
Insights on Metabolic Reprogramming and Its Therapeutic Potential in Acute Leukemia


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Deregulated Metabolic Programs in Leukemia
2.1. Glucose Metabolism: Warburg Effect and Metabolic Flexibility
2.2. One-Carbon Metabolic Network
2.2.1. Folate Cycle: What Have We Learned from the Use of Methotrexate in Leukemia?
2.2.2. Mitochondrial Targeting of the 1C Network as a New Therapeutic Strategy in Acute Leukemia
2.3. Amino Acid Metabolism and Therapeutic Opportunities in Acute Leukemia:
2.3.1. Serine Synthesis Pathway
2.3.2. Metabolic Dependencies as a Result of an Increased Demand for Glutamine
2.3.3. Impaired Synthesis of Asparagine as a Metabolic Vulnerability
2.3.4. Emerging Roles of Tryptophan Metabolism
2.3.5. Branched Chain Amino Acid (BCAA) Metabolism: An Emerging Oncogenic Pathway
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pui, C.H.; Relling, M.V.; Downing, J.R. Acute lymphoblastic leukemia. N. Engl. J. Med. 2004, 350, 1535–1548. [Google Scholar] [CrossRef] [Green Version]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Rashkovan, M.; Ferrando, A. Metabolic dependencies and vulnerabilities in leukemia. Genes Dev. 2019, 33, 1460–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, B.; Hu, J.; Jiang, S.; Liu, Y.; Sahin, E.; Zhuang, L.; Fletcher-Sananikone, E.; Colla, S.; Wang, Y.A.; Chin, L.; et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 2010, 468, 701–704. [Google Scholar] [CrossRef]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Glycolysis versus oxidative phosphorylation. Leuk. Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef]
- Han, L.; Cavazos, A.; Baran, N.; Zhang, Q.; Kuruvilla, V.M.; Gay, J.P.; Feng, N.; Battula, V.L.; Kantarjian, H.M.; Daver, N.G.; et al. Mitochondrial Oxphos As Survival Mechanism of Minimal Residual AML Cells after Induction Chemotherapy: Survival Benefit By Complex I Inhibition with Iacs-010759. Blood 2019, 134 (Suppl 1), 5161. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuis, N.; Poulain, L.; Birsen, R.; Tamburini, J.; Bouscary, D. Rationale for Targeting Deregulated Metabolic Pathways as a Therapeutic Strategy in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Shi, J.; Fu, H.; Jia, Z.; He, K.; Fu, L.; Wang, W. High Expression of CPT1A Predicts Adverse Outcomes: A Potential Therapeutic Target for Acute Myeloid Leukemia. EBioMedicine 2016, 14, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Hurren, R.; MacLean, N.; Gronda, M.; Jitkova, Y.; Sukhai, M.A.; Minden, M.D.; Schimmer, A.D. Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells. Apoptosis Int. J. Program. Cell Death 2015, 20, 1099–1108. [Google Scholar] [CrossRef]
- Dembitz, V.; Gallipoli, P. The Role of Metabolism in the Development of Personalized Therapies in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 665291. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef] [PubMed]
- Boag, J.M.; Beesley, A.H.; Firth, M.J.; Freitas, J.R.; Ford, J.; Hoffmann, K.; Cummings, A.J.; de Klerk, N.H.; Kees, U.R. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia 2006, 20, 1731–1737. [Google Scholar] [CrossRef]
- Jarviaho, T.; Hurme-Niiranen, A.; Soini, H.K.; Niinimaki, R.; Mottonen, M.; Savolainen, E.R.; Hinttala, R.; Harila-Saari, A.; Uusimaa, J. Novel non-neutral mitochondrial DNA mutations found in childhood acute lymphoblastic leukemia. Clin. Genet. 2018, 93, 275–285. [Google Scholar] [CrossRef]
