
Gene-Metabolite Network Analysis Revealed Tissue-Specific Accumulation of Therapeutic Metabolites in Mallotus japonicus

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Mallotus japonicus Metabolome Database Represents Diverse Range of Specialized Metabolites
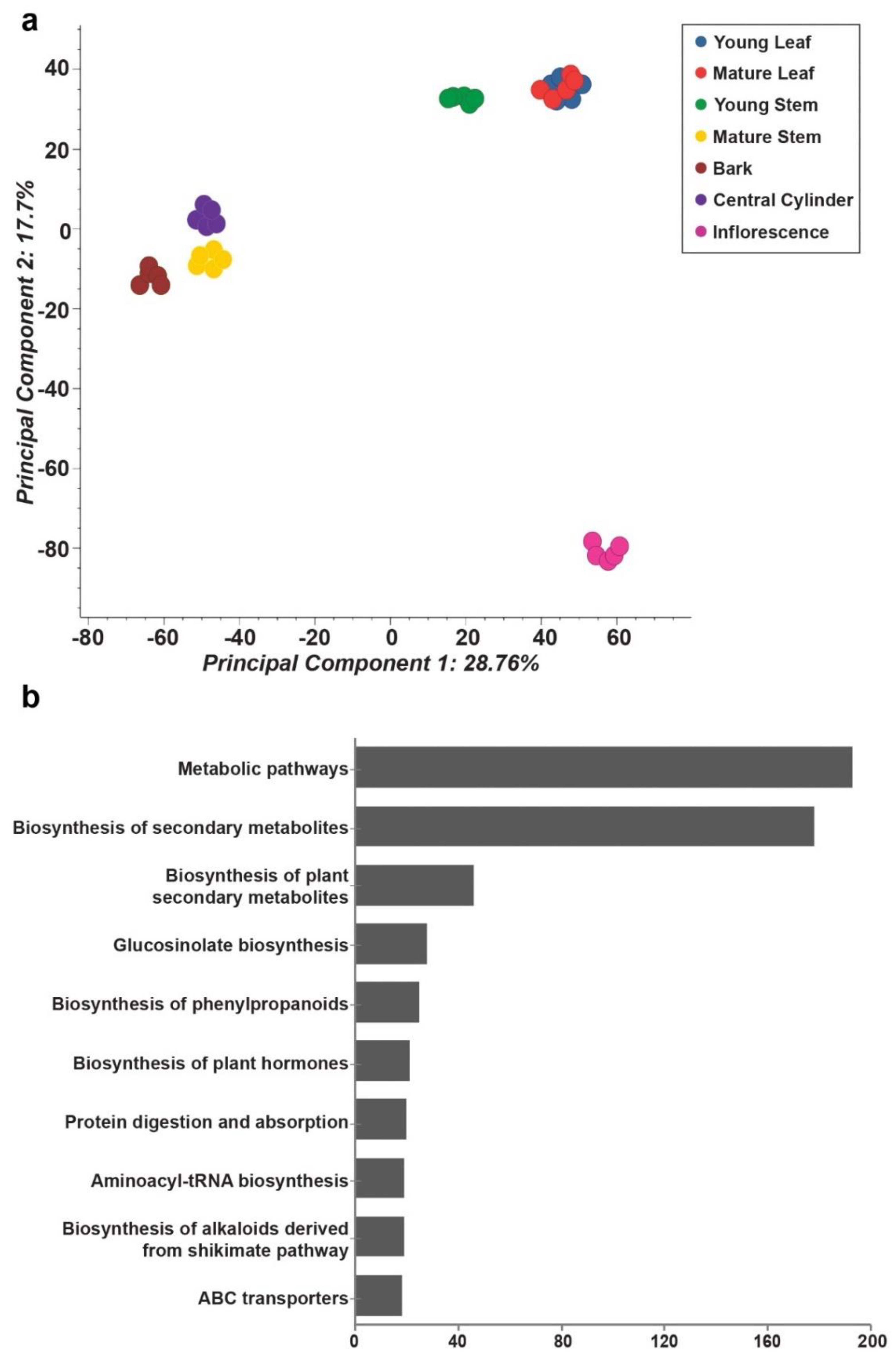
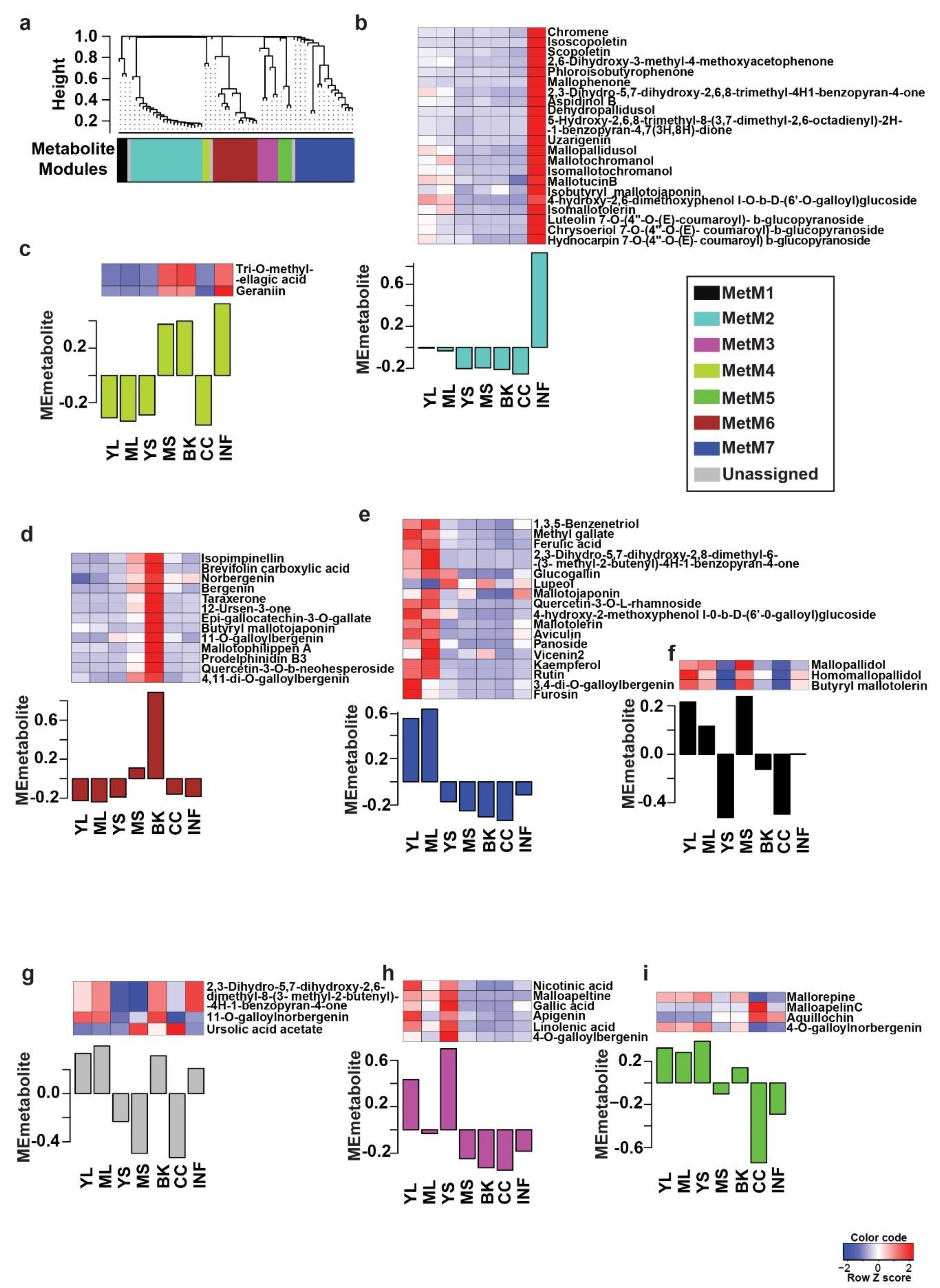
2.2. Tissues of Mallotus japonicus Have Specific Metabolic Signatures Associated with Its Medicinal Use
2.3. Characteristics of Mallotus japonicus De Novo Transcriptome Assembly Derived Using Multiple Tissues
