
CDK4, CDK6/cyclin-D1 Complex Inhibition and Radiotherapy for Cancer Control: A Role for Autophagy

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Molecular Background and Involved Pathways
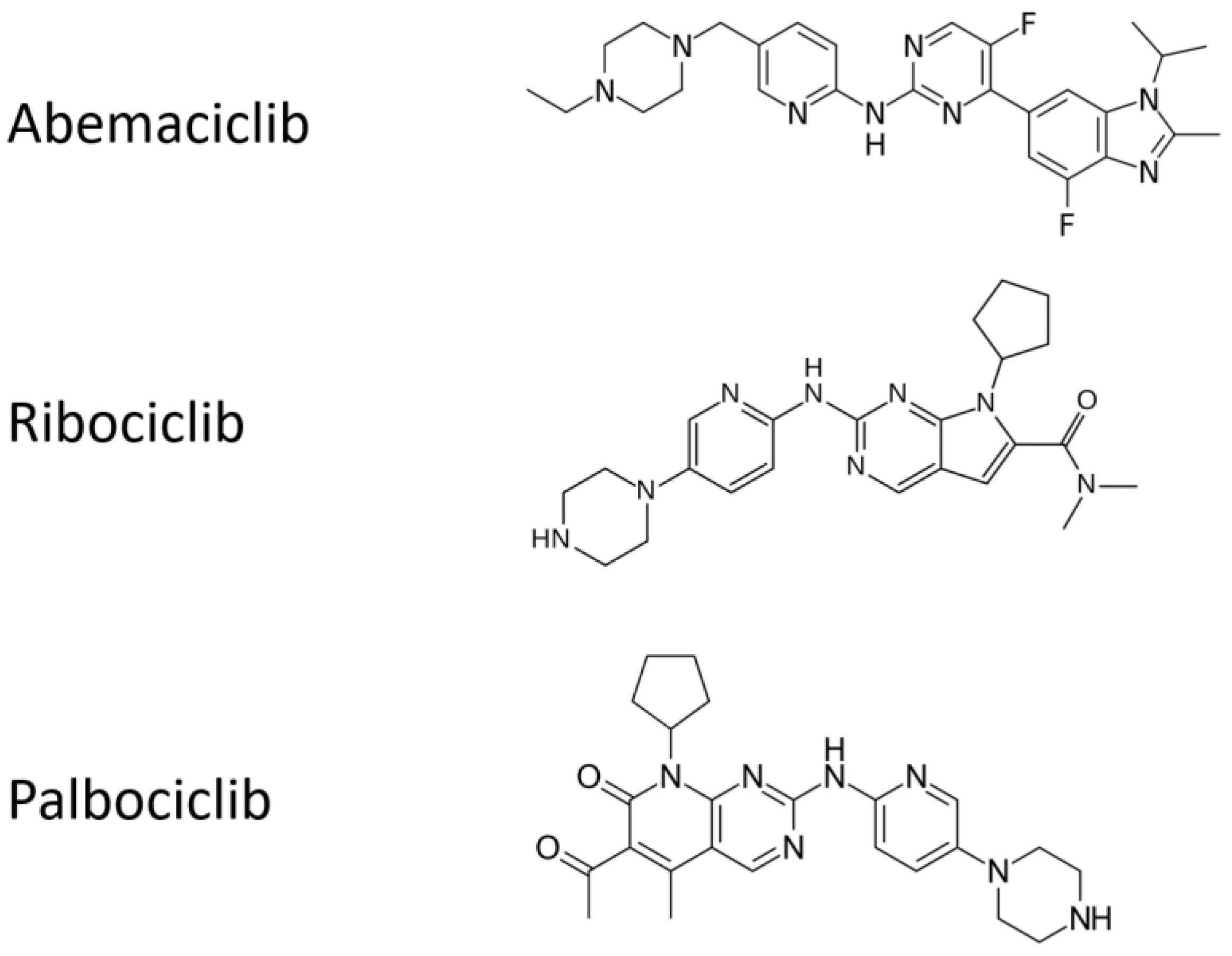
2.1. Antitumor Activity of Cyclin-D1/CDK4 and CDK6 Inhibition
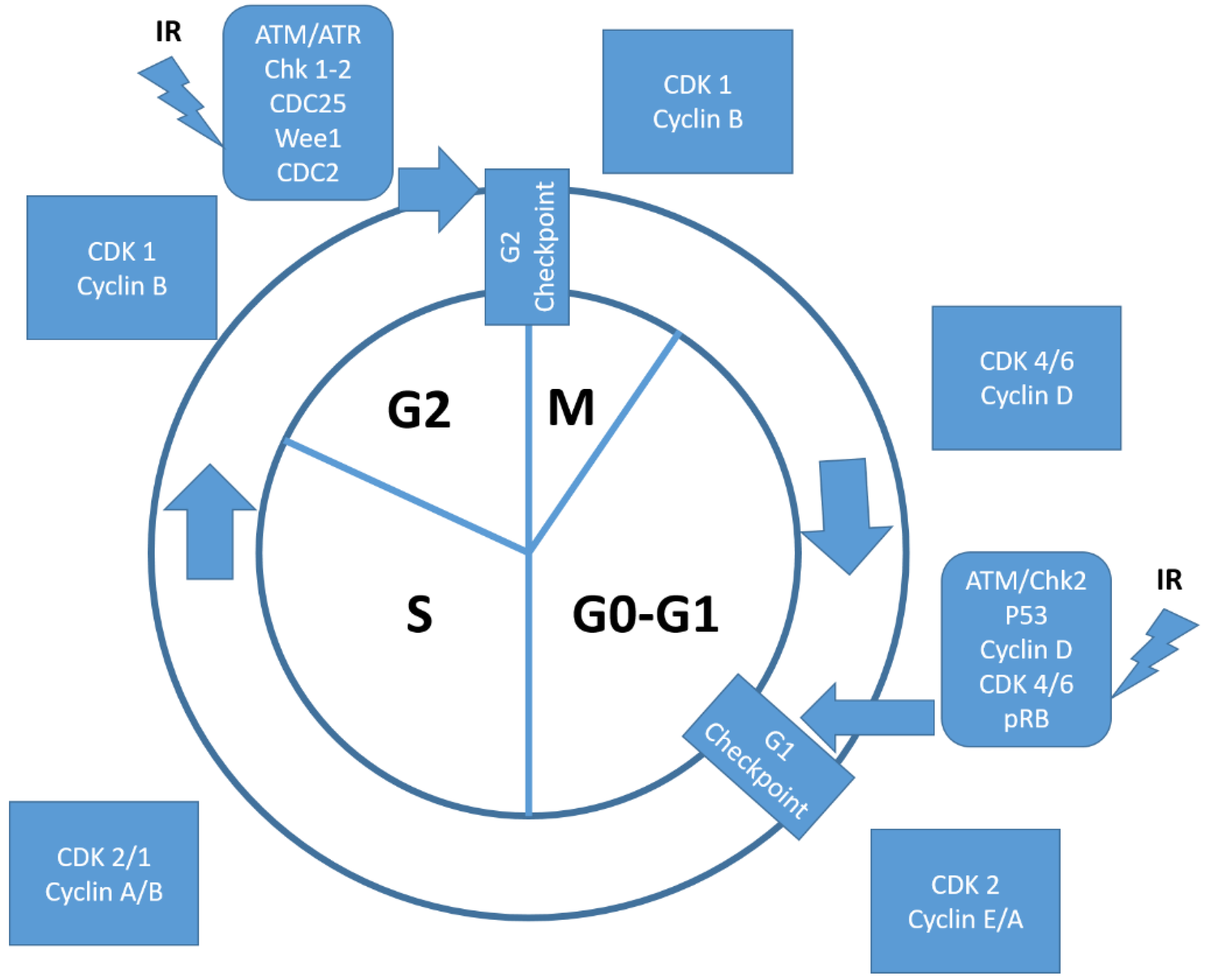
2.2. Ionizing Radiation and the Cyclin-D1/CDK4 and CDK6 Pharmacological Inhibition: Preclinical Evidence
2.3. Autophagy and the Cell Cycle
2.4. The PI3K/AKT/mTOR Pathway in Endocrine Resistant ESR1+/HER2− Breast Cancer, and in other Neoplasms
3. The State of the Art in the Clinical Domain
3.1. Clinical Trials of Cyclin-D1/CDK4 and CDK6 Inhibition in Breast Cancer
3.2. Real-Life Clinical Reports on Cyclin-D1/CDK4 and CDK6 Inhibitors, Including Association with Radiotherapy
4. Molecular Grounds for Future Developments
4.1. CCND1 as a Target of Ionizing Radiation in the CCND1-CDK4, and CDK6 Complex
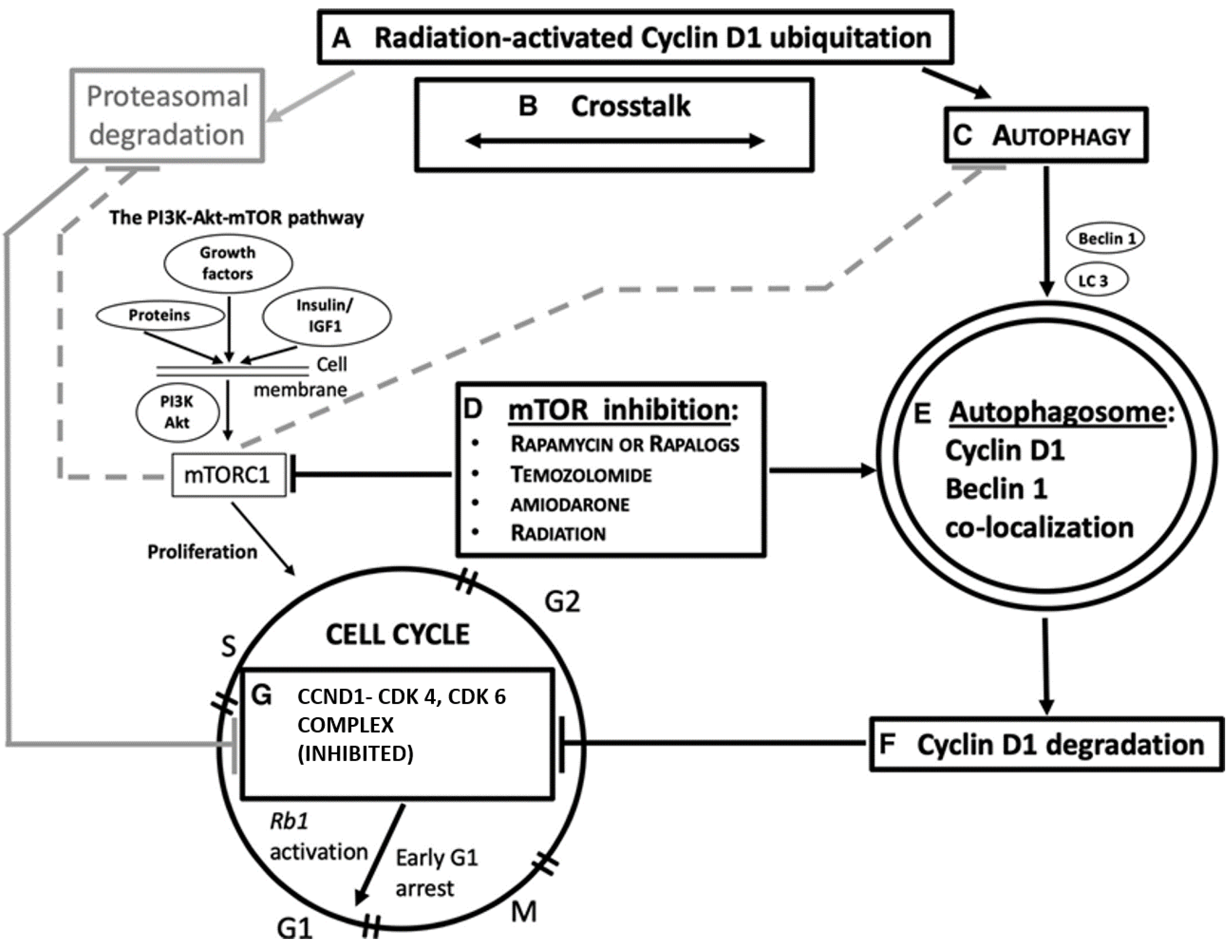
4.2. Ionizing Radiation, Autophagy Enhancement, and CCND1
5. Concluding Remarks, with Special Regard to Immunotherapy Developments
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation resistance of cancer stem cells: The 4 R’s of radiobiology revisited. Stem Cells 2010, 28, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.; Madani, D.; Joshi, S.; Chung, S.A.; Johns, T.; Day, B.; Khasraw, M.; McDonald, K.L. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov. 2017, 3, 17033. [Google Scholar] [CrossRef] [PubMed]
- Nardone, V.; Falivene, S.; Giugliano, F.M.; Gaetano, M.; Giordano, P.; Muto, M.; Daniele, B.; Guida, C. The role of radiation therapy and systemic therapies in elderly with breast cancer. Transl. Cancer Res. 2020, 9, S97–S109. [Google Scholar] [CrossRef]
- Renzulli, M.; Zanotti, S.; Clemente, A.; Mineo, G.; Tovoli, F.; Reginelli, A.; Barile, A.; Cappabianca, S.; Taffurelli, M.; Golfieri, R. Hereditary breast cancer: Screening and risk reducing surgery. Gland. Surg. 2019, 8, S142–S149. [Google Scholar] [CrossRef] [Green Version]
- Sicinska, E.; Aifantis, I.; Le Cam, L.; Swat, W.; Borowski, C.; Yu, Q.; Ferrando, A.A.; Levin, S.D.; Geng, Y.; Von Boehmer, H.; et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 2003, 4, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Y.; Lan, S.H.; Wu, S.R.; Chiu, Y.C.; Lin, X.Z.; Su, I.J.; Tsai, T.F.; Yen, C.J.; Lu, T.H.; Liang, F.W.; et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Haepatology 2018, 68, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [Green Version]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef] [PubMed]
- MacLachlan, T.K.; Sang, N.; Giordano, A. Cyclins, cyclin-dependent kinases and cdk inhibitors: Implications in cell cycle control and cancer. Crit. Rev. Eukaryot. Gene Expr. 1995, 5, 127–156. [Google Scholar] [CrossRef] [PubMed]
- Prokhorova, E.A.; Egorshina, A.Y.; Zhivotovsky, B.; Kopeina, G.S. The DNA-damage response and nuclear events as regulators of nonapoptotic forms of cell death. Oncogene 2019, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010, 18, 63–73. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [Green Version]