- Kodron, A.; Ghanim, M.; Krawczyk, K.K.; Stelmaszczyk-Emmel, A.; Tonska, K.; Demkow, U.; Bartnik, E. Mitochondrial DNA in pediatric leukemia patients. Acta Biochim. Pol. 2017, 64, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Sbirkov, Y.; Burnusuzov, H.; Sarafian, V. Metabolic reprogramming in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2020, 67, e28255. [Google Scholar] [CrossRef]
- Bongiovanni, D.; Saccomani, V.; Piovan, E. Aberrant Signaling Pathways in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2017, 18, 1904. [Google Scholar] [CrossRef] [Green Version]
- Stiles, B.; Wang, Y.; Stahl, A.; Bassilian, S.; Lee, W.P.; Kim, Y.J.; Sherwin, R.; Devaskar, S.; Lesche, R.; Magnuson, M.A.; et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 2082–2087. [Google Scholar] [CrossRef] [Green Version]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.H.; Israelsen, W.J.; Lee, D.; Yu, V.W.C.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Vander Heiden, M.G.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [Green Version]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navas, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
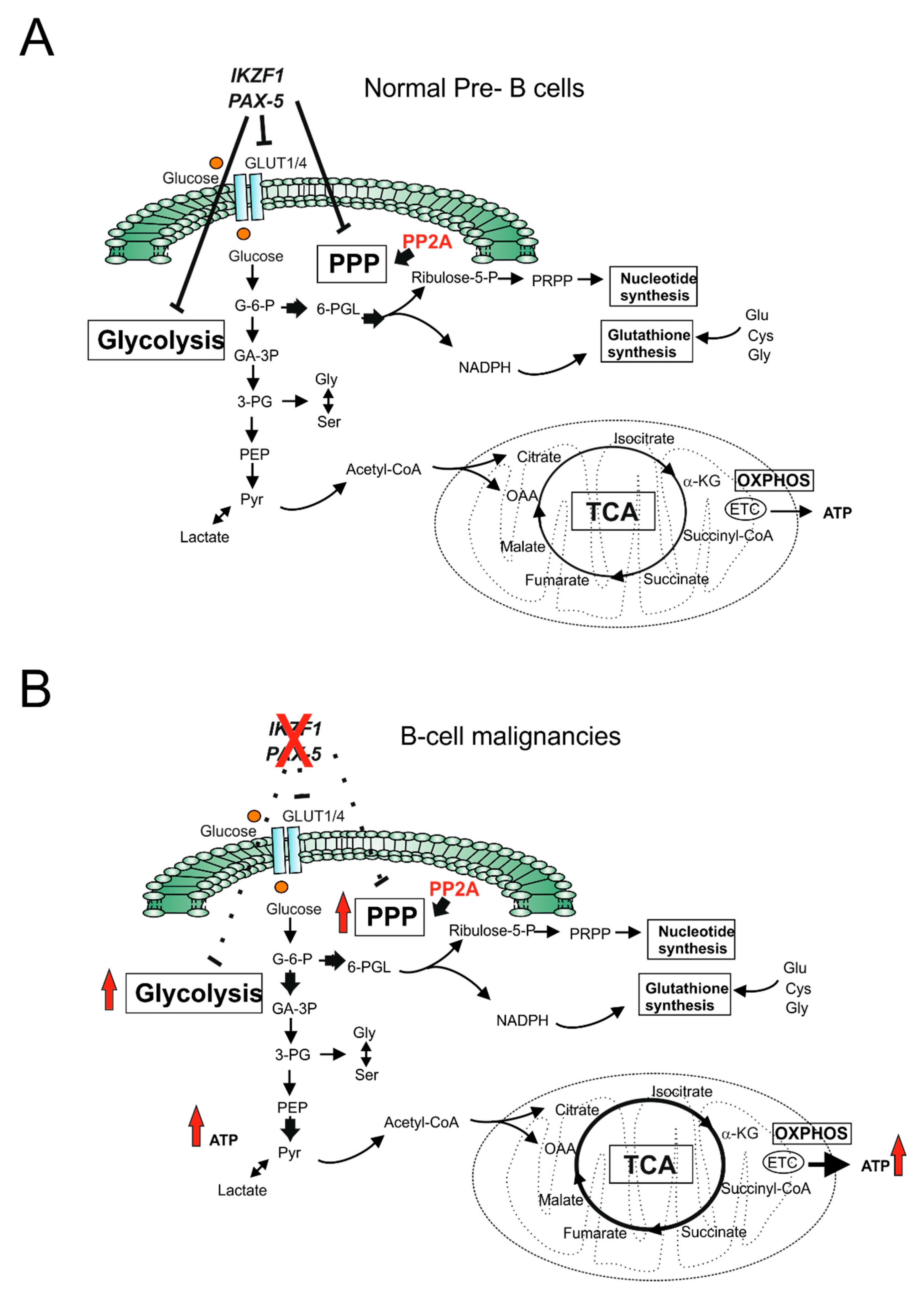
- Chan, L.N.; Chen, Z.; Braas, D.; Lee, J.W.; Xiao, G.; Geng, H.; Cosgun, K.N.; Hurtz, C.; Shojaee, S.; Cazzaniga, V.; et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 2017, 542, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Esplin, E.D.; Li, J.L.; Huang, L.; Gazdar, A.; Minna, J.; Evans, G.A. Alterations of the PPP2R1B gene in human lung and colon cancer. Science 1998, 282, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Chan, L.N.; Klemm, L.; Braas, D.; Chen, Z.; Geng, H.; Zhang, Q.C.; Aghajanirefah, A.; Cosgun, K.N.; Sadras, T.; et al. B-Cell-Specific Diversion of Glucose Carbon Utilization Reveals a Unique Vulnerability in B Cell Malignancies. Cell 2018, 173, 470–484.e18. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Visentin, M.; Zhao, R.; Goldman, I.D. The antifolates. Hematol. Oncol. Clin. N. Am. 2012, 26, 629–648. [Google Scholar] [CrossRef] [Green Version]
- McBurney, M.W.; Whitmore, G.F. Isolation and biochemical characterization of folate deficient mutants of Chinese hamster cells. Cell 1974, 2, 173–182. [Google Scholar] [CrossRef]
- Garrow, T.A.; Shane, B. Purification and general properties of human folylpolyglutamate synthetase. Adv. Exp. Med. Biol. 1993, 338, 659–662. [Google Scholar] [PubMed]
- Anderson, D.D.; Quintero, C.M.; Stover, P.J. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 15163–15168. [Google Scholar] [CrossRef] [Green Version]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- De Beaumais, T.A.; Jacqz-Aigrain, E. Intracellular disposition of methotrexate in acute lymphoblastic leukemia in children. Curr. Drug Metab. 2012, 13, 822–834. [Google Scholar] [CrossRef]
- Izbicka, E.; Diaz, A.; Streeper, R.; Wick, M.; Campos, D.; Steffen, R.; Saunders, M. Distinct mechanistic activity profile of pralatrexate in comparison to other antifolates in in vitro and in vivo models of human cancers. Cancer Chemother. Pharmacol. 2009, 64, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Chabner, B.A.; Allegra, C.J.; Curt, G.A.; Clendeninn, N.J.; Baram, J.; Koizumi, S.; Drake, J.C.; Jolivet, J. Polyglutamation of methotrexate. Is methotrexate a prodrug? J. Clin. Investig. 1985, 76, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Gorlick, R.; Goker, E.; Trippett, T.; Waltham, M.; Banerjee, D.; Bertino, J.R. Intrinsic and acquired resistance to methotrexate in acute leukemia. N. Engl. J. Med. 1996, 335, 1041–1048. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, J.M. Toward a better understanding of methotrexate. Arthritis Rheum. 2004, 50, 1370–1382. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [Green Version]
