
A Michael Acceptor Analogue, SKSI-0412, Down-Regulates Inflammation and Proliferation Factors through Suppressing Signal Transducer and Activator of Transcription 3 Signaling in IL-17A-Induced Human Keratinocyte

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Synthesis of SKSI-0412 (4)
2.2. Fluorescence-Based Analysis of SKSI-0412 (4) Binding to STAT3
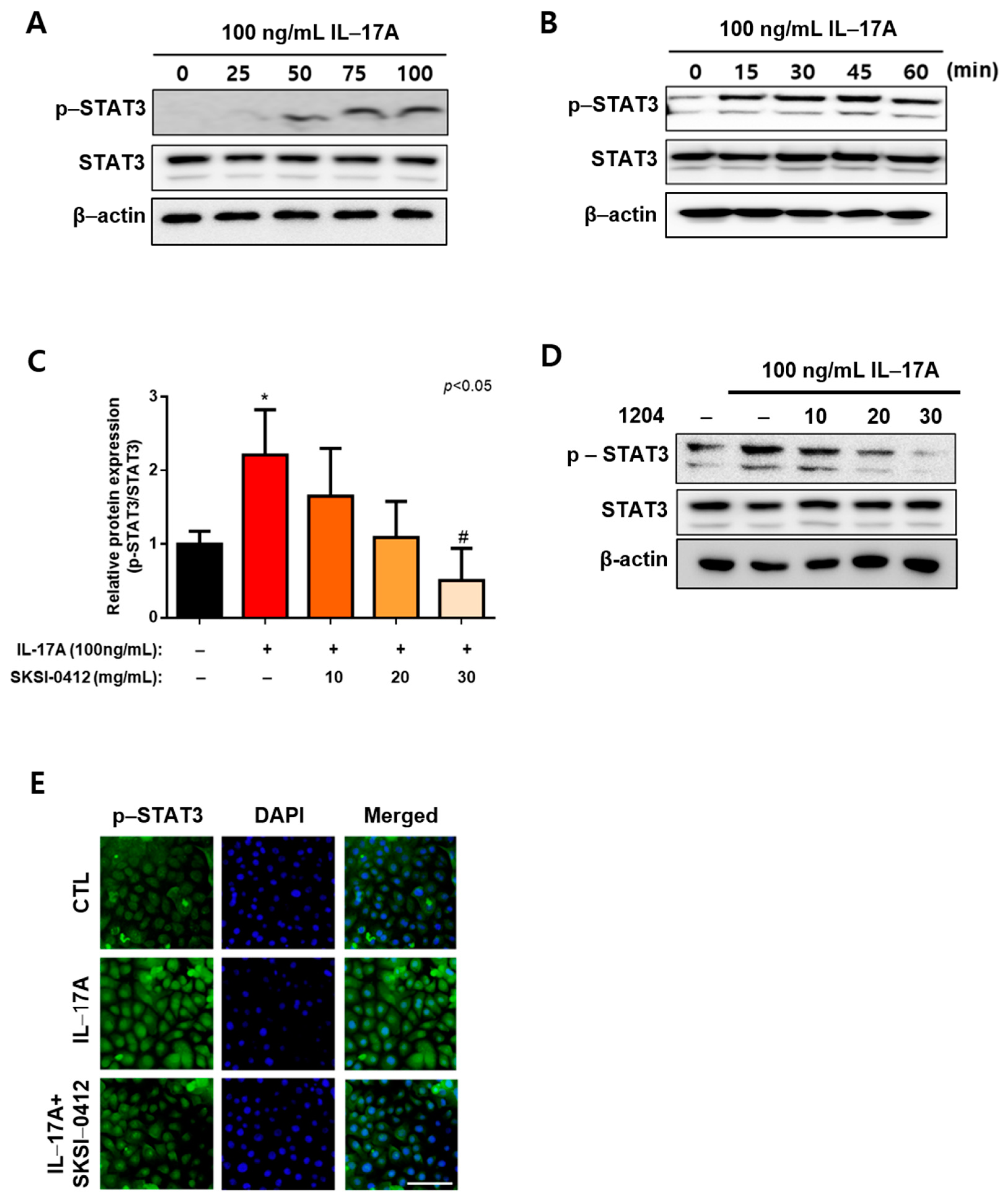
2.3. Inhibition of IL-17A-Induced STAT3 Tyr705 Phosphorylation via SKSI-0412 at Early Time Point
2.4. Inhibition of IκBζ Induction through SKSI-0412 Treatment in IL-17A-Induced Keratinocytes
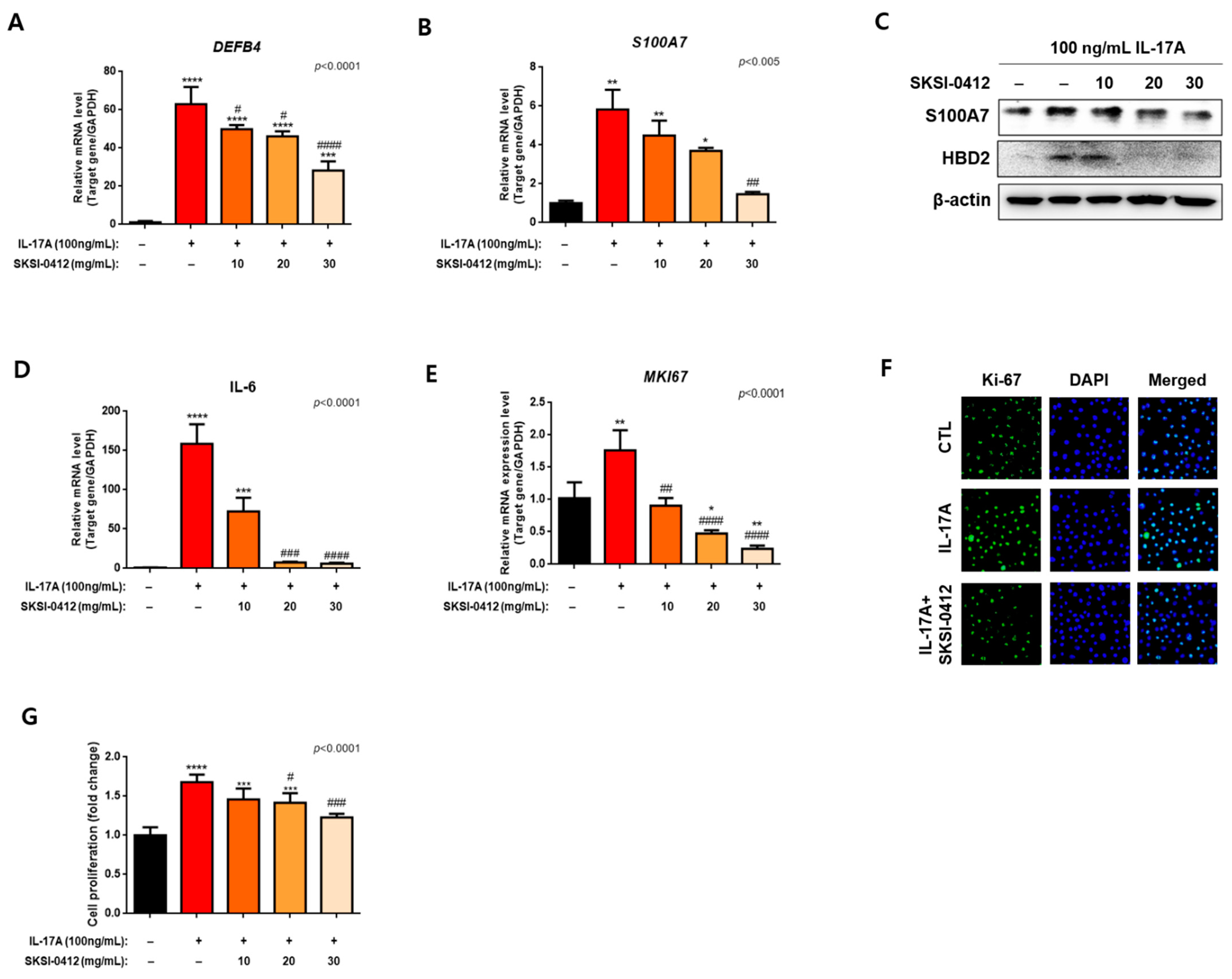
2.5. Improvements in Cellular and Molecular Disease Biomarkers through SKSI-0412 in IL-17A-Induced Keratinocytes
3. Discussion
4. Materials and Methods
4.1. General Methods for Chemistry
4.2. Methyl 2,2-Dimethyl-2H-Chromene-6-Carboxylate (2)
4.3. 2-(3,4-Dimethoxyphenyl)-1-(2,2-Dimethyl-2H-Chromen-6-yl)Ethan-1-One (3)
4.4. 2-(3,4-Dimethoxyphenyl)-1-(2,2-Dimethyl-2H-Chromen-6-yl)Prop-2-en-1-One (4)
4.5. Procedure for Binding Titration Experiments Using Fluorescence Measurements
4.6. Cell Culture and Treatment
4.7. Cell Proliferation Assay
4.8. Quantitative Real-Time PCR
4.9. Western Blot Analysis
4.10. Immunofluorescence (IF) Staining
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lebwohl, M. Psoriasis. Lancet 2003, 361, 1197–1204. [Google Scholar] [CrossRef]
- Bowcock, A.M.; Krueger, J.G. Getting under the skin: The immunogenetics of psoriasis. Nat. Rev. Immunol. 2005, 5, 699–711. [Google Scholar] [CrossRef]
- Coimbra, S.; Figueiredo, A.; Castro, E.; Rocha-Pereira, P.; Santos-Silva, A. The roles of cells and cytokines in the pathogenesis of psoriasis. Int. J. Dermatol. 2012, 51, 389–398. [Google Scholar] [CrossRef]
- Calautti, E.; Avalle, L.; Poli, V. Psoriasis: A STAT3-Centric View. Int. J. Mol. Sci. 2018, 19, 171. [Google Scholar] [CrossRef] [Green Version]
- Muromoto, R.; Tawa, K.; Ohgakiuchi, Y.; Sato, A.; Saino, Y.; Hirashima, K.; Minoguchi, H.; Kitai, Y.; Kashiwakura, J.-I.; Shimoda, K.; et al. IκB-ζ Expression Requires Both TYK2/STAT3 Activity and IL-17-Regulated mRNA Stabilization. ImmunoHorizons 2019, 3, 172–185. [Google Scholar] [CrossRef]
