
Expression of Low Level of VPS35-mCherry Fusion Protein Diminishes Vps35 Depletion Induced Neuron Terminal Differentiation Deficits and Neurodegenerative Pathology, and Prevents Neonatal Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
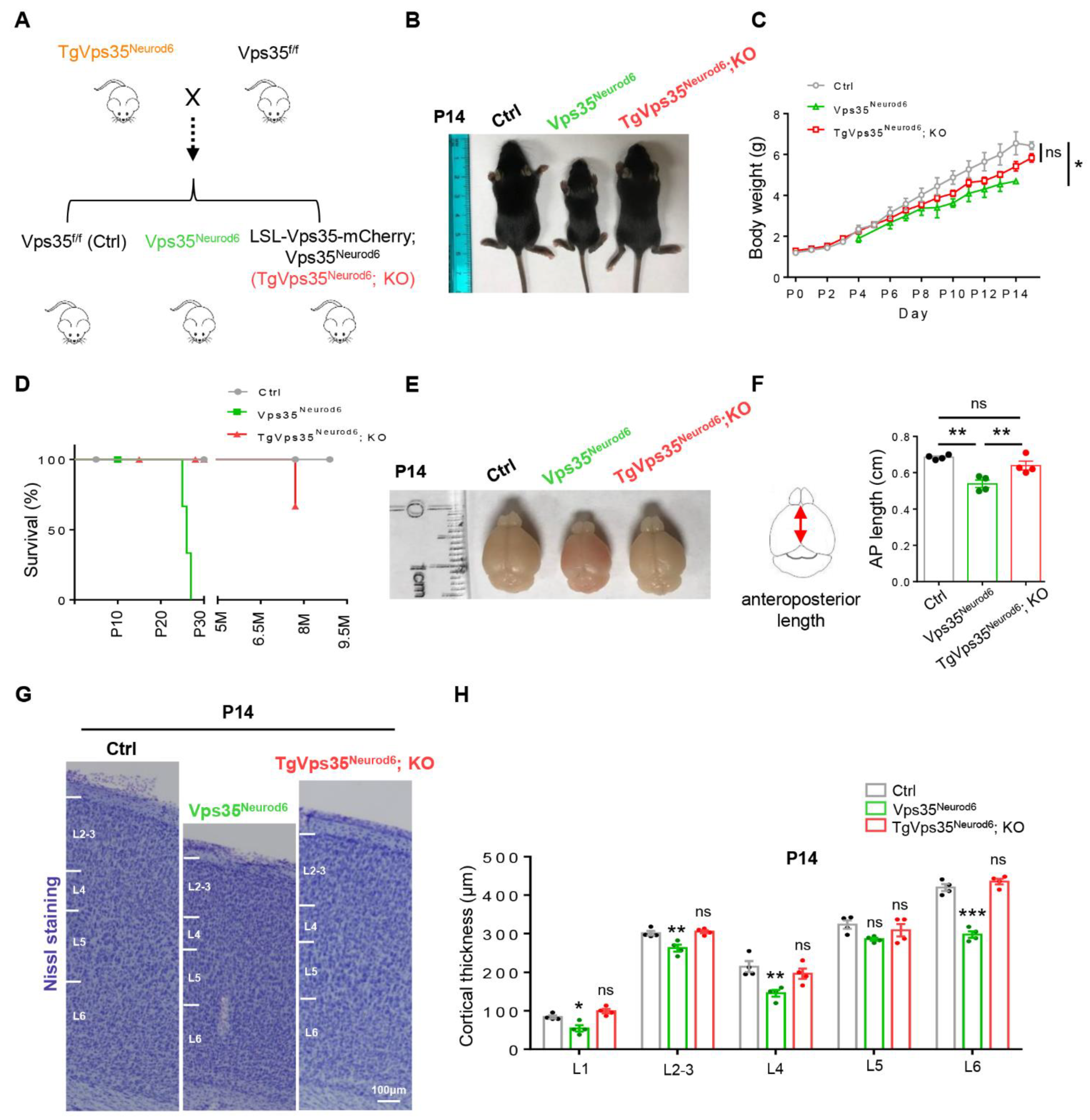
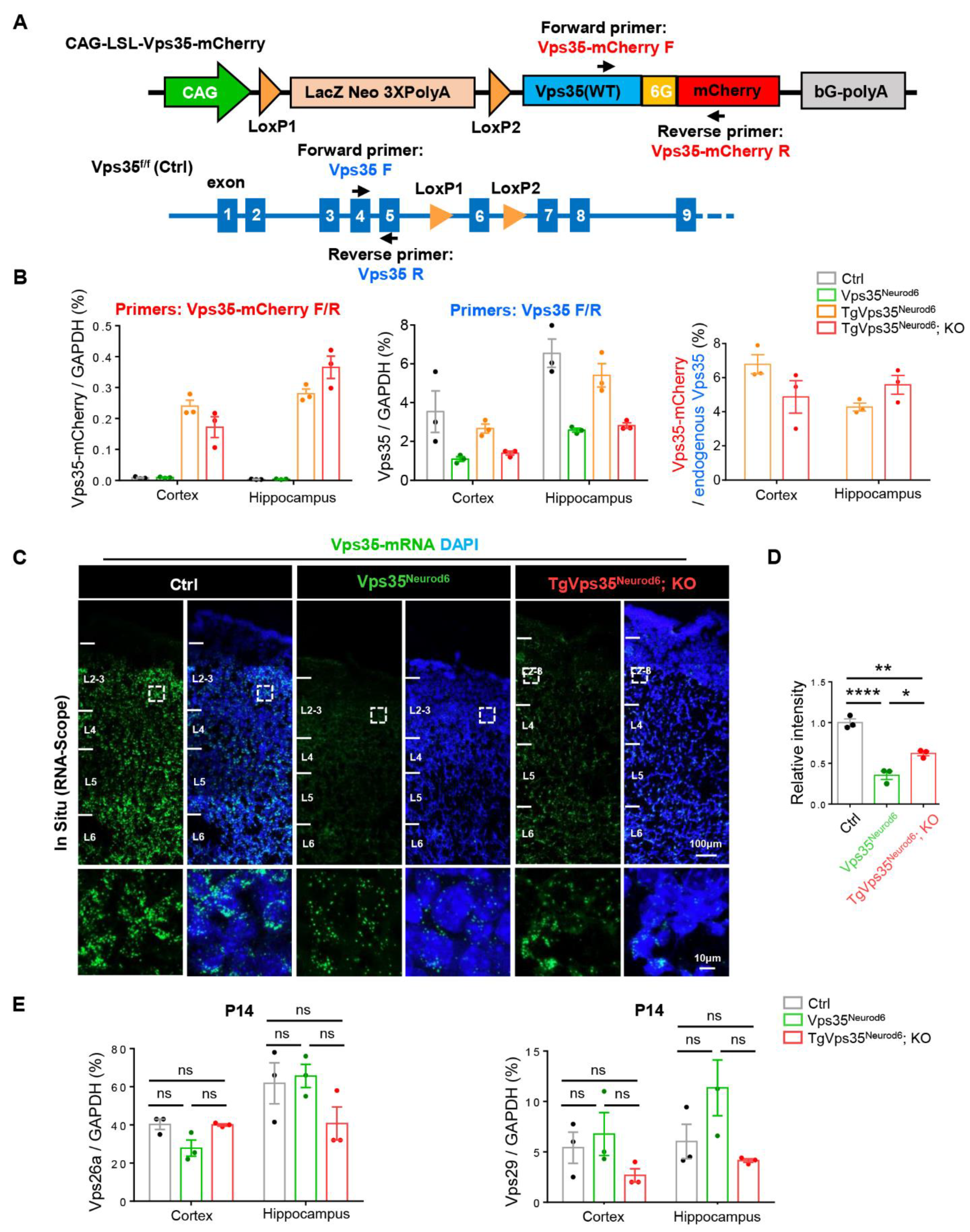