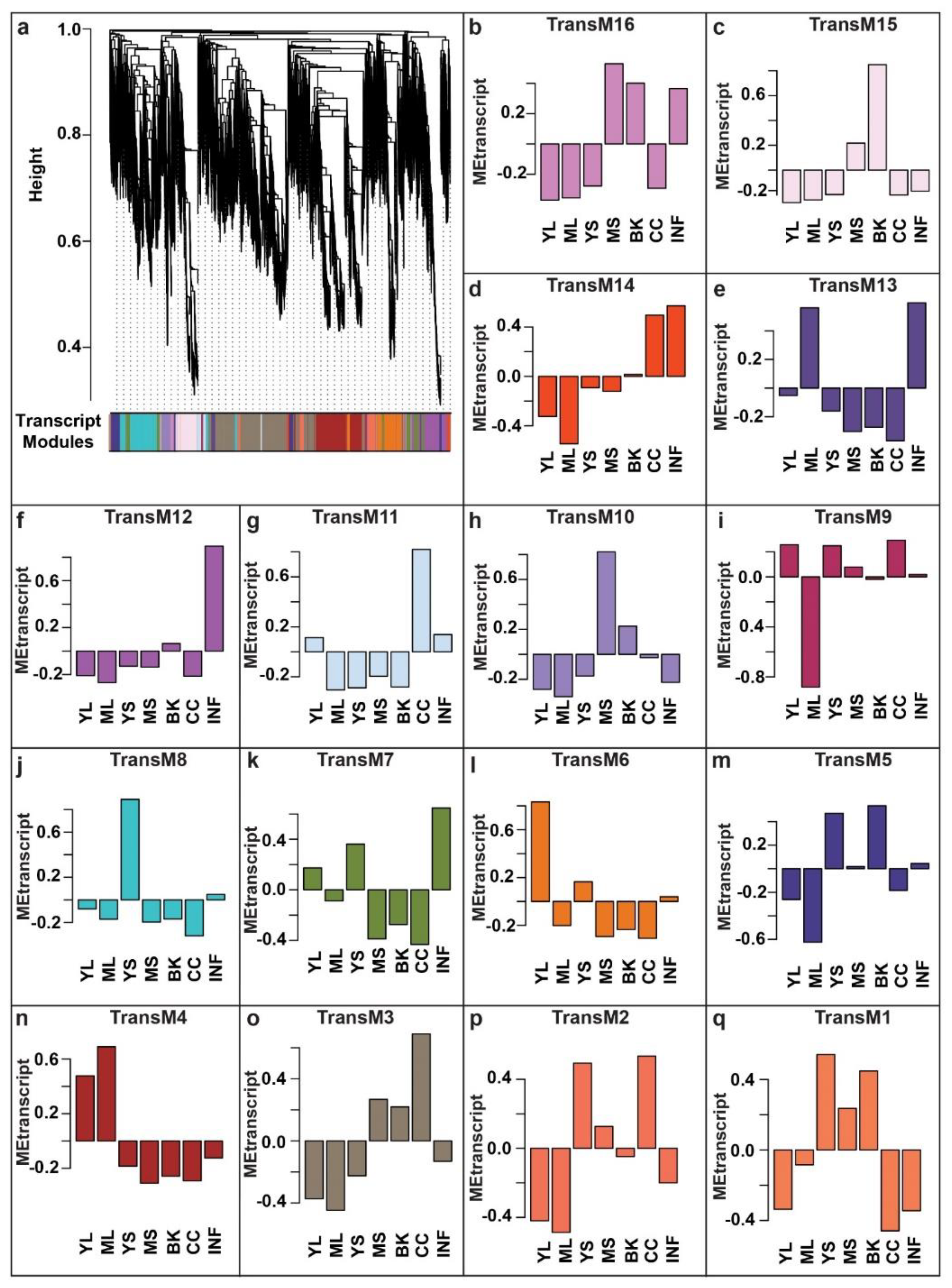
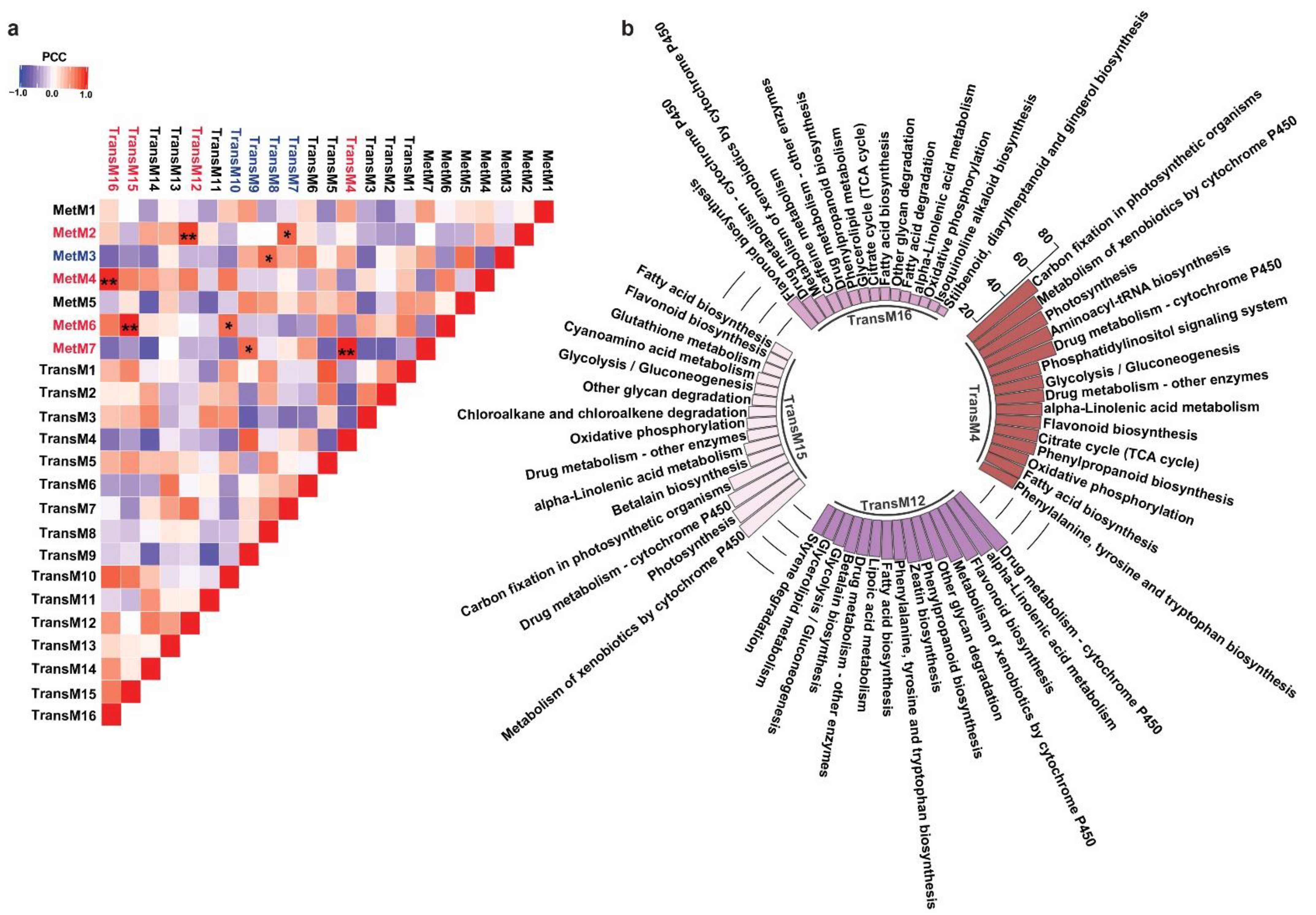
2.4. Network-Based Characterization of Transcriptome and Metabolome Relationships in Mallotus japonicus
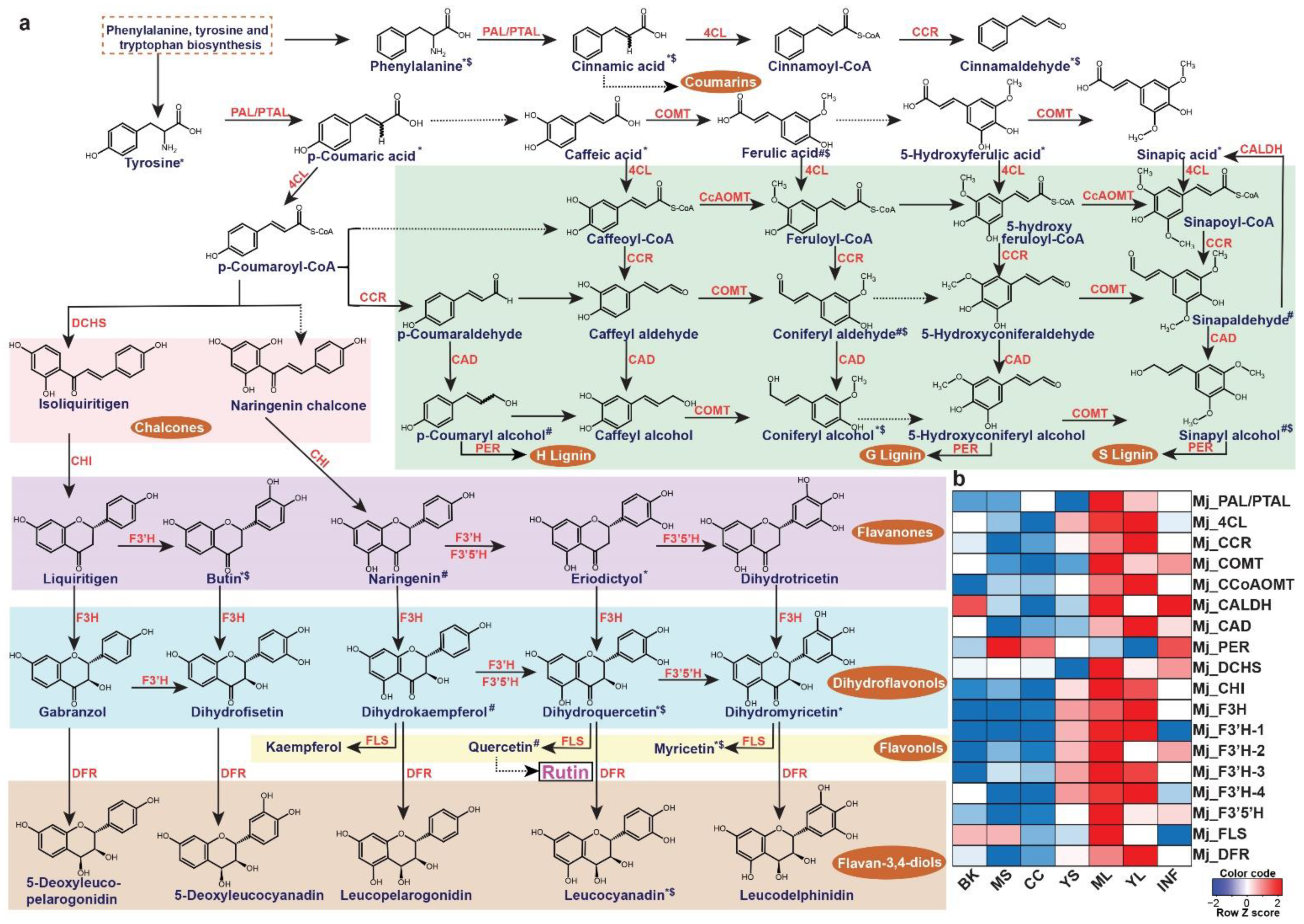
2.5. Metabolome-Assisted Identification of Genes Associated with Rutin Biosynthesis
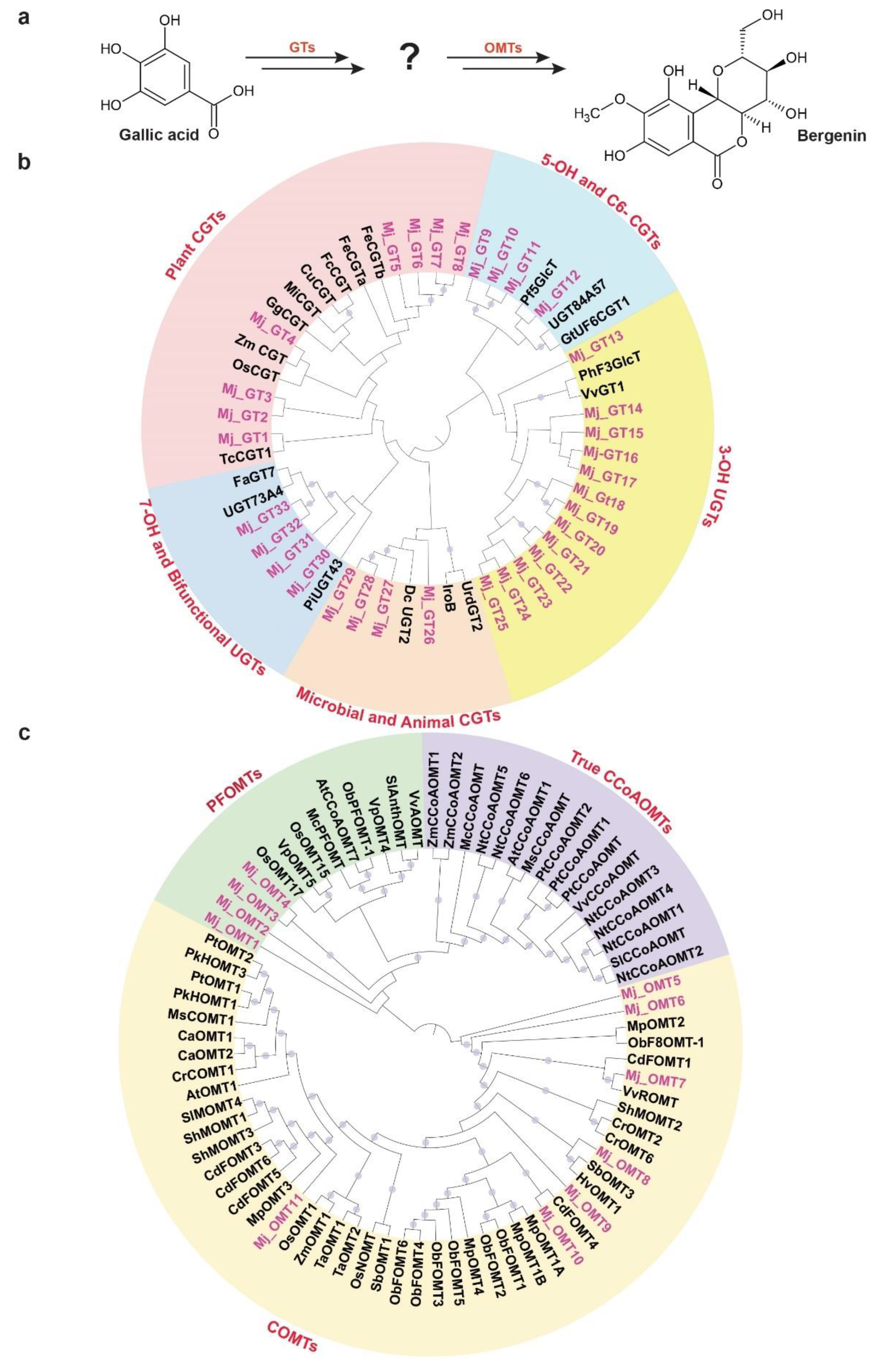
2.6. Phylogenetic Analysis of Glucosyltransferases and O-methyltransferases Reveals Insights into Bergenin Biosynthesis in Mallotus japonicus
3. Materials and Methods
3.1. Plant Materials
3.2. Untargeted Metabolite Profiling Using LC-QTOF-MS