- Hortobagyi, G.; Stemmer, S.; Burris, H.; Yap, Y.-S.; Sonke, G.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.; Winer, E.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 2019, 30, 1842. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, W.K.; Paules, R.S. DNA damage and cell cycle checkpoints. FASEB J. 1996, 10, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Millis, S.Z.; Ross, J.S.; Woo, M.S.; Ali, S.M.; Kurzrock, R. Cyclin Pathway Genomic Alterations Across 190,247 Solid Tumors: Leveraging Large-Scale Data to Inform Therapeutic Directions. Oncologist 2020, 26, e78–e89. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Li, X.; Hydbring, P.; Sanda, T.; Stefano, J.; Christie, A.L.; Signoretti, S.; Look, A.T.; Kung, A.; von Boehmer, H.; et al. The Requirement for Cyclin D Function in Tumor Maintenance. Cancer Cell 2012, 22, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawai, C.M.; Freund, J.; Oh, P.; Ndiaye-Lobry, D.; Bretz, J.C.; Strikoudis, A.; Genesca, L.; Trimarchi, T.; Kelliher, M.A.; Clark, M.; et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell. 2012, 22, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.G.; Deshpande, A.; Enos, M.; Mao, D.; Hinds, E.A.; Hu, G.F.; Chang, R.; Guo, Z.; Dose, M.; Mao, C.; et al. A requirement for cyclin-dependent kinase 6 in thymocyte development and tumorigenesis. Cancer Res. 2009, 69, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.E.; Ichimura, K.; Reifenberger, G.; Collins, V.P. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994, 54, 6321–6324. [Google Scholar]
- Wölfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wolfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Zum Buschenfelde, K.M.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Lui, K.W.; Chi, L.M.; Kuo, Y.C.; Chao, Y.K.; Yeh, C.N.; Lee, L.Y.; Huang, Y.; Lin, T.L.; Huang, M.Y.; et al. Integrated genomic analyses in PDX model reveal a cyclin-dependent kinase inhibitor Palbociclib as a novel candidate drug for nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 233. [Google Scholar] [CrossRef]
- Kaur, G.; Stetler-Stevenson, M.; Sebers, S.; Worland, P.; Sedlacek, H.; Myers, C.; Czech, J.; Naik, R.; Sausville, E. Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone L86-8275. J. Natl. Cancer Inst. 1992, 84, 1736–1740. [Google Scholar] [CrossRef]
- Sammons, S.L.; Topping, D.L.; Blackwell, K.L. HR+, HER2- Advanced Breast Cancer and CDK4/6 Inhibitors: Mode of Action, Clinical Activity, and Safety Profiles. Curr. Cancer Drug Targets 2017, 17, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, W.K.; Morton, R.A. X-ray sensitivity during the cell generation cycle of cultured Chinese hamster cells. Radiat. Res. 1966, 29, 450–474. [Google Scholar] [CrossRef] [PubMed]