- Organista-Nava, J.; Gomez-Gomez, Y.; Illades-Aguiar, B.; Rivera-Ramirez, A.B.; Saavedra-Herrera, M.V.; Leyva-Vazquez, M.A. Overexpression of dihydrofolate reductase is a factor of poor survival in acute lymphoblastic leukemia. Oncol. Lett. 2018, 15, 8405–8411. [Google Scholar] [CrossRef]
- Wojtuszkiewicz, A.; Peters, G.J.; van Woerden, N.L.; Dubbelman, B.; Escherich, G.; Schmiegelow, K.; Sonneveld, E.; Pieters, R.; van de Ven, P.M.; Jansen, G.; et al. Methotrexate resistance in relation to treatment outcome in childhood acute lymphoblastic leukemia. J. Hematol. Oncol. 2015, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Zarou, M.M.; Vazquez, A.; Vignir Helgason, G. Folate metabolism: A re-emerging therapeutic target in haematological cancers. Leukemia 2021, 35, 1539–1551. [Google Scholar] [CrossRef]
- Galpin, A.J.; Schuetz, J.D.; Masson, E.; Yanishevski, Y.; Synold, T.W.; Barredo, J.C.; Pui, C.H.; Relling, M.V.; Evans, W.E. Differences in folylpolyglutamate synthetase and dihydrofolate reductase expression in human B-lineage versus T-lineage leukemic lymphoblasts: Mechanisms for lineage differences in methotrexate polyglutamylation and cytotoxicity. Mol. Pharmacol. 1997, 52, 155–163. [Google Scholar] [CrossRef]
- Castaldo, P.; Magi, S.; Nasti, A.A.; Arcangeli, S.; Lariccia, V.; Alesi, N.; Tocchini, M.; Amoroso, S. Clinical pharmacogenetics of methotrexate. Curr. Drug Metab. 2011, 12, 278–286. [Google Scholar] [CrossRef]
- Morscher, R.J.; Ducker, G.S.; Li, S.H.; Mayer, J.A.; Gitai, Z.; Sperl, W.; Rabinowitz, J.D. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018, 554, 128–132. [Google Scholar] [CrossRef]
- Zhu, Z.; Leung, G.K.K. More Than a Metabolic Enzyme: MTHFD2 as a Novel Target for Anticancer Therapy? Front. Oncol. 2020, 10, 658. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Puissant, A.; Alexe, G.; Furman, A.; Chen, L.M.; Frumm, S.M.; Ross, L.; Fenouille, N.; Bassil, C.F.; Lewis, C.A.; et al. Targeting MTHFD2 in acute myeloid leukemia. J. Exp. Med. 2016, 213, 1285–1306. [Google Scholar] [CrossRef]
- Vazquez, A.; Tedeschi, P.M.; Bertino, J.R. Overexpression of the mitochondrial folate and glycine-serine pathway: A new determinant of methotrexate selectivity in tumors. Cancer Res. 2013, 73, 478–482. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Canaveras, J.C.; Lancho, O.; Ducker, G.S.; Ghergurovich, J.M.; Xu, X.; da Silva-Diz, V.; Minuzzo, S.; Indraccolo, S.; Kim, H.; Herranz, D.; et al. SHMT inhibition is effective and synergizes with methotrexate in T-cell acute lymphoblastic leukemia. Leukemia 2021, 35, 377–388. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Hanson, R.W. Resurgence of serine: An often neglected but indispensable amino Acid. J. Biol. Chem. 2012, 287, 19786–19791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 2011, 43, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12. [Google Scholar] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Mattaini, K.R.; Sullivan, M.R.; Lau, A.N.; Fiske, B.P.; Bronson, R.T.; Vander Heiden, M.G. Increased PHGDH expression promotes aberrant melanin accumulation. BMC Cancer 2019, 19, 723. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019, 29, 1410–1421.