3.3. Feature-Based Molecular Networking
3.4. RNA Isolation and cDNA Synthesis
3.5. RNA-Sequencing and Processing of Raw Data to Generate Transcriptome Assembly
3.6. Functional Classification, KEGG Pathway Mapping, and Expression Analysis of the Assembled Transcripts of Mallotus japonicus
3.7. Module Construction Using Transcriptome Dataset
3.8. Correlation Calculation between the Identified Metabolite and Transcript Modules
3.9. Gene Ontology Enrichment Analysis
3.10. Phylogenetic Analysis of GTs and O-Methyltransferases
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arisawa, M. A review of the biological activity and chemistry of Mallotus japonicus (Euphorbiaceae). Phytomedicine 1994, 1, 261–269. [Google Scholar] [CrossRef]
- Rivière, C.; Nguyen Thi Hong, V.; Tran Hong, Q.; Chataigné, G.; Nguyen Hoai, N.; Dejaegher, B.; Tistaert, C.; Nguyen Thi Kim, T.; Vander Heyden, Y.; Chau Van, M.; et al. Mallotus species from Vietnamese mountainous areas: Phytochemistry and pharmacological activities. Phytochem. Rev. 2010, 9, 217–253. [Google Scholar] [CrossRef]
- Li, D.Z.; Tang, C.; Quinn, R.J.; Feng, Y.; Ke, C.Q.; Yao, S.; Ye, Y. ent-Labdane diterpenes from the stems of Mallotus japonicus. J. Nat. Prod. 2013, 76, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.P.; Zhang, X.F.; Zhu, Z.X.; Wang, H.F. Complete plastome sequence of Mallotus japonicus (Linn. f.) Mull. Arg. (Euphorbiaceae): A medicinal plant species endemic in East Asia. Mitochondrial DNA B Resour. 2021, 6, 1409–1410. [Google Scholar] [CrossRef]
- Chiang Su New Medical College. Dictionary of Chinese Crude Drugs; Shanghai People’ Publishing House: Shanghai, China, 1977.