- Gurley, L.R.; Walters, R.A.; Tobey, R.A. Cell cycle-specific changes in histone phosphorylation associated with cell proliferation and chromosome condensation. J. Cell. Biol. 1974, 60, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Terzoudi, G.I.; Pantelias, G.E. Conversion of DNA damage into chromosome damage in response to cell cycle regulation of chromatin condensation after irradiation. Mutagenesis 1997, 12, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Luo, J.; Chen, X.; Yang, Z.; Mei, X.; Ma, J.; Zhang, Z.; Guo, X.; Yu, X. CDK4/6 inhibitors: A novel strategy for tumor radiosensitization. J. Exp. Clin. Cancer Res. 2020, 39, 188. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.A.; Kim, W.; Jo, H.-R.; Shin, Y.-J.; Kim, M.-H.; Jeong, J.-H. Anticancer and radiosensitizing effects of the cyclin-dependent kinase inhibitors, AT7519 and SNS-032, on cervical cancer. Int. J. Oncol. 2018, 53, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.P.; Zhu, Y.L.; Ratner, E.S. Targeting Cyclin-Dependent Kinases for Treatment of Gynecologic Cancers. Front. Oncol. 2018, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.L.; Oh, P.; Xu, E.S.; Ma, Y.; Kim, Y.; Daniel, A.R.; Kirsch, D.G. Blocking Cyclin-Dependent Kinase 4/6 During Single Dose Versus Fractionated Radiation Therapy Leads to Opposite Effects on Acute Gastrointestinal Toxicity in Mice. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 1569–1576. [Google Scholar] [CrossRef]
- Naz, S.; Sowers, A.; Choudhuri, R.; Wissler, M.; Gamson, J.; Mathias, A.; Cook, J.A.; Mitchell, J.B. Abemaciclib, a Selective CDK4/6 Inhibitor, Enhances the Radiosensitivity of Non-Small Cell Lung Cancer In Vitro and In Vivo. Clin. Cancer Res. 2018, 24, 3994–4005. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.-Y.; Nam, K.-Y.; Park, J.-I.; Song, K.-H.; Ahn, J.; Park, J.K.; Um, H.-D.; Hwang, S.-G.; Choi, S.U.; Song, J.-Y. Radiosensitizing Effect of Novel Phenylpyrimidine Derivatives on Human Lung Cancer Cells via Cell Cycle Perturbation. J. Pharmacol. Exp. Ther. 2019, 370, 514–527. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y.; Naderi, S.; Chen, T.T. Role of retinoblastoma tumor suppressor protein in DNA damage response. Acta. Oncol. 2001, 40, 689–695. [Google Scholar] [PubMed]
- Juric, V.; Murphy, B. Cyclin-dependent kinase inhibitors in brain cancer: Current state and future directions. Cancer Drug Resist. 2020, 3, 48–65. [Google Scholar] [CrossRef] [Green Version]
- Naz, S.; Cook, J.A.; Mitchell, J.B. Abemaciclib: A multi-functional radiation modifier. Oncotarget 2019, 10, 1230–1232. [Google Scholar] [CrossRef]
- Johnson, S.M.; Torrice, C.D.; Bell, J.F.; Monahan, K.B.; Jiang, Q.; Wang, Y.; Ramsey, M.R.; Jin, J.; Wong, K.K.; Su, L.; et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J. Clin. Investig. 2010, 120, 2528–2536. [Google Scholar] [CrossRef]
- Wei, L.; Leibowitz, B.J.; Wang, X.; Epperly, M.; Greenberger, J.; Zhang, L.; Yu, J. Inhibition of CDK4/6 protects against radiation-induced intestinal injury in mice. J. Clin. Investig. 2016, 126, 4076–4087. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.L.; Hill, G.A.; Klein, R.R.; Arnett, D.G.; Burd, R.; Limesand, K.H. Prevention of radiation-induced salivary gland dysfunction utilizing a CDK inhibitor in a mouse model. PLoS ONE 2012, 7, e51363. [Google Scholar] [CrossRef] [Green Version]