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zheng, A.; Hydbring, P.; Ambroise, G.; Ouchida, A.T.; Goiny, M.; Vakifahmetoglu-Norberg, H.; Norberg, E. PHGDH Defines a Metabolic Subtype in Lung Adenocarcinomas with Poor Prognosis. Cell Rep. 2017, 19, 2289–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X.; et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampen, K.R.; Fancello, L.; Girardi, T.; Rinaldi, G.; Planque, M.; Sulima, S.O.; Loayza-Puch, F.; Verbelen, B.; Vereecke, S.; Verbeeck, J.; et al. Translatome analysis reveals altered serine and glycine metabolism in T-cell acute lymphoblastic leukemia cells. Nat. Commun. 2019, 10, 2542. [Google Scholar] [CrossRef]
- Polet, F.; Corbet, C.; Pinto, A.; Rubio, L.I.; Martherus, R.; Bol, V.; Drozak, X.; Gregoire, V.; Riant, O.; Feron, O. Reducing the serine availability complements the inhibition of the glutamine metabolism to block leukemia cell growth. Oncotarget 2016, 7, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Savino, A.M.; Chirayil, R.; Barin, E.; Cheng, Y.; Park, S.M.; Schurer, A.; Mullarky, E.; Cantley, L.C.; Kharas, M.G.; et al. High Fructose Drives the Serine Synthesis Pathway in Acute Myeloid Leukemic Cells. Cell Metab. 2021, 33, 145–159.e6. [Google Scholar] [CrossRef]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mates, J.M.; Campos-Sandoval, J.A.; Santos-Jimenez, J.L.; Marquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [Green Version]
- Ni, F.; Yu, W.M.; Li, Z.; Graham, D.K.; Jin, L.; Kang, S.; Rossi, M.R.; Li, S.; Broxmeyer, H.E.; Qu, C.K. Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat. Metab. 2019, 1, 390–403. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Katt, W.P.; Lukey, M.J.; Cerione, R.A. A tale of two glutaminases: Homologous enzymes with distinct roles in tumorigenesis. Future Med. Chem. 2017, 9, 223–243. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef] [Green Version]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Sudderth, J.; Sanchez-Martin, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef]
- Hu, J.; Wang, T.; Xu, J.; Wu, S.; Wang, L.; Su, H.; Jiang, J.; Yue, M.; Wang, J.; Wang, D.; et al. WEE1 inhibition induces glutamine addiction in T-cell acute lymphoblastic leukemia. Haematologica 2021, 106, 1816–1827. [Google Scholar] [CrossRef] [Green Version]
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.S.H.; Vohra, S.; Medina-Perez, P.; Basheer, F.; Marando, L.; Di Lisio, L.; Dias, J.M.L.; et al. Glutaminolysis is a metabolic dependency in FLT3(ITD) acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018, 131, 1639–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gastel, N.; Spinelli, J.B.; Sharda, A.; Schajnovitz, A.; Baryawno, N.; Rhee, C.; Oki, T.; Grace, E.; Soled, H.J.; Milosevic, J.; et al. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metab. 2020, 32, 391–403.e6. [Google Scholar] [CrossRef] [PubMed]
- Guerra, V.; Dinardo, C.D.; Konopleva, M.; Burger, J.A.; Borthakur, G.; Jabbour, E.; Pemmaraju, N.; Sheppard, K.; Garcia-Manero, G.; Kantarjian, H.M. Interim results from a phase Ib/II clinical study of the glutaminase inhibitor telaglenastat (CB-839) in combination with azacitidine in patients with advanced myelodysplastic syndrome (MDS). J. Clin. Oncol. 2019, 37 (Suppl. 15), 7037. [Google Scholar] [CrossRef]
- Tabe, Y.; Lorenzi, P.L.; Konopleva, M. Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood 2019, 134, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [Green Version]