- Lim, H.-K.; Kim, H.-S.; Choi, H.-S.; Oh, S.; Choi, J. Hepatoprotective effects of bergenin, a major constituent of Mallotus japonicus, on carbon tetrachloride-intoxicated rats. J. Ethnopharmacol. 2000, 72, 469–474. [Google Scholar] [CrossRef]
- Jahromi, M.A.F.; Chansouria, J.P.N.; Ray, A.B. Hypolipidaemic Activity in Rats of Bergenin, the Major Constituent of Flueggea microcarpa. Phytother. Res. 1992, 6, 180–183. [Google Scholar] [CrossRef]
- Okada, T.; Suzuki, T.; Hasobe, S.; Kisara, K. Bergenin. 1. Antiulcerogenic activities of bergenin. Nihon Yakurigaku Zasshi 1973, 69, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.K.; Kim, H.S.; Choi, H.S.; Choi, J.; Kim, S.H.; Chang, M.J. Effects of Bergenin, the Major Constituent of Mallotus japonicus against D-Galactosamine-Induced Hepatotoxicity in Rats. Pharmacology 2001, 63, 71–75. [Google Scholar] [CrossRef]
- Abe, K.; Sakai, K.; Uchida, M. Effects of bergenin on experimental ulcers—Prevention of stress induced ulcers in rats. Gen. Pharmacol. Vasc. Syst. 1980, 11, 361–368. [Google Scholar] [CrossRef]
- Yoshida, T.; Seno, K.; Takama, Y.; Okuda, T. Bergenin derivatives from Mallotus japonicus. Phytochemistry 1982, 21, 1180–1182. [Google Scholar] [CrossRef]
- Rai, A.; Saito, K. Omics data input for metabolic modeling. Curr. Opin. Biotechnol. 2016, 37, 127–134. [Google Scholar] [CrossRef]
- Rai, A.; Saito, K.; Yamazaki, M. Integrated omics analysis of specialized metabolism in medicinal plants. Plant J. 2017, 90, 764–787. [Google Scholar] [CrossRef]
- Gertsman, I.; Barshop, B.A. Promises and pitfalls of untargeted metabolomics. J. Inherit. Metab. Dis. 2018, 41, 355–366. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Nothias, L.F.; Petras, D.; Schmid, R.; Duhrkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Nakamura, Y.; Afendi, F.M.; Parvin, A.K.; Ono, N.; Tanaka, K.; Hirai Morita, A.; Sato, T.; Sugiura, T.; Altaf-Ul-Amin, M.; Kanaya, S. KNApSAcK Metabolite Activity Database for retrieving the relationships between metabolites and biological activities. Plant Cell Physiol. 2014, 55, e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ferrer, J.L.; Austin, M.B.; Stewart, C., Jr.; Noel, J.P. Structure and function of enzymes involved in the biosynthesis of phenylpropanoids. Plant Physiol. Biochem. 2008, 46, 356–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mierziak, J.; Kostyn, K.; Kulma, A. Flavonoids as important molecules of plant interactions with the environment. Molecules 2014, 19, 16240–16265. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Ramawat, K.G. Isoflavonoids. In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, K.G., Mérillon, J.-M., Eds.; Springer: Berlin/Heidelberg, Gremany, 2013; pp. 1849–1865. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- DiLeo, M.V.; Strahan, G.D.; den Bakker, M.; Hoekenga, O.A. Weighted correlation network analysis (WGCNA) applied to the tomato fruit metabolome. PLoS ONE 2011, 6, e26683. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Chen, L.; Wang, J.; Qiao, J.; Zhang, W. Protein Network Signatures Associated with Exogenous Biofuels Treatments in Cyanobacterium Synechocystis sp. PCC 6803. Front. Bioeng. Biotechnol. 2014, 2, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taira, J.; Tsuchida, E.; Uehara, M.; Ohhama, N.; Ohmine, W.; Ogi, T. The leaf extract of Mallotus japonicus and its major active constituent, rutin, suppressed on melanin production in murine B16F1 melanoma. Asian Pac. J. Trop. Biomed. 2015, 5, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Tabata, H.; Katsube, T.; Tsuma, T.; Ohta, Y.; Imawaka, N.; Utsumi, T. Isolation and evaluation of the radical-scavenging activity of the antioxidants in the leaves of an edible plant, Mallotus japonicus. Food Chem. 2008, 109, 64–71. [Google Scholar] [CrossRef]
- Rai, A.; Hirakawa, H.; Nakabayashi, R.; Kikuchi, S.; Hayashi, K.; Rai, M.; Tsugawa, H.; Nakaya, T.; Mori, T.; Nagasaki, H.; et al. Chromosome-level genome assembly of Ophiorrhiza pumila reveals the evolution of camptothecin biosynthesis. Nat. Commun. 2021, 12, 405. [Google Scholar] [CrossRef]
- One Thousand Plant Transcriptomes Initiative. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Rai, A.; Nakaya, T.; Shimizu, Y.; Rai, M.; Nakamura, M.; Suzuki, H.; Saito, K.; Yamazaki, M. De Novo Transcriptome Assembly and Characterization of Lithospermum officinale to Discover Putative Genes Involved in Specialized Metabolites Biosynthesis. Planta Med. 2018, 84, 920–934. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Rai, A.; Rai, M.; Nakamura, M.; Kawano, N.; Yoshimatsu, K.; Suzuki, H.; Kawahara, N.; Saito, K.; Yamazaki, M. Comparative transcriptome analyses of three medicinal Forsythia species and prediction of candidate genes involved in secondary metabolisms. J. Nat. Med. 2018, 72, 867–881. [Google Scholar] [CrossRef]
- Rai, M.; Rai, A.; Kawano, N.; Yoshimatsu, K.; Takahashi, H.; Suzuki, H.; Kawahara, N.; Saito, K.; Yamazaki, M. De Novo RNA Sequencing and Expression Analysis of Aconitum carmichaelii to Analyze Key Genes Involved in the Biosynthesis of Diterpene Alkaloids. Molecules 2017, 22, 2155. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [Green Version]