- Janji, B.; Viry, E.; Baginska, J.; Van Moer, K.; Berchem, G. Role of Autophagy in Cancer and Tumor Progression. In Autophagy—A Double-Edged Sword—Cell Survival or Death? Bailly, Y., Ed.; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Toscano, M.; Palumbo, S.; Tini, P.; Miracco, C.; Gravina, G.; Comincini, S. Cell Death Pathways, with Special Regard to Ionizing Radiation and Temozolomide. In Radiobiology of Glioblastoma-Recent Advances and Related Pathobiology; Pirtoli, L., Gravina, G.L., Giordano, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 209–224. [Google Scholar] [CrossRef]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell. 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Andò, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef] [Green Version]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in life and death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef]
- Polager, S.; Ofir, M.; Ginsberg, D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008, 27, 4860–4864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Martin, V.; Gomez-Manzano, C.; Johnson, D.G.; Alonso, M.; White, E.; Xu, J.; McDonnell, T.J.; Shinojima, N.; Fueyo, J. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010, 70, 7882–7893. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; He, Z.; Kitazato, K.; Wang, Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics 2019, 9, 104–125. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iriyama, N.; Hino, H.; Moriya, S.; Hiramoto, M.; Hatta, Y.; Takei, M.; Miyazawa, K. The cyclin-dependent kinase 4/6 inhibitor, abemaciclib, exerts dose-dependent cytostatic and cytocidal effects and induces autophagy in multiple myeloma cells. Leuk. Lymphoma 2018, 59, 1439–1450. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Ding, X.F.; Bouamar, H.; Pressley, K.; Sun, L.Z. Everolimus induces G(1) cell cycle arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells. Am. J. Physiol. Cell. Physiol. 2019, 317, C244–C252. [Google Scholar] [CrossRef] [PubMed]
- Pirtoli, L.; Belmonte, G.; Toscano, M.; Tini, P.; Miracco, C. Cyclin D1 Co-localizes with Beclin-1 in Glioblastoma Recurrences: A Clue to a Therapy-induced, Autophagy-mediated Degradative Mechanism? Anticancer Res. 2016, 36, 4057–4062. [Google Scholar] [PubMed]
- Palumbo, S.; Pirtoli, L.; Tini, P.; Cevenini, G.; Calderaro, F.; Toscano, M.; Miracco, C.; Comincini, S. Different involvement of autophagy in human malignant glioma cell lines undergoing irradiation and temozolomide combined treatments. J. Cell. Biochem. 2012, 113, 2308–2318. [Google Scholar] [CrossRef]
- Bostner, J.; Skoog, L.; Fornander, T.; Nordenskjöld, B.; Stål, O. Estrogen receptor-alpha phosphorylation at serine 305, nuclear p21-activated kinase 1 expression, and response to tamoxifen in postmenopausal breast cancer. Clin. Cancer Res. 2010, 16, 1624–1633. [Google Scholar] [CrossRef] [Green Version]