- Sano, S.; Chan, K.S.; Carbajal, S.; Clifford, J.L.; Peavey, M.; Kiguchi, K.; Itami, S.; Nickoloff, B.J.; DiGiovanni, J. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat. Med. 2004, 11, 43–49. [Google Scholar] [CrossRef]
- Miyoshi, K.; Takaishi, M.; Nakajima, K.; Ikeda, M.; Kanda, T.; Tarutani, M.; Iiyama, T.; Asao, N.; DiGiovanni, J.; Sano, S. Stat3 as a Therapeutic Target for the Treatment of Psoriasis: A Clinical Feasibility Study with STA-21, a Stat3 Inhibitor. J. Investig. Dermatol. 2011, 131, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Leonard, W.J.; O’Shea, J.J. Jaks and Stats: Biological Implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.; Darnell, J.E. STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Jin, L.; Dang, E.; Chang, T.; Feng, Z.; Liu, Y.; Wang, G. IL-17A Upregulates Keratin 17 Expression in Keratinocytes through STAT1- and STAT3-Dependent Mechanisms. J. Investig. Dermatol. 2011, 131, 2401–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, H.C.; Jeong, S.H.; Kim, J.H.; Lee, H.; Ryu, W.-I.; Kim, M.-G.; Son, E.D.; Lee, T.R.; Son, S.W. RIP4 upregulates CCL20 expression through STAT3 signaling in cultured keratinocytes. Exp. Dermatol. 2018, 27, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Hennig, A.; Lorscheid, S.; Grondona, P.; Schulze-Osthoff, K.; Hailfinger, S.; Kramer, D. IκBζ is a key transcriptional regulator of IL-36–driven psoriasis-related gene expression in keratinocytes. Proc. Natl. Acad. Sci. USA 2018, 115, 10088–10093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Luo, D.; Wang, D.; Ma, L.; Zhao, Y.; Li, L. IL-17 Activates the IL-6/STAT3 Signal Pathway in the Proliferation of Hepatitis B Virus-Related Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2017, 43, 2379–2390. [Google Scholar] [CrossRef]
