; Muratov, E.N.; Golbraikh, A.; Zhu, H.; Tropsha, A. Does rational selection of training and test sets improve the outcome of QSAR modeling? J. Chem. Inf. Model. 2012, 52, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.P.; Kovarich, S.; Gramatica, P. QSAR model reproducibility and applicability: A case study of rate constants of hydroxyl radical reaction models applied to polybrominated diphenyl ethers and (benzo-)triazoles. J. Comput. Chem. 2011, 32, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models: How to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
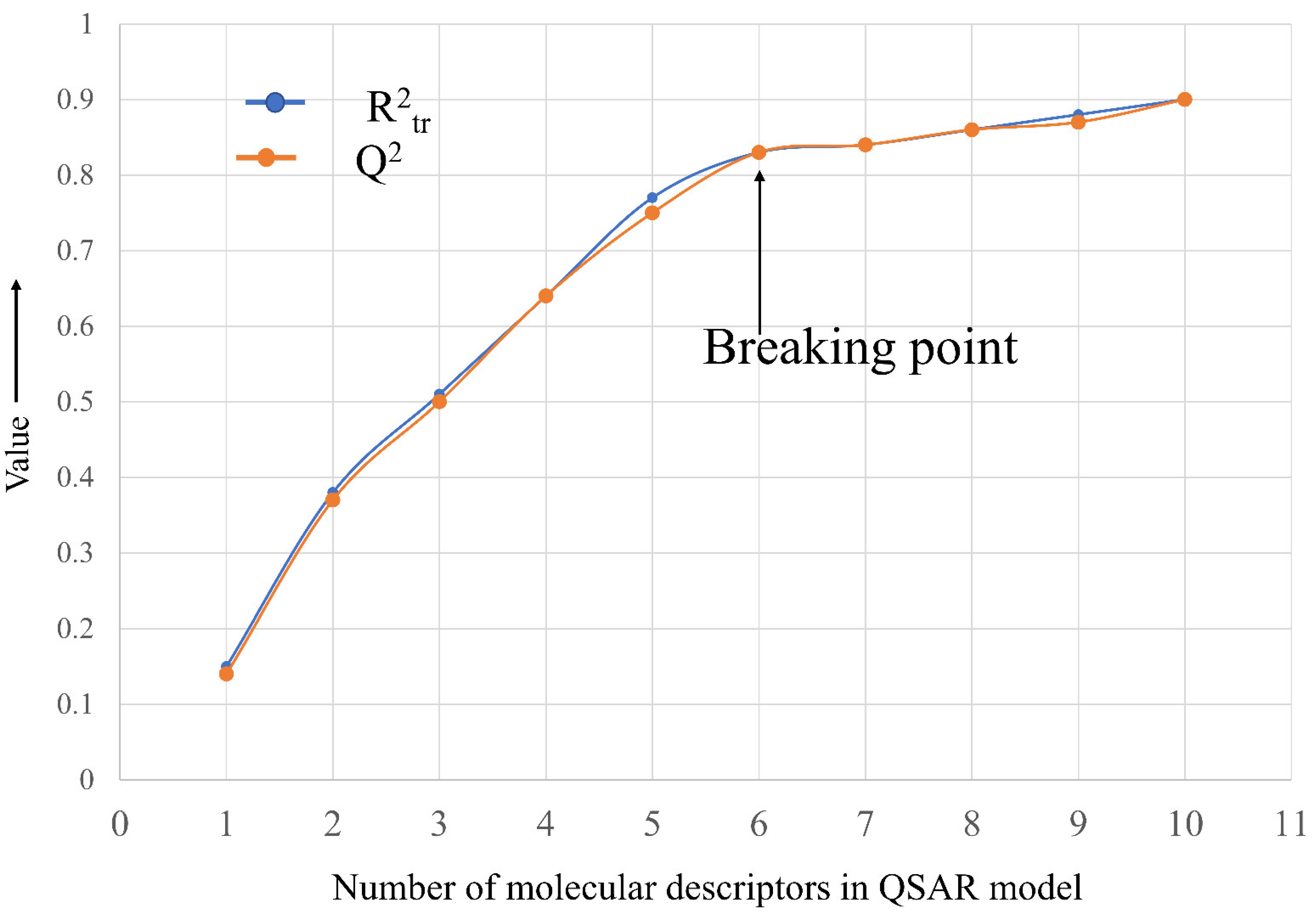{kind=link}
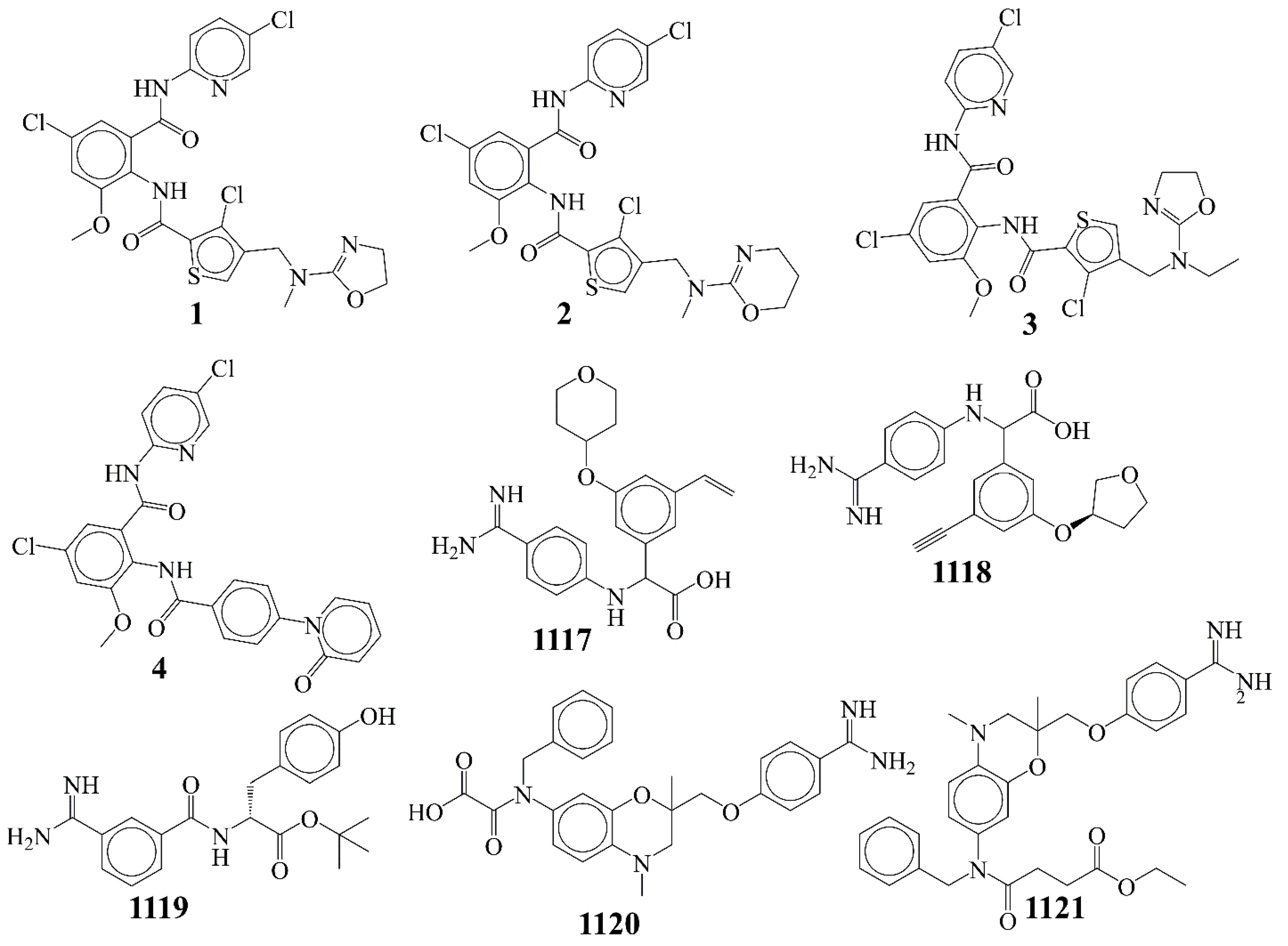
| S.N. | SMILES Notation | Ki (nM) | pKi (M) |
| 1 | COc1cc(Cl)cc(C(=O)Nc2ccc(Cl)cn2)c1NC(=O)c1scc(CN(C)C2=NCCO2)c1Cl | 0.007 | 11.155 |
| 2 | COc1cc(Cl)cc(C(=O)Nc2ccc(Cl)cn2)c1NC(=O)c1scc(CN(C)C2=NCCCO2)c1Cl | 0.012 | 10.921 |
| 3 | CCN(Cc1csc(C(=O)Nc2c(OC)cc(Cl)cc2C(=O)Nc2ccc(Cl)cn2)c1Cl)C1=NCCO1 | 0.012 | 10.921 |
| 4 | COc1cc(Cl)cc(C(=O)Nc2ccc(Cl)cn2)c1NC(=O)c1ccc(-n2ccccc2=O)cc1 | 0.013 | 10.886 |
| 5 | COc1cc(Cl)cc(C(=O)Nc2ccc(Cl)cn2)c1NC(=O)c1scc(CN(C)C2=NCCS2)c1Cl | 0.024 | 10.62 |
| 1117 | C=Cc1cc(OC2CCOCC2)cc(C(Nc2ccc(C(=N)N)cc2)C(=O)O)c1 | 13,300 | 4.876 |
| 1118 | C#Cc1cc(O[C@@H]2CCOC2)cc(C(Nc2ccc(C(=N)N)cc2)C(=O)O)c1 | 15,300 | 4.815 |