- Presti, D.; Quaquarini, E. The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2- Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers 2019, 11, 1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulany, M.; Rodemann, H.P. Potential of Akt mediated DNA repair in radioresistance of solid tumors overexpressing erbB-PI3K-Akt pathway. Transl. Cancer Res. 2013, 2, 190–202. [Google Scholar]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef] [Green Version]
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 4760–4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2020, 382, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Rossi, V.; Berchialla, P. Should All Patients with HR-Positive HER2-Negative Metastatic Breast Cancer Receive CDK 4/6 Inhibitor as First-Line Based Therapy? A Network Meta-Analysis of Data from the PALOMA 2, MONALEESA 2, MONALEESA 7, MONARCH 3, FALCON, SWOG and FACT trials. Cancers 2019, 11, 1661. [Google Scholar] [CrossRef] [Green Version]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Li, J.; Huo, X.; Zhao, F.; Ren, D.; Ahmad, R.; Yuan, X.; Du, F.; Zhao, J. Association of Cyclin-Dependent Kinases 4 and 6 Inhibitors with Survival in Patients with Hormone Receptor-Positive Metastatic Breast Cancer: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e2020312. [Google Scholar] [CrossRef]
- Meattini, I.; Desideri, I.; Scotti, V.; Simontacchi, G.; Livi, L. Ribociclib plus letrozole and concomitant palliative radiotherapy for metastatic breast cancer. Breast 2018, 42, 1–2. [Google Scholar] [CrossRef]
- Ratosa, I.; Orazem, M.; Scoccimarro, E.; Steinacher, M.; Dominici, L.; Aquilano, M.; Cerbai, C.; Desideri, I.; Ribnikar, D.; Marinko, T.; et al. Cyclin-Dependent Kinase 4/6 Inhibitors Combined with Radiotherapy for Patients with Metastatic Breast Cancer. Clin. Breast Cancer 2020, 20, 495–502. [Google Scholar] [CrossRef]
- Ippolito, E.; Greco, C.; Silipigni, S.; Dell’Aquila, E.; Petrianni, G.M.; Tonini, G.; Fiore, M.; D’Angelillo, R.M.; Ramella, S. Concurrent radiotherapy with palbociclib or ribociclib for metastatic breast cancer patients: Preliminary assessment of toxicity. Breast 2019, 46, 70–74. [Google Scholar] [CrossRef]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casimiro, M.C.; Di Sante, G.; Di Rocco, A.; Loro, E.; Pupo, C.; Pestell, T.G.; Bisetto, S.; Velasco-Velázquez, M.A.; Jiao, X.; Li, Z.; et al. Cyclin D1 Restrains Oncogene-Induced Autophagy by Regulating the AMPK-LKB1 Signaling Axis. Cancer Res. 2017, 77, 3391–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, J.; Zhao, W.; Gu, W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol. Cell 2009, 36, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, D.W.; Baek, H.J.; Motoyama, N.; Cho, K.H.; Kim, H.S.; Kim, S.S. ATM is required for rapid degradation of cyclin D1 in response to gamma-irradiation. Biochem. Biophys. Res. Commun. 2009, 378, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Paglin, S.; Lee, N.Y.; Nakar, C.; Fitzgerald, M.; Plotkin, J.; Deuel, B.; Hackett, N.; McMahill, M.; Sphicas, E.; Lampen, N.; et al. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005, 65, 11061–11070. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Moretti, L.; Mitchell, L.R.; Jung, D.K.; Lu, B. Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2alpha in caspase-3/7-deficient cells. Oncogene 2010, 29, 3241–3251. [Google Scholar] [CrossRef] [Green Version]