- Madonna, S.; Girolomoni, G.; Dinarello, C.A.; Albanesi, C. The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. Int. J. Mol. Sci. 2019, 20, 3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, J.G.; Brunner, P. Interleukin-17 alters the biology of many cell types involved in the genesis of psoriasis, systemic inflammation and associated comorbidities. Exp. Dermatol. 2017, 27, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Miao, X.; Xiang, Y.; Mao, W.; Chen, Y.; Li, Q.; Fan, B. TRIM27 promotes IL-6-induced proliferation and inflammation factor production by activating STAT3 signaling in HaCaT cells. Am. J. Physiol. Physiol. 2020, 318, C272–C281. [Google Scholar] [CrossRef] [PubMed]
- Slowikowski, K.; Nguyen, H.N.; Noss, E.H.; Simmons, D.P.; Mizoguchi, F.; Watts, G.F.M.; Gurish, M.F.; Brenner, M.B.; Raychaudhuri, S. CUX1 and IκBζ (NFKBIZ) mediate the synergistic inflammatory response to TNF and IL-17A in stromal fibroblasts. Proc. Natl. Acad. Sci. USA 2020, 117, 5532–5541. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, S.-J.; Han, Y.T.; Hong, S.-J.; An, H.; Chang, D.-J.; Kim, T.; Lim, B.; Lee, J.; Surh, Y.-J.; et al. Identification of small molecule inhibitors of the STAT3 signaling pathway: Insights into their structural features and mode of action. Bioorg. Med. Chem. Lett. 2015, 25, 5444–5448. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-J.; Kim, J.-T.; Kim, S.-J.; Cho, N.-C.; Kim, K.; Lee, S.; Suh, Y.-G.; Cho, K.-C.; Kim, K.P.; Surh, Y.-J. An Electrophilic Deguelin Analogue Inhibits STAT3 Signaling in H-Ras-Transformed Human Mammary Epithelial Cells: The Cysteine 259 Residue as a Potential Target. Biomedicines 2020, 8, 407. [Google Scholar] [CrossRef]
- Park, S.K.; Byun, W.S.; Lee, S.; Han, Y.T.; Jeong, Y.-S.; Jang, K.; Chung, S.-J.; Lee, J.; Suh, Y.-G.; Lee, S.K. A novel small molecule STAT3 inhibitor SLSI-1216 suppresses proliferation and tumor growth of triple-negative breast cancer cells through apoptotic induction. Biochem. Pharmacol. 2020, 178, 114053. [Google Scholar] [CrossRef]