- Kidd, J.G. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. I. Course of transplanted cancers of various kinds in mice and rats given guinea pig serum, horse serum, or rabbit serum. J. Exp. Med. 1953, 98, 565–582. [Google Scholar] [CrossRef]
- Kidd, J.G. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. II. Studies on the nature of the active serum constituent: Histological mechanism of the regression: Tests for effects of guinea pig serum on lymphoma cells in vitro: Discussion. J. Exp. Med. 1953, 98, 583–606. [Google Scholar]
- Broome, J.D. Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. II. Lymphoma 6C3HED cells cultured in a medium devoid of L-asparagine lose their susceptibility to the effects of guinea pig serum in vivo. J. Exp. Med. 1963, 118, 121–148. [Google Scholar] [CrossRef] [Green Version]
- Broome, J.D. Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. I. Properties of the L-asparaginase of guinea pig serum in relation to those of the antilymphoma substance. J. Exp. Med. 1963, 118, 99–120. [Google Scholar] [CrossRef] [Green Version]
- Parmentier, J.H.; Maggi, M.; Tarasco, E.; Scotti, C.; Avramis, V.I.; Mittelman, S.D. Glutaminase activity determines cytotoxicity of L-asparaginases on most leukemia cell lines. Leuk. Res. 2015, 39, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Beckett, A.; Gervais, D. What makes a good new therapeutic L-asparaginase? World J. Microbiol. Biotechnol. 2019, 35, 152. [Google Scholar] [CrossRef]
- Gupta, S.; Wang, C.; Raetz, E.A.; Schore, R.; Salzer, W.L.; Larsen, E.C.; Maloney, K.W.; Mattano, L.A., Jr.; Carroll, W.L.; Winick, N.J.; et al. Impact of Asparaginase Discontinuation on Outcome in Childhood Acute Lymphoblastic Leukemia: A Report From the Children’s Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1897–1905. [Google Scholar] [CrossRef]
- Su, N.; Pan, Y.X.; Zhou, M.; Harvey, R.C.; Hunger, S.P.; Kilberg, M.S. Correlation between asparaginase sensitivity and asparagine synthetase protein content, but not mRNA, in acute lymphoblastic leukemia cell lines. Pediatric Blood Cancer 2008, 50, 274–279. [Google Scholar] [CrossRef]
- Hermanova, I.; Zaliova, M.; Trka, J.; Starkova, J. Low expression of asparagine synthetase in lymphoid blasts precludes its role in sensitivity to L-asparaginase. Exp. Hematol. 2012, 40, 657–665. [Google Scholar] [CrossRef]
- Watanabe, A.; Miyake, K.; Nordlund, J.; Syvanen, A.C.; van der Weyden, L.; Honda, H.; Yamasaki, N.; Nagamachi, A.; Inaba, T.; Ikawa, T.; et al. Association of aberrant ASNS imprinting with asparaginase sensitivity and chromosomal abnormality in childhood BCP-ALL. Blood 2020, 136, 2319–2333. [Google Scholar] [CrossRef]
- Touzart, A.; Lengline, E.; Latiri, M.; Belhocine, M.; Smith, C.; Thomas, X.; Spicuglia, S.; Puthier, D.; Pflumio, F.; Leguay, T.; et al. Epigenetic Silencing Affects l-Asparaginase Sensitivity and Predicts Outcome in T-ALL. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2483–2493. [Google Scholar] [CrossRef] [Green Version]
- Haskell, C.M.; Canellos, G.P. l-asparaginase resistance in human leukemia--asparagine synthetase. Biochem. Pharmacol. 1969, 18, 2578–2580. [Google Scholar] [CrossRef]
- Horowitz, B.; Madras, B.K.; Meister, A.; Old, L.J.; Boyes, E.A.; Stockert, E. Asparagine synthetase activity of mouse leukemias. Science 1968, 160, 533–535. [Google Scholar] [CrossRef]
- Appel, I.M.; den Boer, M.L.; Meijerink, J.P.; Veerman, A.J.; Reniers, N.C.; Pieters, R. Up-regulation of asparagine synthetase expression is not linked to the clinical response L-asparaginase in pediatric acute lymphoblastic leukemia. Blood 2006, 107, 4244–4249. [Google Scholar] [CrossRef] [Green Version]