2.1. Generation of Conditional Transgenic Mice That Express VPS35-mCherry Fusion Protein in a Cre-Dependent Manner
2.2. Preventing Vps35Neurod6 Mice from Neonatal Death by Expressing VPS35-mCherry Fusion Protein
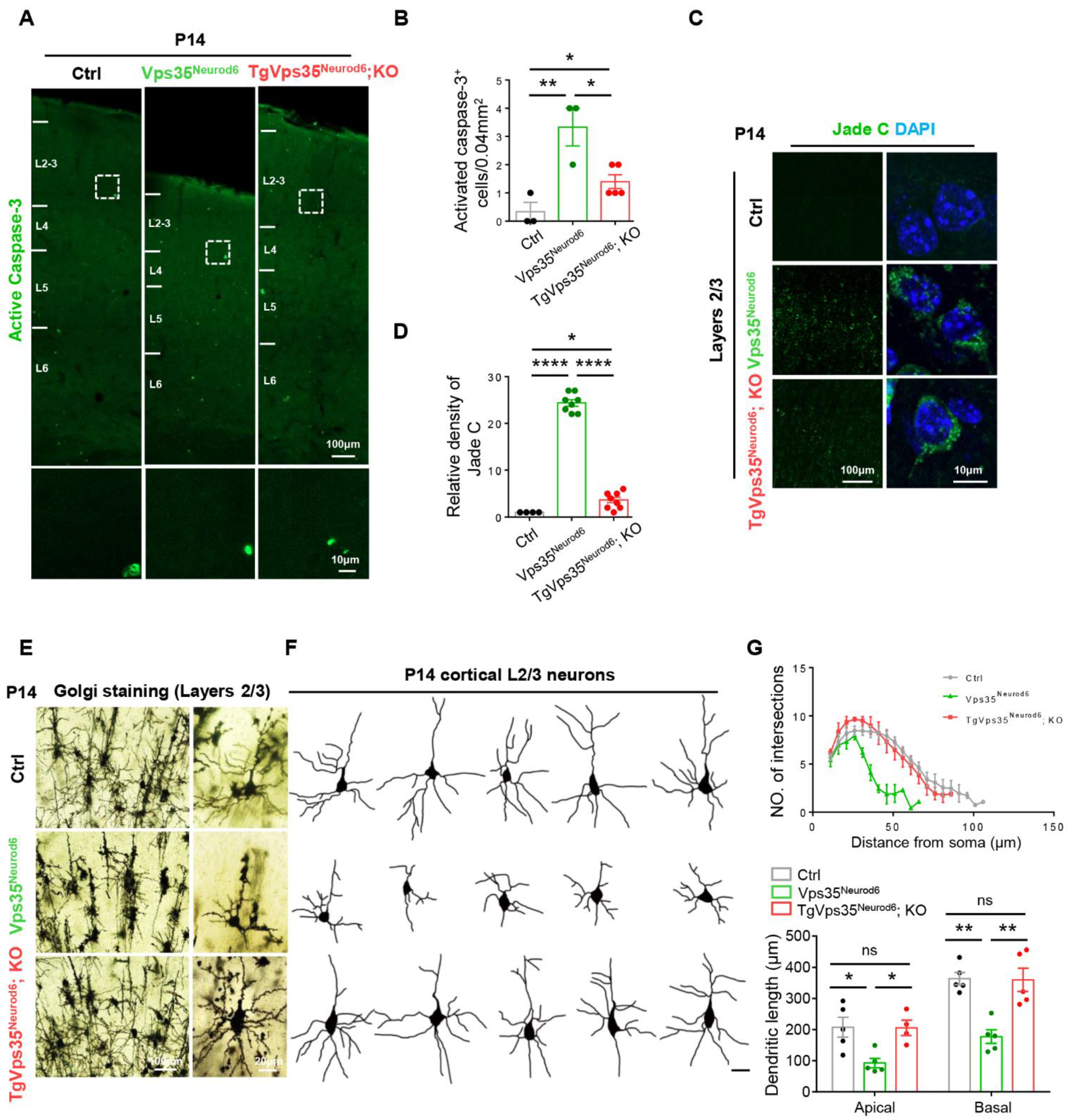
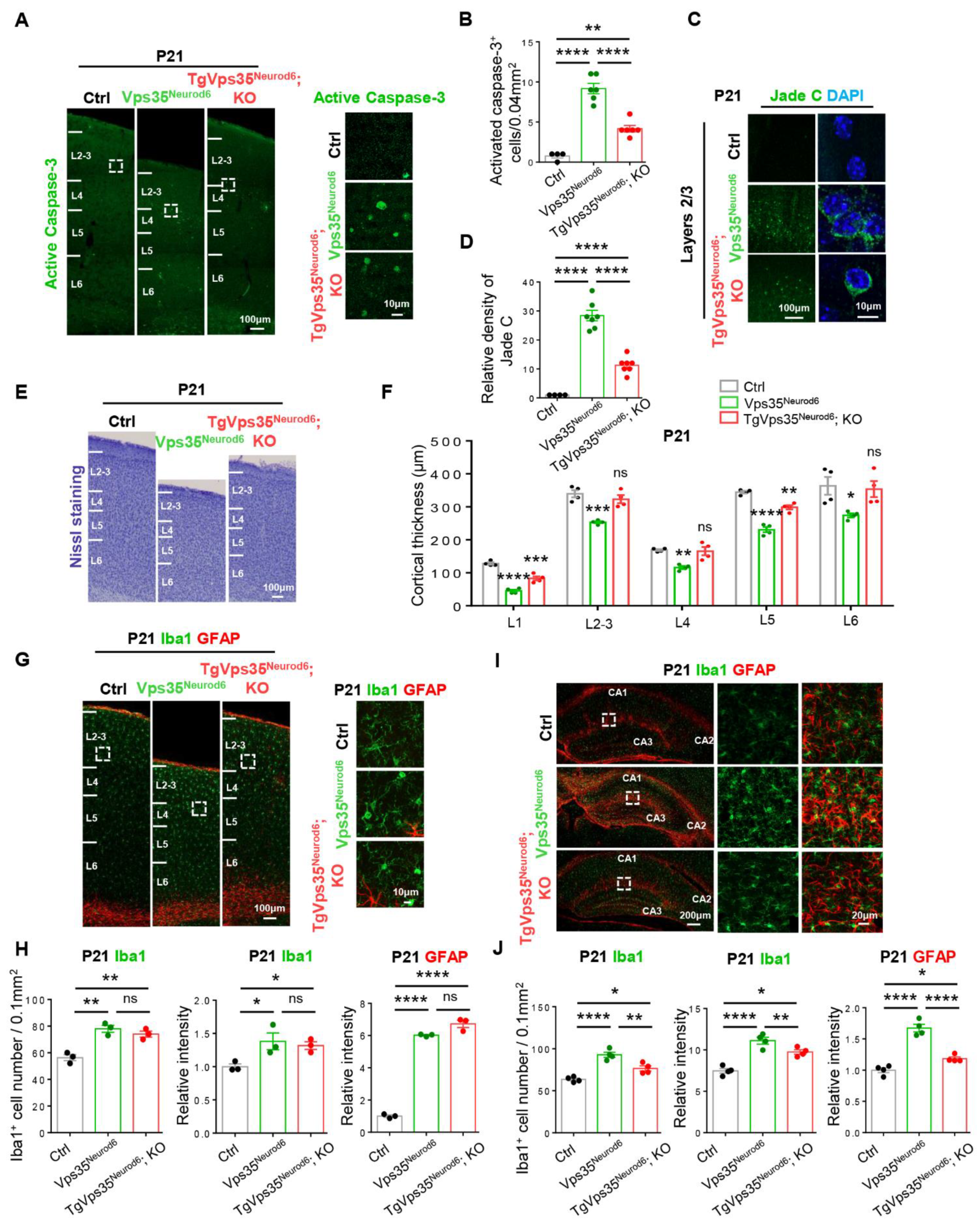
2.3. Decrease of Cell Death and Restoring of Terminal Differentiation of Neuronal