- De Cataldo, C.; Bruno, F.; Palumbo, P.; Di Sibio, A.; Arrigoni, F.; Clemente, A.; Bafile, A.; Gravina, G.L.; Cappabianca, S.; Barile, A.; et al. Apparent diffusion coefficient magnetic resonance imaging (ADC-MRI) in the axillary breast cancer lymph node metastasis detection: A narrative review. Gland Surg. 2020, 9, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Reginelli, A.; Silvestro, G.; Fontanella, G.; Sangiovanni, A.; Conte, M.; Nuzzo, I.; Calvanese, M.; Traettino, M.; Ferraioli, P.; Grassi, R.; et al. Validation of DWI in assessment of radiotreated bone metastases in elderly patients. Int. J. Surg. 2016, 33, S148–S153. [Google Scholar] [CrossRef]
- Brown, N.E.; Jeselsohn, R.; Bihani, T.; Hu, M.G.; Foltopoulou, P.; Kuperwasser, C.; Hinds, P.W. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012, 72, 6477–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirtoli, L.; Belmonte, G.; Toscano, M.; Tini, P.; Miracco, C. Comment on “Everolimus induces G1 cell cycle arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells”. Am. J. Physiol. Physiol. 2020, 318, C448–C449. [Google Scholar] [CrossRef] [Green Version]
- Pirtoli, L.; Cevenini, G.; Tini, P.; Vannini, M.; Oliveri, G.; Marsili, S.; Mourmouras, V.; Rubino, G.; Miracco, C. The prognostic role of Beclin 1 protein expression in high-grade gliomas. Autophagy 2009, 5, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Matthew-Onabanjo, A.N.; Janusis, J.; Mercado-Matos, J.; Carlisle, A.; Kim, D.; Levine, F.; Cruz-Gordillo, P.; Richards, R.; Lee, M.J.; Shaw, L.M. Beclin 1 Promotes Endosome Recruitment of Hepatocyte Growth Factor Tyrosine Kinase Substrate to Suppress Tumor Proliferation. Cancer Res. 2019, 80, 249–262. [Google Scholar] [CrossRef]
- Schreiber, K.H.; Apelo, S.I.A.; Yu, D.; Brinkman, J.A.; Velarde, M.C.; Syed, F.A.; Liao, C.-Y.; Baar, E.L.; Carbajal, K.A.; Sherman, D.S.; et al. A novel rapamycin analog is highly selective for mTORC1 in vivo. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef] [PubMed]
- Tini, P.; Nardone, V.; Pastina, P.; Pirtoli, L.; Correale, P.; Giordano, A. The effects of radiotherapy on the survival of patients with unresectable non-small cell lung cancer. Expert Rev. Anticancer Ther. 2018, 18, 593–602. [Google Scholar] [CrossRef]
- Nardone, V.; Pastina, P.; Giannicola, R.; Agostino, R.; Croci, S.; Tini, P.; Pirtoli, L.; Giordano, A.; Tagliaferri, P.; Correale, P. How to Increase the Efficacy of Immunotherapy in NSCLC and HNSCC: Role of Radiation Therapy, Chemotherapy, and Other Strategies. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Phipps, A.I.; Peters, U.; Milner, D.A., Jr.; Giovannucci, E.L.; Nishihara, R.; Giannakis, M.; Garrett, W.S.; et al. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut 2018, 67, 1168–1180. [Google Scholar] [CrossRef]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Milner, D.A., Jr.; Nishihara, R. Insights into Pathogenic Interactions Among Environment, Host, and Tumor at the Crossroads of Molecular Pathology and Epidemiology. Annu. Rev. Pathol. 