- Rai, A.; Rai, M.; Kamochi, H.; Mori, T.; Nakabayashi, R.; Nakamura, M.; Suzuki, H.; Saito, K.; Yamazaki, M. Multiomics-based characterization of specialized metabolites biosynthesis in Cornus Officinalis. DNA Res. 2020, 27, dsaa009. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Gamboa-Tuz, S.D.; Pereira-Santana, A.; Zamora-Briseno, J.A.; Castano, E.; Espadas-Gil, F.; Ayala-Sumuano, J.T.; Keb-Llanes, M.A.; Sanchez-Teyer, F.; Rodriguez-Zapata, L.C. Transcriptomics and co-expression networks reveal tissue-specific responses and regulatory hubs under mild and severe drought in papaya (Carica papaya L.). Sci. Rep. 2018, 8, 14539. [Google Scholar] [CrossRef]
- Greenham, K.; Guadagno, C.R.; Gehan, M.A.; Mockler, T.C.; Weinig, C.; Ewers, B.E.; McClung, C.R. Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in Brassica rapa. eLife 2017, 6, e29655. [Google Scholar] [CrossRef]
- Shahan, R.; Zawora, C.; Wight, H.; Sittmann, J.; Wang, W.; Mount, S.M.; Liu, Z. Consensus Coexpression Network Analysis Identifies Key Regulators of Flower and Fruit Development in Wild Strawberry. Plant Physiol. 2018, 178, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Wang, B.; Yang, J.; Wang, W.; Liu, A.; Leng, L.; Xiang, L.; Song, C.; Chen, S. Weighted Gene Co-Expression Network Analysis of the Dioscin Rich Medicinal Plant Dioscorea nipponica. Front. Plant Sci. 2017, 8, 789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.; Liu, C.; Yu, S.; Yang, H.; Sun, J.; Guo, C.; Huang, B.; Liu, Z.; Yuan, Y.; Xia, E.; et al. Gene co-expression network analysis reveals coordinated regulation of three characteristic secondary biosynthetic pathways in tea plant (Camellia sinensis). BMC Genom. 2018, 19, 616. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zeng, X.; Song, Q.; Sun, Y.; Feng, Y.; Lai, Y. Identification of key genes and modules in response to Cadmium stress in different rice varieties and stem nodes by weighted gene co-expression network analysis. Sci. Rep. 2020, 10, 9525. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Magwanga, R.O.; Yang, X.; Jin, D.; Cai, X.; Hou, Y.; Wei, Y.; Zhou, Z.; Wang, K.; Liu, F. Genetic regulatory networks for salt-alkali stress in Gossypium hirsutum with differing morphological characteristics. BMC Genom. 2020, 21, 15. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; He, C.; Qin, Y.; Lin, G.; Park, W.D.; Sun, M.; Li, J.; Lu, X.; Zhang, C.; Yeh, C.T.; et al. Co-expression analysis aids in the identification of genes in the cuticular wax pathway in maize. Plant J. 2019, 97, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, C.J.; Kohane, I.S.; Butte, A.J. Systematic survey reveals general applicability of “guilt-by-association” within gene coexpression networks. BMC Bioinform. 2005, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.; Dixon, R.A. Co-expression networks for plant biology: Why and how. Acta Biochim. Biophys. Sin. 2019, 51, 981–988. [Google Scholar] [CrossRef]
- Saito, K.; Hirai, M.Y.; Yonekura-Sakakibara, K. Decoding genes with coexpression networks and metabolomics—‘majority report by precogs’. Trends Plant Sci. 2008, 13, 36–43. [Google Scholar] [CrossRef]
- Higashi, Y.; Saito, K. Network analysis for gene discovery in plant-specialized metabolism. Plant Cell Environ. 2013, 36, 1597–1606. [Google Scholar] [CrossRef] [Green Version]
- Scossa, F.; Benina, M.; Alseekh, S.; Zhang, Y.; Fernie, A.R. The Integration of Metabolomics and Next-Generation Sequencing Data to Elucidate the Pathways of Natural Product Metabolism in Medicinal Plants. Planta Med. 2018, 84, 855–873. [Google Scholar] [CrossRef] [Green Version]
- Shinozaki, Y.; Beauvoit, B.P.; Takahara, M.; Hao, S.; Ezura, K.; Andrieu, M.H.; Nishida, K.; Mori, K.; Suzuki, Y.; Kuhara, S.; et al. Fruit setting rewires central metabolism via gibberellin cascades. Proc. Natl. Acad. Sci. USA 2020, 117, 23970–23981. [Google Scholar] [CrossRef]
- Lang, X.; Li, N.; Li, L.; Zhang, S. Integrated Metabolome and Transcriptome Analysis Uncovers the Role of Anthocyanin Metabolism in Michelia maudiae. Int. J. Genom. 2019, 2019, 4393905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rischer, H.; Oresic, M.; Seppanen-Laakso, T.; Katajamaa, M.; Lammertyn, F.; Ardiles-Diaz, W.; Van Montagu, M.C.; Inze, D.; Oksman-Caldentey, K.M.; Goossens, A. Gene-to-metabolite networks for terpenoid indole alkaloid biosynthesis in Catharanthus roseus cells. Proc. Natl. Acad. Sci. USA 2006, 103, 5614–5619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohge, T.; Nishiyama, Y.; Hirai, M.Y.; Yano, M.; Nakajima, J.; Awazuhara, M.; Inoue, E.; Takahashi, H.; Goodenowe, D.B.; Kitayama, M.; et al. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 2005, 42, 218–235. [Google Scholar] [CrossRef]