- Stams, W.A.; den Boer, M.L.; Beverloo, H.B.; Meijerink, J.P.; Stigter, R.L.; van Wering, E.R.; Janka-Schaub, G.E.; Slater, R.; Pieters, R. Sensitivity to L-asparaginase is not associated with expression levels of asparagine synthetase in t(12;21)+ pediatric ALL. Blood 2003, 101, 2743–2747. [Google Scholar] [CrossRef]
- Williams, R.T.; Guarecuco, R.; Gates, L.A.; Barrows, D.; Passarelli, M.C.; Carey, B.; Baudrier, L.; Jeewajee, S.; La, K.; Prizer, B.; et al. ZBTB1 Regulates Asparagine Synthesis and Leukemia Cell Response to L-Asparaginase. Cell Metab. 2020, 31, 852–861.e6. [Google Scholar] [CrossRef]
- Jiang, J.; Srivastava, S.; Seim, G.; Pavlova, N.N.; King, B.; Zou, L.; Zhang, C.; Zhong, M.; Feng, H.; Kapur, R.; et al. Promoter demethylation of the asparagine synthetase gene is required for ATF4-dependent adaptation to asparagine depletion. J. Biol. Chem. 2019, 294, 18674–18684. [Google Scholar] [CrossRef] [PubMed]
- Siggs, O.M.; Li, X.; Xia, Y.; Beutler, B. ZBTB1 is a determinant of lymphoid development. J. Exp. Med. 2012, 209, 19–27. [Google Scholar] [CrossRef]
- Punwani, D.; Simon, K.; Choi, Y.; Dutra, A.; Gonzalez-Espinosa, D.; Pak, E.; Naradikian, M.; Song, C.H.; Zhang, J.; Bodine, D.M.; et al. Transcription factor zinc finger and BTB domain 1 is essential for lymphocyte development. J. Immunol. 2012, 189, 1253–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Kang, S.; Wang, X.; Rosales, J.L.; Gao, X.; Byun, H.G.; Jin, Y.; Fu, S.; Wang, J.; Lee, K.Y. HAP1 loss confers l-asparaginase resistance in ALL by downregulating the calpain-1-Bid-caspase-3/12 pathway. Blood 2019, 133, 2222–2232. [Google Scholar] [CrossRef] [Green Version]
- Hinze, L.; Pfirrmann, M.; Karim, S.; Degar, J.; McGuckin, C.; Vinjamur, D.; Sacher, J.; Stevenson, K.E.; Neuberg, D.S.; Orellana, E.; et al. Synthetic Lethality of Wnt Pathway Activation and Asparaginase in Drug-Resistant Acute Leukemias. Cancer Cell 2019, 35, 664–676.e7. [Google Scholar] [CrossRef] [Green Version]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. IJTR 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Liu, X.Q.; Lu, K.; Feng, L.L.; Ding, M.; Gao, J.M.; Ge, X.L.; Wang, X. Up-regulated expression of indoleamine 2,3-dioxygenase 1 in non-Hodgkin lymphoma correlates with increased regulatory T-cell infiltration. Leuk. Lymphoma 2014, 55, 405–414. [Google Scholar] [CrossRef]
- Curti, A.; Trabanelli, S.; Salvestrini, V.; Baccarani, M.; Lemoli, R.M. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: Focus on hematology. Blood 2009, 113, 2394–2401. [Google Scholar] [CrossRef]
- Curti, A.; Trabanelli, S.; Onofri, C.; Aluigi, M.; Salvestrini, V.; Ocadlikova, D.; Evangelisti, C.; Rutella, S.; De Cristofaro, R.; Ottaviani, E.; et al. Indoleamine 2,3-dioxygenase-expressing leukemic dendritic cells impair a leukemia-specific immune response by inducing potent T regulatory cells. Haematologica 2010, 95, 2022–2030. [Google Scholar] [CrossRef] [Green Version]
- Venkateswaran, N.; Lafita-Navarro, M.C.; Hao, Y.H.; Kilgore, J.A.; Perez-Castro, L.; Braverman, J.; Borenstein-Auerbach, N.; Kim, M.; Lesner, N.P.; Mishra, P.; et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019, 33, 1236–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishnupuri, K.S.; Alvarado, D.M.; Khouri, A.N.; Shabsovich, M.; Chen, B.; Dieckgraefe, B.K.; Ciorba, M.A. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res. 2019, 79, 1138–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, G.; Kennedy, P.T.; Dahal, L.N. Investigating the Role of Indoleamine 2,3-Dioxygenase in Acute Myeloid Leukemia: A Systematic Review. Front. Immunol. 2021, 12, 651687. [Google Scholar] [CrossRef]