| 1119 | CC(C)(C)OC(=O)[C@@H](Cc1ccc(O)cc1)NC(=O)c1cccc(C(=N)N)c1 | 16,000 | 4.796 |
| 1120 | CN1CC(C)(COc2ccc(C(=N)N)cc2)Oc2cc(N(Cc3ccccc3)C(=O)C(=O)O)ccc21 | 16,600 | 4.78 |
| 1121 | CCOC(=O)CCC(=O)N(Cc1ccccc1)c1ccc2c(c1)OC(C)(COc1ccc(C(=N)N)cc1)CN2C | 18,000 | 4.745 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaki, M.E.A.; Al-Hussain, S.A.; Masand, V.H.; Sabnani, M.K.; Samad, A. Mechanistic and Predictive QSAR Analysis of Diverse Molecules to Capture Salient and Hidden Pharmacophores for Anti-Thrombotic Activity. Int. J. Mol. Sci. 2021, 22, 8352. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158352
Zaki MEA, Al-Hussain SA, Masand VH, Sabnani MK, Samad A. Mechanistic and Predictive QSAR Analysis of Diverse Molecules to Capture Salient and Hidden Pharmacophores for Anti-Thrombotic Activity. International Journal of Molecular Sciences. 2021; 22(15):8352. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158352
Chicago/Turabian StyleZaki, Magdi E. A., Sami A. Al-Hussain, Vijay H. Masand, Manoj K. Sabnani, and Abdul Samad. 2021. "Mechanistic and Predictive QSAR Analysis of Diverse Molecules to Capture Salient and Hidden Pharmacophores for Anti-Thrombotic Activity" International Journal of Molecular Sciences 22, no. 15: 8352. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158352