Dendrites in Vps35Neurod6 Pups by Expressing VPS35-mCherry Fusion Protein
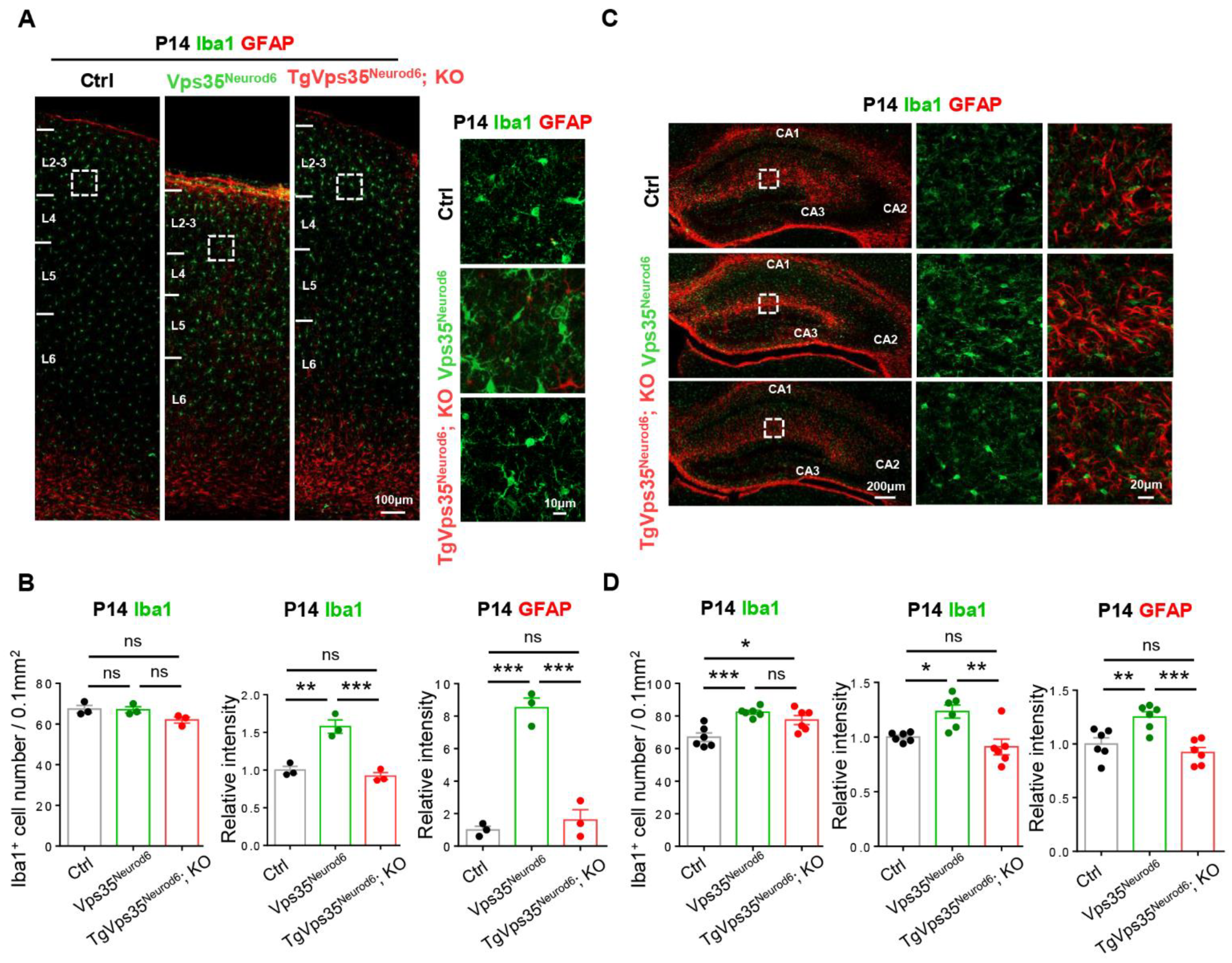
2.4. Attenuation of Gliosis in Vps35Neurod6 Pups by Expressing VPS35-mCherry Fusion Protein
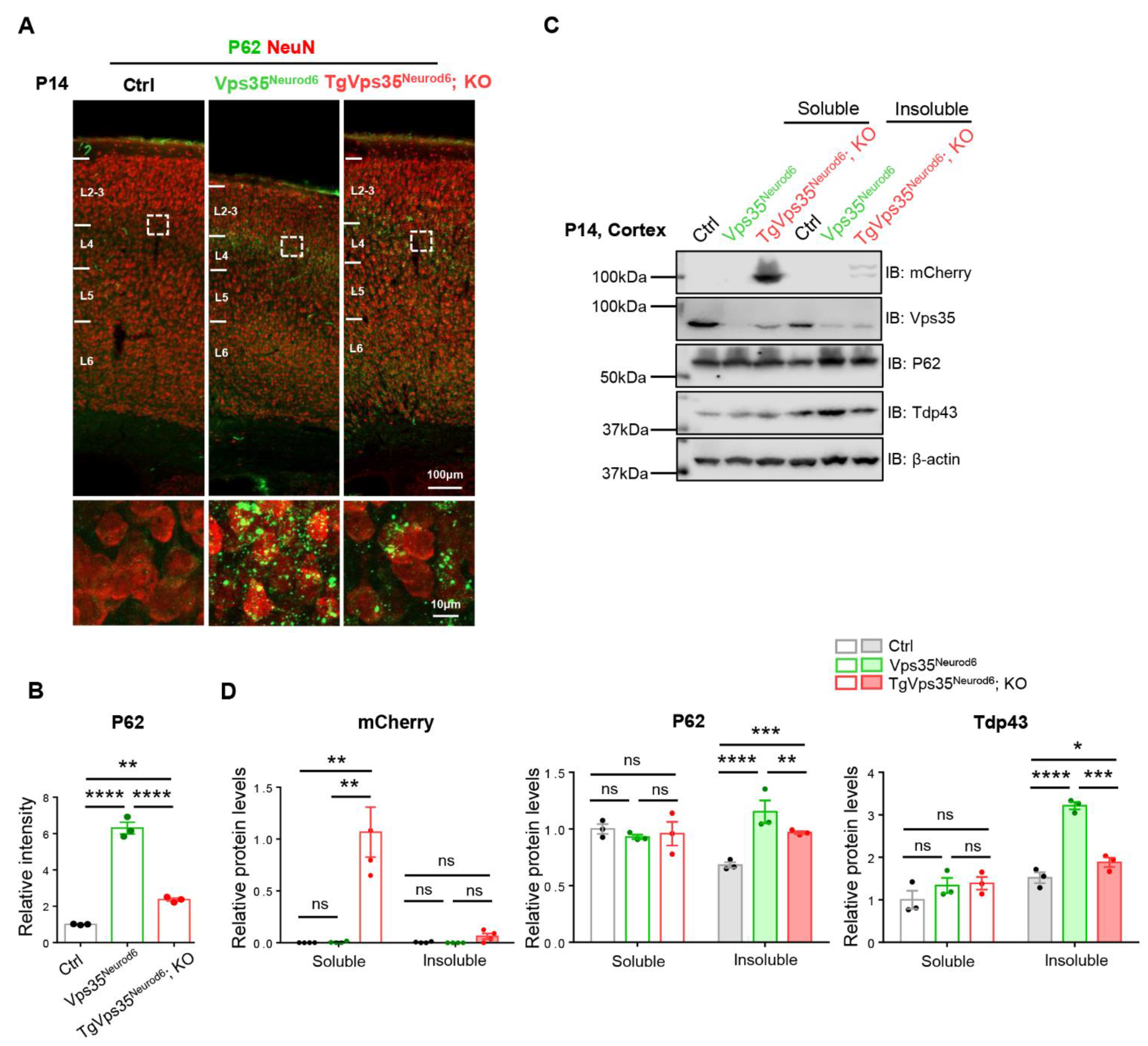
2.5. Partial Reduction of P62 and Tdp43 Proteins in TgVps35Neurod6; KO Pubs
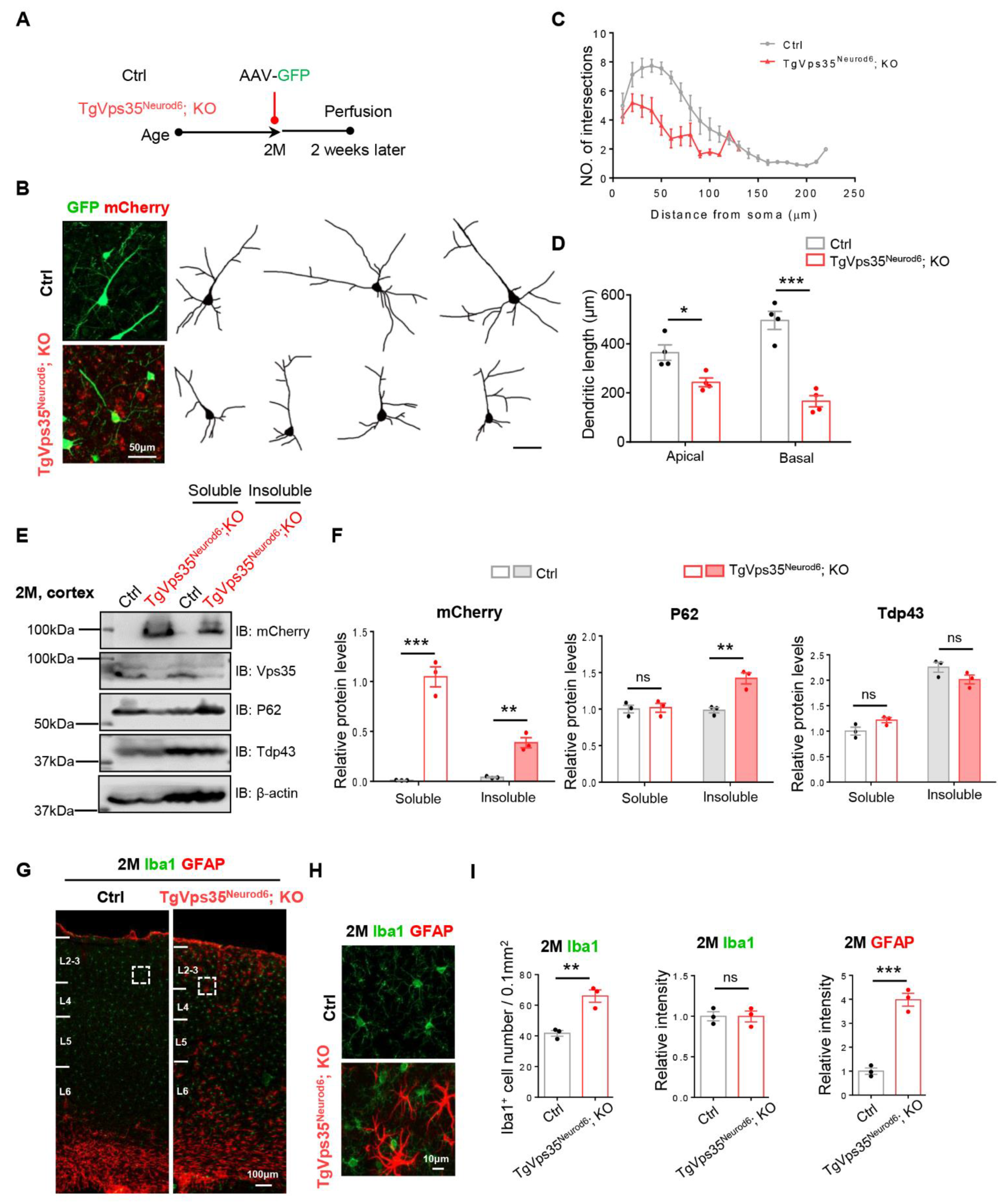