2019, 14, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, Y.; Gao, X.; Li, Y.; Lin, J.; Chen, L.; Chang, L.; Chen, G.; Guan, Y.; Pan, L.K.; et al. CCND1 Amplification Contributes to Immunosuppression and Is Associated with a Poor Prognosis to Immune Checkpoint Inhibitors in Solid Tumors. Front. Immunol. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Scheme. | INTERVENTION | PATIENT CHARACTERISTICS | MEDIAN PFS(Months) | Clinical Gain/Approval |
|---|---|---|---|---|
| PALOMA-1 | Palbociclib + letrozole vs. letrozole alone | Post-menopausal women with untreated ER+/HER2−advanced breast cancer | 20.2 vs. 10.2 | Significant gain in terms of median PFS (10 Months). FDA approval in 2015 |
| PALOMA-2 | Palbociclib + letrozole vs. letrozole alone | ESR1+/HER2−advanced breast cancer | 27.6 vs. 14.5 | Delayed ChT: 40.4 vs. 29.9 months |
| PALOMA-3 | Palbociclib + fulvestrant vs. placebo + fulvestrant | ESR1+/HER2− metastatic breast cancer after hormone therapy (30% received prior ChT) | 9.5 (11.2) vs. 4.6 | Significant gain in terms of median PFS |
| MONALEESA-2 | Ribociclib + letrozole vs. letrozole alone | Postmenopausal women with ESR1+/HER2 advanced breast cancer | Not reached vs. 14.7 | FDA approval in 2017 |
| MONARCH-3 | Abemaciclib + aromatase inhib. vs. placebo + aromatase inhib. | Postmenopausal women with ESR1+/HER2 locoregionally recurrent or metastatic breast cancer with no prior systemic therapy | 28.1 vs. 14.7 | Significantly prolonged PFSFDA approval in 2018 |
| NCT Number | Study Type | Cancer Type | Trial Title | Sponsors and Collaborators |
|---|---|---|---|---|
| NCT02290145 | Interventional, Phase II | Squamous cell carcinoma of mouth | CCND1 Based TPF Induction Chemotherapy for Oral Squamous Cell Carcinoma Patients at Clinical N2 Stage | •Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, Shanghai, China |
| NCT04585724 | Interventional, Phase I | Metastatic breast carcinoma | Stereotactic Radiosurgery with Abemaciclib, Ribociclib, or Palbociclib in Treating Patients with Hormone Receptor Positive Breast Cancer with Brain Metastases | •Grady Health System, Atlanta, GA, United States |
| NCT03024489 | Interventional, Phase II | Head and neck cancer | Palbociclib with Cetuximab and IMRT for Locally Advanced Squamous Cell Carcinoma | •Faculty of Medicine, Ramathibodi Hospital, Bangkok, Thailand |
| NCT02607124 | Interventional, Phase II | High grade glioma | A Phase I/II Study of Ribociclib, a CDK4 and CDK6 Inhibitor, Following Radiation Therapy | •Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States |
| NCT04298983 | Interventional, Phase II | Prostate cancer | Abemaciclib in Combination with Androgen Deprivation Therapy for Locally Advanced Prostate Cancer | University of Alabama at Birmingham |
| NCT04923542 | Interventional, Phase II | Brain metastases from breast cancer | Stereotactic Radiation and Abemaciclib in the Management of ESR1+/HER2- Breast Cancer Brain Metastases | •H. Lee Moffitt Cancer Centerand Research Institute |