- Sobash, P.T.; Kolhe, R.; Karim, N.A.; Guddati, A.K.; Jillella, A.; Kota, V. Role of indoleamine 2,3-dioxygenase in acute myeloid leukemia. Future Oncol. 2020, 16, 3085–3094. [Google Scholar]
- Corm, S.; Berthon, C.; Imbenotte, M.; Biggio, V.; Lhermitte, M.; Dupont, C.; Briche, I.; Quesnel, B. Indoleamine 2,3-dioxygenase activity of acute myeloid leukemia cells can be measured from patients’ sera by HPLC and is inducible by IFN-gamma. Leuk. Res. 2009, 33, 490–494. [Google Scholar] [CrossRef]
- Mabuchi, R.; Hara, T.; Matsumoto, T.; Shibata, Y.; Nakamura, N.; Nakamura, H.; Kitagawa, J.; Kanemura, N.; Goto, N.; Shimizu, M.; et al. High serum concentration of L-kynurenine predicts unfavorable outcomes in patients with acute myeloid leukemia. Leuk. Lymphoma 2016, 57, 92–98. [Google Scholar] [CrossRef]
- Hara, T.; Matsumoto, T.; Shibata, Y.; Nakamura, N.; Nakamura, H.; Ninomiya, S.; Kitagawa, J.; Nannya, Y.; Shimizu, M.; Ito, H.; et al. Prognostic value of the combination of serum l-kynurenine level and indoleamine 2,3-dioxygenase mRNA expression in acute myeloid leukemia. Leuk. Lymphoma 2016, 57, 2208–2211. [Google Scholar] [CrossRef]
- Hoshi, M.; Ito, H.; Fujigaki, H.; Takemura, M.; Takahashi, T.; Tomita, E.; Ohyama, M.; Tanaka, R.; Saito, K.; Seishima, M. Indoleamine 2,3-dioxygenase is highly expressed in human adult T-cell leukemia/lymphoma and chemotherapy changes tryptophan catabolism in serum and reduced activity. Leuk. Res. 2009, 33, 39–45. [Google Scholar] [CrossRef]
- Masaki, A.; Ishida, T.; Maeda, Y.; Suzuki, S.; Ito, A.; Takino, H.; Ogura, H.; Totani, H.; Yoshida, T.; Kinoshita, S.; et al. Prognostic Significance of Tryptophan Catabolism in Adult T-cell Leukemia/Lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2830–2839. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Hara, T.; Matsumoto, T.; Nakamura, N.; Nakamura, H.; Ninomiya, S.; Kitagawa, J.; Goto, N.; Nannya, Y.; Ito, H.; et al. Serum concentrations of l-kynurenine predict clinical outcomes of patients with peripheral T-cell lymphoma, not otherwise specified. Hematol. Oncol. 2017, 35, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Hara, T.; Tsurumi, H.; Goto, N.; Hoshi, M.; Kitagawa, J.; Kanemura, N.; Kasahara, S.; Ito, H.; Takemura, M.; et al. Serum concentration of L-kynurenine predicts the clinical outcome of patients with diffuse large B-cell lymphoma treated with R-CHOP. Eur. J. Haematol. 2010, 84, 304–309. [Google Scholar] [CrossRef]
- Folgiero, V.; Goffredo, B.M.; Filippini, P.; Masetti, R.; Bonanno, G.; Caruso, R.; Bertaina, V.; Mastronuzzi, A.; Gaspari, S.; Zecca, M.; et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity in leukemia blasts correlates with poor outcome in childhood acute myeloid leukemia. Oncotarget 2014, 5, 2052–2064. [Google Scholar] [CrossRef] [Green Version]
- Fukuno, K.; Hara, T.; Tsurumi, H.; Shibata, Y.; Mabuchi, R.; Nakamura, N.; Kitagawa, J.; Shimizu, M.; Ito, H.; Saito, K.; et al. Expression of indoleamine 2,3-dioxygenase in leukemic cells indicates an unfavorable prognosis in acute myeloid leukemia patients with intermediate-risk cytogenetics. Leuk. Lymphoma 2015, 56, 1398–1405. [Google Scholar] [CrossRef]