2.6. Neurodegenerative Pathology in Adult TgVps35Neurod6; KO Mice
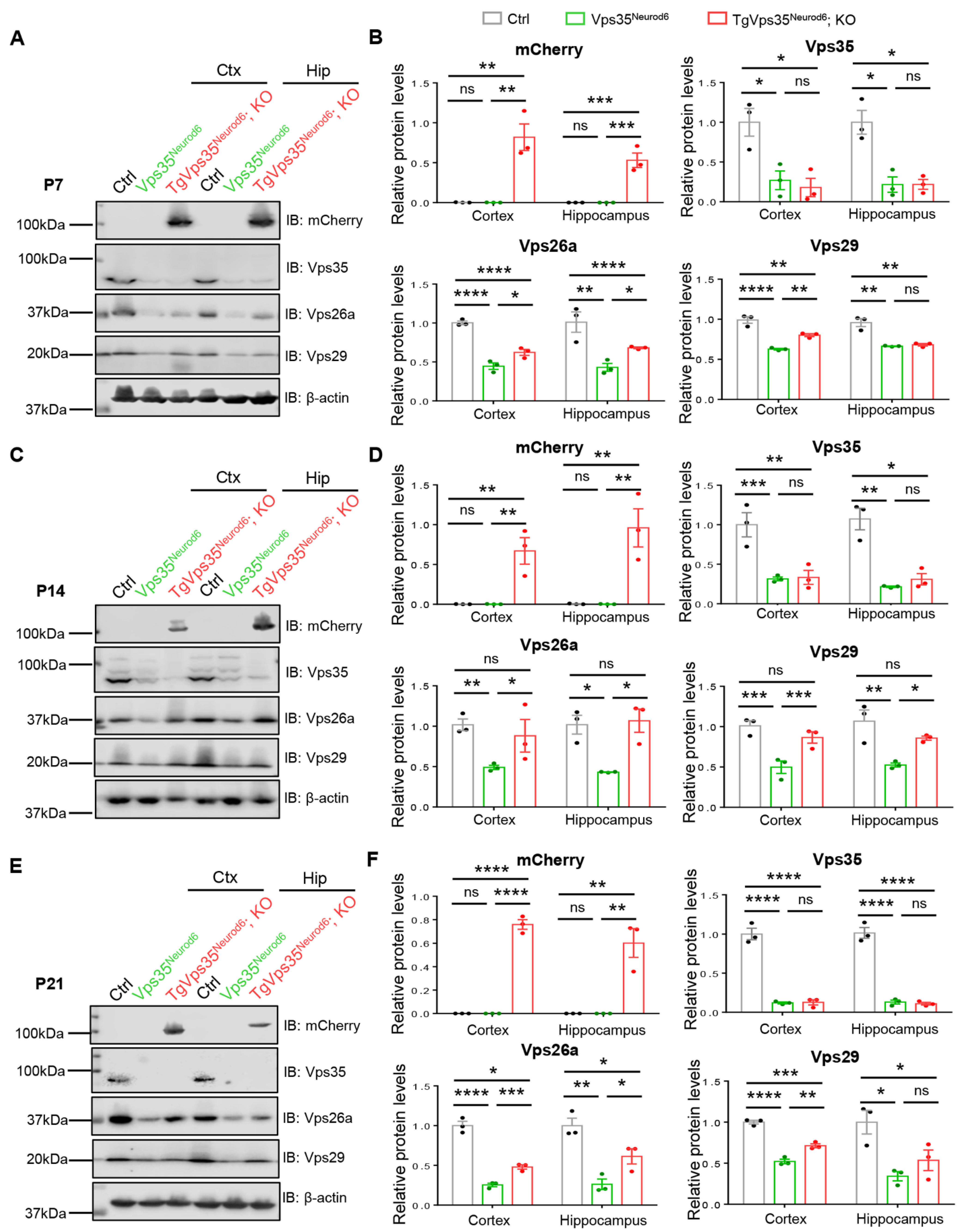
2.7. Age-Dependent Restoration of Retromer Complex in Vps35Neurod6 Mice Expressing VPS35-mCherry Fusion Protein
2.8. Low Level of Vps35-mCherry’s Expression
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Antibodies for Immunostaining and Western Blotting
4.3. Nissl and Golgi Staining
4.4. Adeno-Associated Virus (AAV) Injection
4.5. Tissue Processing and Immunofluorescence
4.6. Western Blotting
4.7. Jade C Staining
4.8. Quantitative Real-Time RT-PCR (qRT-PCR)
- Vps35-mCherry, forward: 5′ AGAGTCTGAGGGGCCAATCT 3′,
- reverse: 5′ CCTTGAAGCGCATGAACTCC 3′;
- Vps35, forward: 5′ TTGGTAGAAATGTGCCGTGGTGT 3′,
- reverse: 5′ CATCCGCACCCAGAGCTTATTCA 3′;
- Vps26a, forward: 5′ CTCGTGCTTGTTGATGAGGAGG 3′,
- reverse: 5′ CGCTGGTGAAAGTTCGTCCTCT 3′;
- Vps29, forward: 5′ TGCACCAAGGAGAGCTACGACT 3′,
- reverse: 5′ ACTTGGTGTCCGTGGATCAGAC 3′;
- GAPDH, forward: 5′ AAG GTC ATC CCA GAG CTG AA 3′,
- reverse: 5′ CTG CTT CAC CAC CTT CTT GA 3′.