| NCT04220892 | Interventional, Phase I | High grade glioma | Pilot Study of Pembrolizumab Combined with Pemetrexed or Abemaciclib for High Grade Glioma | •Jose Carrillo •Eli Lilly and Company •John Wayne Cancer Institute |
| NCT03355794 | Interventional, Phase I | Cerebral glioma | A Study of Ribociclib and Everolimus Following Radiation Therapy in Children with Newly Diagnosed Non-biopsied Diffuse Pontine Gliomas (DIPG) and RB+ Biopsied DIPG and High Grade Gliomas (HGG) | Children’s Hospital Medical Center, Cincinnati, OH, United States •Novartis |
| NCT03691493 | Interventional, Phase II | Bone metastases from breast cancer | Radiation Therapy, Palbociclib, and Hormone Therapy in Treating Breast Cancer Patients with Bone Metastasis | •Emory University •Pfizer |
| NCT03870919 | Interventional, Phase I | Breast cancer stage IV | Locoregional Treatment and Palbociclib in de Novo, Treatment Naive, Stage IV ESR1+, HER2- Breast Cancer Patients | •UNICANCER •Pfizer |
| NCT03024489 | Interventional, Phase II | Head and neck cancer | Palbociclib with Cetuximab and IMRT for Locally Advanced Squamous Cell Carcinoma | •Mahidol University |
| NCT04563507 | Interventional, Phase II | Breast cancer stage IV | Combined Immunotherapies in Metastatic ESR1+ Breast Cancer | •Weill Medical College of Cornell University |
| NCT03389477 | Interventional, Phase II | Head and neck cancer | Los Tres Paso: Neoadjuvant Palbociclib Monotherapy, Concurrent Chemoradiation Therapy, Adjuvant Palbociclib Monotherapy in Patients with p16INK4a Negative, HPV- Unrelated Head and Neck Squamous Cell Carcinoma | •Washington University School of Medicine •Pfizer |
| NCT04605562 | Interventional, Phase II | Nasopharyngeal carcinoma | Umbrella Biomarker-Guided Therapy in NPC | •Sun Yat-sen University |
| NCT02624973 | Interventional, Phase II | Breast cancer | PErsonalized TREatment of High-risk MAmmary Cancer—the PETREMAC Trial | •Haukeland University Hospital •Helse Vest •Pfizer •AstraZeneca |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardone, V.; Barbarino, M.; Angrisani, A.; Correale, P.; Pastina, P.; Cappabianca, S.; Reginelli, A.; Mutti, L.; Miracco, C.; Giannicola, R.; et al. CDK4, CDK6/cyclin-D1 Complex Inhibition and Radiotherapy for Cancer Control: A Role for Autophagy. Int. J. Mol. Sci. 2021, 22, 8391. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168391
Nardone V, Barbarino M, Angrisani A, Correale P, Pastina P, Cappabianca S, Reginelli A, Mutti L, Miracco C, Giannicola R, et al. CDK4, CDK6/cyclin-D1 Complex Inhibition and Radiotherapy for Cancer Control: A Role for Autophagy. International Journal of Molecular Sciences. 2021; 22(16):8391. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168391
Chicago/Turabian StyleNardone, Valerio, Marcella Barbarino, Antonio Angrisani, Pierpaolo Correale, Pierpaolo Pastina, Salvatore Cappabianca, Alfonso Reginelli, Luciano Mutti, Clelia Miracco, Rocco Giannicola, and et al. 2021. "CDK4, CDK6/cyclin-D1 Complex Inhibition and Radiotherapy for Cancer Control: A Role for Autophagy" International Journal of Molecular Sciences 22, no. 16: 8391. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168391