- Nishihara, M.; Ogura, H.; Ueda, N.; Tsuruoka, M.; Kitabayashi, C.; Tsuji, F.; Aono, H.; Ishihara, K.; Huseby, E.; Betz, U.A.K.; et al. IL-6–gp130–STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int. Immunol. 2007, 19, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.; Leonard, W.J.; Littman, D.R. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Ma, C.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef] [Green Version]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nat. Cell Biol. 2008, 452, 773–776. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.O.; Panopoulos, A.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 Regulates Cytokine-mediated Generation of Inflammatory Helper T Cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cua, D.J.; Sherlock, J.P.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.W.P.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nat. Cell Biol. 2003, 421, 744–748. [Google Scholar] [CrossRef]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonderegger, I.; Röhn, T.A.; Kurrer, M.O.; Iezzi, G.; Zou, Y.; Kastelein, R.A.; Bachmann, M.F.; Kopf, M. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur. J. Immunol. 2006, 36, 2849–2856. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Danilenko, D.; Valdez, P.A.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nat. Cell Biol. 2006, 445, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.S.; Lamb, P.; Seidelm, H.M.; Stein, R.B.; Rosen, J. Rapid activation of the STAT3 transcription factor by granulocyte colony-stimulating factor. Blood 1994, 84, 1760–1764. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Meyer, D.; Campbell, G.; Larner, A.; Carter-Su, C.; Schwartz, J.; Jove, R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 1995, 269, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhong, Z.; Darnell, J.E. Maximal activation of transcription by statl and stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Schlessinger, K.; Zhu, X.; Meffre, E.; Quimby, F.; Levy, D.; Darnell, J.E. Essential Role of STAT3 in Postnatal Survival and Growth Revealed by Mice Lacking STAT3 Serine 727 Phosphorylation. Mol. Cell. Biol. 2004, 24, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016, 31, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Yamasaki, K. Psoriasis and Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 6791. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wang, M.; Liu, M.; Zhou, E.; Ren, T.; Chang, X.; He, M.; Zeng, K.; Guo, Y.; Wu, J. Efficacy and safety of IL-17 inhibitors for the treatment of ankylosing spondylitis: A systematic review and meta-analysis. Arthritis Res. 2020, 22, 1–11. [Google Scholar] [CrossRef]
- Johansen, C.; Mose, M.; Ommen, P.; Bertelsen, T.; Vinter, H.; Hailfinger, S.; Lorscheid, S.; Schulze-Osthoff, K.; Iversen, L. IκBζ is a key driver in the development of psoriasis. Proc. Natl. Acad. Sci. USA 2015, 112, E5825–E5833. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Iwai, Y.; Oh-Hora, M.; Yamamoto, M.; Morio, T.; Aoki, K.; Ohya, K.; Jetten, A.; Akira, S.; Muta, T.; et al. IκBζ regulates TH17 development by cooperating with ROR nuclear receptors. Nat. Cell Biol. 2010, 464, 1381–1385. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, A.-R.; Lee, S.; Shin, J.U.; Seok, S.H.; Suh, Y.-G.; Kim, D.H. A Michael Acceptor Analogue, SKSI-0412, Down-Regulates Inflammation and Proliferation Factors through Suppressing Signal Transducer and Activator of Transcription 3 Signaling in IL-17A-Induced Human Keratinocyte. Int. J. Mol. Sci. 2021, 22, 8813. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168813
Kim A-R, Lee S, Shin JU, Seok SH, Suh Y-G, Kim DH. A Michael Acceptor Analogue, SKSI-0412, Down-Regulates Inflammation and Proliferation Factors through Suppressing Signal Transducer and Activator of Transcription 3 Signaling in IL-17A-Induced Human Keratinocyte. International Journal of Molecular Sciences. 2021; 22(16):8813. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168813
Chicago/Turabian StyleKim, A-Ram, Seungbeom Lee, Jung U Shin, Seung Hui Seok, Young-Ger Suh, and Dong Hyun Kim. 2021. "A Michael Acceptor Analogue, SKSI-0412, Down-Regulates Inflammation and Proliferation Factors through Suppressing Signal Transducer and Activator of Transcription 3 Signaling in IL-17A-Induced Human Keratinocyte" International Journal of Molecular Sciences 22, no. 16: 8813. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168813