4.9. RNA Scope
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seaman, M.N. The retromer complex—Endosomal protein recycling and beyond. J. Cell Sci. 2012, 125, 4693–4702. [Google Scholar] [CrossRef] [Green Version]
- Chamberland, J.P.; Ritter, B. Retromer revisited: Evolving roles for retromer in endosomal sorting. J. Cell Biol. 2017, 216, 3433–3436. [Google Scholar] [CrossRef]
- Burd, C.; Cullen, P.J. Retromer: A master conductor of endosome sorting. Cold Spring Harb. Perspect. Biol. 2014, 6, a016774. [Google Scholar] [CrossRef]
- Kim, E.; Lee, Y.; Lee, H.J.; Kim, J.S.; Song, B.S.; Huh, J.W.; Lee, S.R.; Kim, S.U.; Kim, S.H.; Hong, Y.; et al. Implication of mouse Vps26b-Vps29-Vps35 retromer complex in sortilin trafficking. Biochem. Biophys. Res. Commun. 2010, 403, 167–171. [Google Scholar] [CrossRef]
- Bugarcic, A.; Zhe, Y.; Kerr, M.C.; Griffin, J.; Collins, B.M.; Teasdale, R.D. Vps26A and Vps26B subunits define distinct retromer complexes. Traffic 2011, 12, 1759–1773. [Google Scholar] [CrossRef]
- Seaman, M.N. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J. Cell Biol. 2004, 165, 111–122. [Google Scholar] [CrossRef]
- Arighi, C.N.; Hartnell, L.M.; Aguilar, R.C.; Haft, C.R.; Bonifacino, J.S. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 2004, 165, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belenkaya, T.Y.; Wu, Y.; Tang, X.; Zhou, B.; Cheng, L.; Sharma, Y.V.; Yan, D.; Selva, E.M.; Lin, X. The retromer complex influences Wnt secretion by recycling wntless from endosomes to the trans-Golgi network. Dev. Cell 2008, 14, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, S. Retromer retrieves wntless. Dev. Cell 2008, 14, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Canuel, M.; Lefrancois, S.; Zeng, J.; Morales, C.R. AP-1 and retromer play opposite roles in the trafficking of sortilin between the Golgi apparatus and the lysosomes. Biochem. Biophys. Res. Commun. 2008, 366, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Zhang, W.; Peterhoff, C.; Hwang, J.C.; Nixon, R.A.; Ryu, S.H.; Kim, T.W. Proteomic identification of sorting nexin 6 as a negative regulator of BACE1-mediated APP processing. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2783–2794. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, A.; Flores, I.; Zhang, H.; Yu, R.; Staniszewski, A.; Planel, E.; Herman, M.; Ho, L.; Kreber, R.; Honig, L.S.; et al. Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc. Natl. Acad. Sci. USA 2008, 105, 7327–7332. [Google Scholar] [CrossRef] [Green Version]
- Hermey, G. The Vps10p-domain receptor family. Cell. Mol. Life Sci. CMLS 2009, 66, 2677–2689. [Google Scholar] [CrossRef] [PubMed]
- Vieira, S.I.; Rebelo, S.; Esselmann, H.; Wiltfang, J.; Lah, J.; Lane, R.; Small, S.A.; Gandy, S.; Silva, E.F.C.; Silva, O.A.B.C. Retrieval of the Alzheimer’s amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol. Neurodegener. 2010, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Mecozzi, V.J.; Berman, D.E.; Simoes, S.; Vetanovetz, C.; Awal, M.R.; Patel, V.M.; Schneider, R.T.; Petsko, G.A.; Ringe, D.; Small, S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014, 10, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Tang, F.L.; Hong, Y.; Luo, S.W.; Wang, C.L.; He, W.; Shen, C.; Jung, J.U.; Xiong, F.; Lee, D.H.; et al. VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J. Cell Biol. 2011, 195, 765–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.L.; Tang, F.L.; Peng, Y.; Shen, C.Y.; Mei, L.; Xiong, W.C. VPS35 regulates developing mouse hippocampal neuronal morphogenesis by promoting retrograde trafficking of BACE1. Biol. Open 2012, 1, 1248–1257. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef]
- Yin, J.; Liu, X.; He, Q.; Zhou, L.; Yuan, Z.; Zhao, S. Vps35-dependent recycling of Trem2 regulates microglial function. Traffic 2016, 17, 1286–1296. [Google Scholar] [CrossRef]
- Xia, W.F.; Tang, F.L.; Xiong, L.; Xiong, S.; Jung, J.U.; Lee, D.H.; Li, X.S.; Feng, X.; Mei, L.; Xiong, W.C. Vps35 loss promotes hyperresorptive osteoclastogenesis and osteoporosis via sustained RANKL signaling. J. Cell Biol. 2013, 200, 821–837. [Google Scholar] [CrossRef] [Green Version]
- Rademakers, R.; Neumann, M.; Mackenzie, I.R. Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 2012, 8, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernay, A.; Sellal, F.; Rene, F. Evaluating Behavior in Mouse Models of the Behavioral Variant of Frontotemporal Dementia: Which Test for Which Symptom? Neuro Degener. Dis. 2016, 16, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.N. Recycle your receptors with retromer. Trends Cell Biol. 2005, 15, 68–75. [Google Scholar] [CrossRef]
- Li, J.G.; Chiu, J.; Pratico, D. Full recovery of the Alzheimer’s disease phenotype by gain of function of vacuolar protein sorting 35. Mol. Psychiatry 2020, 25, 2630–2640. [Google Scholar] [CrossRef]
- Small, S.A.; Kent, K.; Pierce, A.; Leung, C.; Kang, M.S.; Okada, H.; Honig, L.; Vonsattel, J.P.; Kim, T.W. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann. Neurol. 2005, 58, 909–919. [Google Scholar] [CrossRef]
- Tang, F.L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for alpha-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.L.; Zhao, L.; Zhao, Y.; Sun, D.; Zhu, X.J.; Mei, L.; Xiong, W.C. Coupling of terminal differentiation deficit with neurodegenerative pathology in Vps35-deficient pyramidal neurons. Cell Death Differ. 2020, 27, 2099–2116. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.L.; Liu, W.; Hu, J.X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.C. VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Bellen, H.J. The retromer complex in development and disease. Development 2015, 142, 2392–2396. [Google Scholar] [CrossRef] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Goebbels, S.; Bormuth, I.; Bode, U.; Hermanson, O.; Schwab, M.H.; Nave, K.A. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 2006, 44, 611–621. [Google Scholar] [CrossRef]