- Mangaonkar, A.; Mondal, A.K.; Fulzule, S.; Pundkar, C.; Park, E.J.; Jillella, A.; Kota, V.; Xu, H.; Savage, N.M.; Shi, H.; et al. A novel immunohistochemical score to predict early mortality in acute myeloid leukemia patients based on indoleamine 2,3 dioxygenase expression. Sci. Rep. 2017, 7, 12892. [Google Scholar] [CrossRef] [PubMed]
- Dolsak, A.; Gobec, S.; Sova, M. Indoleamine and tryptophan 2,3-dioxygenases as important future therapeutic targets. Pharmacol. Ther. 2021, 221, 107746. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Fontenay, M.; Corm, S.; Briche, I.; Allorge, D.; Hennart, B.; Lhermitte, M.; Quesnel, B. Metabolites of tryptophan catabolism are elevated in sera of patients with myelodysplastic syndromes and inhibit hematopoietic progenitor amplification. Leuk. Res. 2013, 37, 573–579. [Google Scholar] [CrossRef]
- Neinast, M.; Murashige, D.; Arany, Z. Branched Chain Amino Acids. Annu. Rev. Physiol. 2019, 81, 139–164. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Wang, Y.; Luo, W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 2020, 39, 6747–6756. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Vander Heiden, M.G. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell 2020, 37, 147–156. [Google Scholar] [CrossRef]
- Bonvini, A.; Coqueiro, A.Y.; Tirapegui, J.; Calder, P.C.; Rogero, M.M. Immunomodulatory role of branched-chain amino acids. Nutr. Rev. 2018, 76, 840–856. [Google Scholar] [CrossRef] [Green Version]
- Ananieva, E.A.; Powell, J.D.; Hutson, S.M. Leucine Metabolism in T Cell Activation: mTOR Signaling and Beyond. Adv. Nutr. 2016, 7, 798S–805S. [Google Scholar] [CrossRef]
- Ikeda, K.; Kinoshita, M.; Kayama, H.; Nagamori, S.; Kongpracha, P.; Umemoto, E.; Okumura, R.; Kurakawa, T.; Murakami, M.; Mikami, N.; et al. Slc3a2 Mediates Branched-Chain Amino-Acid-Dependent Maintenance of Regulatory T Cells. Cell Rep. 2017, 21, 1824–1838. [Google Scholar] [CrossRef] [Green Version]
- Ananieva, E.A.; Patel, C.H.; Drake, C.H.; Powell, J.D.; Hutson, S.M. Cytosolic branched chain aminotransferase (BCATc) regulates mTORC1 signaling and glycolytic metabolism in CD4+ T cells. J. Biol. Chem. 2014, 289, 18793–18804. [Google Scholar] [CrossRef] [Green Version]
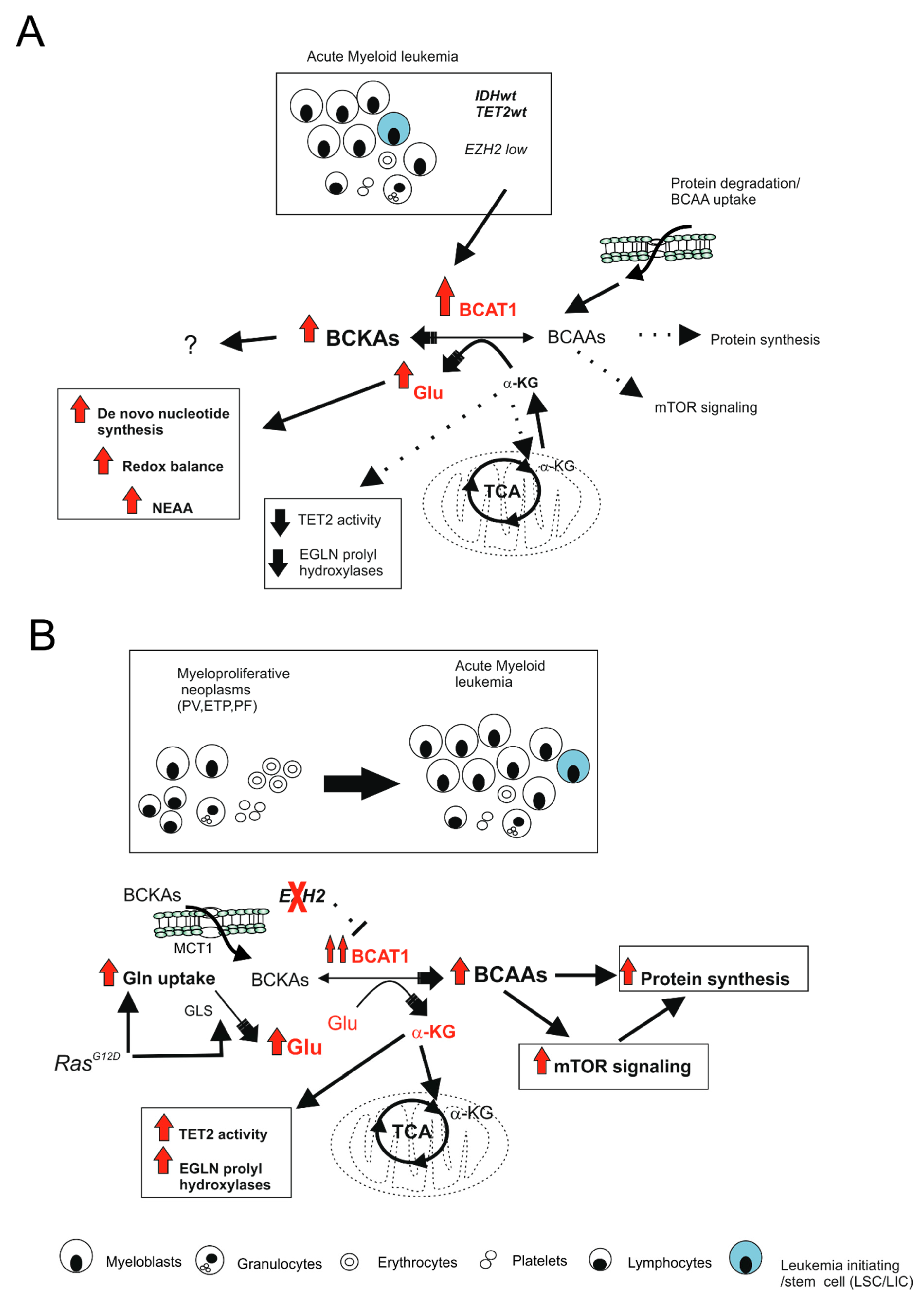
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Liu, Y.; Cai, F.; Patrick, M.; Zmajkovic, J.; Cao, H.; Zhang, Y.; Tasdogan, A.; Chen, M.; Qi, L.; et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov. 2019, 9, 1228–1247. [Google Scholar] [CrossRef]
- Tonjes, M.; Barbus, S.; Park, Y.J.; Wang, W.; Schlotter, M.; Lindroth, A.M.; Pleier, S.V.; Bai, A.H.C.; Karra, D.; Piro, R.M.; et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med. 2013, 19, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Martino, L.; Tosello, V.; Peroni, E.; Piovan, E. Insights on Metabolic Reprogramming and Its Therapeutic Potential in Acute Leukemia. Int. J. Mol. Sci. 2021, 22, 8738. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168738
Di Martino L, Tosello V, Peroni E, Piovan E. Insights on Metabolic Reprogramming and Its Therapeutic Potential in Acute Leukemia. International Journal of Molecular Sciences. 2021; 22(16):8738. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168738
Chicago/Turabian StyleDi Martino, Ludovica, Valeria Tosello, Edoardo Peroni, and Erich Piovan. 2021. "Insights on Metabolic Reprogramming and Its Therapeutic Potential in Acute Leukemia" International Journal of Molecular Sciences 22, no. 16: 8738. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168738