- Su, J.H.; Nichol, K.E.; Sitch, T.; Sheu, P.; Chubb, C.; Miller, B.L.; Tomaselli, K.J.; Kim, R.C.; Cotman, C.W. DNA damage and activated caspase-3 expression in neurons and astrocytes: Evidence for apoptosis in frontotemporal dementia. Exp. Neurol. 2000, 163, 9–19. [Google Scholar] [CrossRef]
- Kasu, Y.; Alemu, A.S.; Lamari, A.A.; Loew, B.N.; Browera, A. The N Termini of TAR DNA-Binding Protein 43 (TDP43) C-Terminal Fragments Influence Degradation, Aggregation Propensity, and Morphology. Mol. Cell. Biol. 2018, 38, e00243-18. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, B.M.; Hartmann, H.; Serdaroglu, A.; Schludi, M.H.; Hornburg, D.; Meissner, F.; Orozco, D.; Colombo, A.; Tahirovic, S.; Michaelsen, M.; et al. TDP-43 loss of function inhibits endosomal trafficking and alters trophic signaling in neurons. EMBO J. 2016, 35, 2350–2370. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusten, T.E.; Stenmark, H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- Roberson, E.D. Mouse models of frontotemporal dementia. Ann. Neurol. 2012, 72, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Fuse, A.; Furuya, N.; Kakuta, S.; Inose, A.; Sato, M.; Koike, M.; Saiki, S.; Hattori, N. VPS29-VPS35 intermediate of retromer is stable and may be involved in the retromer complex assembly process. FEBS Lett. 2015, 589, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Pratico, D. The retromer complex system in a transgenic mouse model of AD: Influence of age. Neurobiol. Aging 2017, 52, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.E.; Ringe, D.; Petsko, G.A.; Small, S.A. The use of pharmacological retromer chaperones in Alzheimer’s disease and other endosomal-related disorders. Neurotherapeutics 2015, 12, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Young, J.E.; Fong, L.K.; Frankowski, H.; Petsko, G.A.; Small, S.A.; Goldstein, L.S.B. Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer’s Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem Cell Rep. 2018, 10, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Muzio, L.; Sirtori, R.; Gornati, D.; Eleuteri, S.; Fossaghi, A.; Brancaccio, D.; Manzoni, L.; Ottoboni, L.; Feo, L.; Quattrini, A.; et al. Retromer stabilization results in neuroprotection in a model of Amyotrophic Lateral Sclerosis. Nat. Commun. 2020, 11, 3848. [Google Scholar] [CrossRef]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases—An update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef]
- Clayton, K.A.; Van Enoo, A.A.; Ikezu, T. Alzheimer’s Disease: The Role of Microglia in Brain Homeostasis and Proteopathy. Front. Neurosci. 2017, 11, 680. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Gotzl, J.K.; Lang, C.M.; Haass, C.; Capell, A. Impaired protein degradation in FTLD and related disorders. Ageing Res. Rev. 2016, 32, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, P.; Komatsu, M. p62/SQSTM1—Steering the cell through health and disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appel, J.R.; Ye, S.; Tang, F.; Sun, D.; Zhang, H.; Mei, L.; Xiong, W.C. Increased Microglial Activity, Impaired Adult Hippocampal Neurogenesis, and Depressive-like Behavior in Microglial VPS35-Depleted Mice. J. Neurosci. 2018, 38, 5949–5968. [Google Scholar] [CrossRef]
- Tian, Y.; Tang, F.L.; Sun, X.; Wen, L.; Mei, L.; Tang, B.S.; Xiong, W.C. VPS35-deficiency results in an impaired AMPA receptor trafficking and decreased dendritic spine maturation. Mol. Brain 2015, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Sun, X.D.; Zhao, L.; Lee, D.H.; Hu, J.X.; Tang, F.L.; Pan, J.X.; Mei, L.; Zhu, X.J.; Xiong, W.C. Neogenin, a regulator of adult hippocampal neurogenesis, prevents depressive-like behavior. Cell Death Dis. 2018, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Vivar, C.; Potter, M.C.; Choi, J.; Lee, J.Y.; Stringer, T.P.; Callaway, E.M.; Gage, F.H.; Suh, H.; van Praag, H. Monosynaptic inputs to new neurons in the dentate gyrus. Nat. Commun. 2012, 3, 1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohara, K.; Pignatelli, M.; Rivest, A.J.; Jung, H.Y.; Kitamura, T.; Suh, J.; Frank, D.; Kajikawa, K.; Mise, N.; Obata, Y.; et al. Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nat. Neurosci. 2014, 17, 269–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehara, A.; Ueda, S. Application of Fluoro-Jade C in acute and chronic neurodegeneration models: Utilities and staining differences. Acta Histochem. Cytochem. 2009, 42, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Tang, F.L.; Erion, J.; Xiao, H.; Ye, J.; Xiong, W.C. Vps35 haploinsufficiency results in degenerative-like deficit in mouse retinal g anglion neurons and impairment of optic nerve injury-induced gliosis. Mol. Brain 2014, 7, 10. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Tang, F.; Lee, D.; Xiong, W.-C. Expression of Low Level of VPS35-mCherry Fusion Protein Diminishes Vps35 Depletion Induced Neuron Terminal Differentiation Deficits and Neurodegenerative Pathology, and Prevents Neonatal Death. Int. J. Mol. Sci. 2021, 22, 8394. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168394
Zhao Y, Tang F, Lee D, Xiong W-C. Expression of Low Level of VPS35-mCherry Fusion Protein Diminishes Vps35 Depletion Induced Neuron Terminal Differentiation Deficits and Neurodegenerative Pathology, and Prevents Neonatal Death. International Journal of Molecular Sciences. 2021; 22(16):8394. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168394
Chicago/Turabian StyleZhao, Yang, Fulei Tang, Daehoon Lee, and Wen-Cheng Xiong. 2021. "Expression of Low Level of VPS35-mCherry Fusion Protein Diminishes Vps35 Depletion Induced Neuron Terminal Differentiation Deficits and Neurodegenerative Pathology, and Prevents Neonatal Death" International Journal of Molecular Sciences 22, no. 16: 8394. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168394