- Yan, J.; Qian, L.; Zhu, W.; Qiu, J.; Lu, Q.; Wang, X.; Wu, Q.; Ruan, S.; Huang, Y. Integrated analysis of the transcriptome and metabolome of purple and green leaves of Tetrastigma hemsleyanum reveals gene expression patterns involved in anthocyanin biosynthesis. PLoS ONE 2020, 15, e0230154. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Kim, Y.B.; Kim, Y.; Zhao, S.; Kim, H.H.; Chung, E.; Lee, J.H.; Park, S.U. Differential stress-response expression of two flavonol synthase genes and accumulation of flavonols in tartary buckwheat. J. Plant Physiol. 2013, 170, 1630–1636. [Google Scholar] [CrossRef]
- Liang, W.; Ni, L.; Carballar-Lejarazu, R.; Zou, X.; Sun, W.; Wu, L.; Yuan, X.; Mao, Y.; Huang, W.; Zou, S. Comparative transcriptome among Euscaphis konishii Hayata tissues and analysis of genes involved in flavonoid biosynthesis and accumulation. BMC Genom. 2019, 20, 24. [Google Scholar] [CrossRef]
- Shi, J.; Li, W.; Gao, Y.; Wang, B.; Li, Y.; Song, Z. Enhanced rutin accumulation in tobacco leaves by overexpressing the NtFLS2 gene. Biosci. Biotechnol. Biochem. 2017, 81, 1721–1725. [Google Scholar] [CrossRef] [Green Version]
- Franz, G.; Grun, M. Chemistry, occurrence and biosynthesis of C-glycosyl compounds in plants. Planta Med. 1983, 47, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Taneyama, M.; Yoshida, S. Studies on C-glycosides in higher plants. The Botanical Magazine = Shokubutsu-Gaku-Zasshi 1979, 92, 69–73. [Google Scholar] [CrossRef]
- Gutmann, A.; Nidetzky, B. Switching between O- and C-glycosyltransferase through exchange of active-site motifs. Angew. Chem. Int. Ed. Engl. 2012, 51, 12879–12883. [Google Scholar] [CrossRef]
- He, J.B.; Zhao, P.; Hu, Z.M.; Liu, S.; Kuang, Y.; Zhang, M.; Li, B.; Yun, C.H.; Qiao, X.; Ye, M. Molecular and Structural Characterization of a Promiscuous C-Glycosyltransferase from Trollius chinensis. Angew. Chem. Int. Ed. Engl. 2019, 58, 11513–11520. [Google Scholar] [CrossRef] [PubMed]
- Putkaradze, N.; Teze, D.; Fredslund, F.; Welner, D.H. Natural product C-glycosyltransferases—A scarcely characterised enzymatic activity with biotechnological potential. Nat. Prod. Rep. 2021, 38, 432–443. [Google Scholar] [CrossRef]
- Hao, B.; Caulfield, J.C.; Hamilton, M.L.; Pickett, J.A.; Midega, C.A.; Khan, Z.R.; Wang, J.; Hooper, A.M. Biosynthesis of natural and novel C-glycosylflavones utilising recombinant Oryza sativa C-glycosyltransferase (OsCGT) and Desmodium incanum root proteins. Phytochemistry 2016, 125, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Brazier-Hicks, M.; Evans, K.M.; Gershater, M.C.; Puschmann, H.; Steel, P.G.; Edwards, R. The C-glycosylation of flavonoids in cereals. J. Biol. Chem. 2009, 284, 17926–17934. [Google Scholar] [CrossRef] [Green Version]
- Joshi, C.P.; Chiang, V.L. Conserved sequence motifs in plant S-adenosyl-L-methionine-dependent methyltransferases. Plant Mol. Biol. 1998, 37, 663–674. [Google Scholar] [CrossRef]
- Kim, B.-G.; Sung, S.H.; Chong, Y.; Lim, Y.; Ahn, J.-H. Plant Flavonoid O-Methyltransferases: Substrate Specificity and Application. J. Plant Biol. 2010, 53, 321–329. [Google Scholar] [CrossRef]
- Ibdah, M.; Zhang, X.H.; Schmidt, J.; Vogt, T. A novel Mg(2+)-dependent O-methyltransferase in the phenylpropanoid metabolism of Mesembryanthemum crystallinum. J. Biol. Chem. 2003, 278, 43961–43972. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhao, C.; Gong, Q.; Wang, Y.; Cao, J.; Li, X.; Grierson, D.; Sun, C. Characterization of a caffeoyl-CoA O-methyltransferase-like enzyme involved in biosynthesis of polymethoxylated flavones in Citrus reticulata. J. Exp. Bot. 2020, 71, 3066–3079. [Google Scholar] [CrossRef]
- Widiez, T.; Hartman, T.G.; Dudai, N.; Yan, Q.; Lawton, M.; Havkin-Frenkel, D.; Belanger, F.C. Functional characterization of two new members of the caffeoyl CoA O-methyltransferase-like gene family from Vanilla planifolia reveals a new class of plastid-localized O-methyltransferases. Plant Mol. Biol. 2011, 76, 475–488. [Google Scholar] [CrossRef]
- Mohimani, H.; Gurevich, A.; Mikheenko, A.; Garg, N.; Nothias, L.F.; Ninomiya, A.; Takada, K.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of peptidic natural products through database search of mass spectra. Nat. Chem. Biol. 2017, 13, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Yamazaki, M.; Takahashi, H.; Nakamura, M.; Kojoma, M.; Suzuki, H.; Saito, K. RNA-Seq Transcriptome Analysis of Panax japonicus, and Its Comparison with Other Panax Species to Identify Potential Genes Involved in the Saponins Biosynthesis. Front. Plant Sci. 2016, 7, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, A.; Nakamura, M.; Takahashi, H.; Suzuki, H.; Saito, K.; Yamazaki, M. High-throughput sequencing and de novo transcriptome assembly of Swertia japonica to identify genes involved in the biosynthesis of therapeutic metabolites. Plant Cell Rep. 2016, 35, 2091–2111. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembler | Kmer | No. of Contigs | N50 | Mean Length | Median Length | Max Length | n: >500 | n: >1000 | Total Size of Contigs |
|---|---|---|---|---|---|---|---|---|---|
| CLC | 20 | 108,863 | 769 | 592 | 366 | 17,150 | 35,538 (32.6%) | 14,771 (13.6%) | 64,424,765 |
| Trinity | 25 | 321,190 | 1503 | 879 | 486 | 17,097 | 156,853 (48.8%) | 90,563 (28.2%) | 282,445,544 |
| SOAPdenovo | 31 | 278,339 | 595 | 322 | 156 | 17,202 | 34,941 (12.6%) | 17,967 (6.5%) | 89,684,117 |
| 41 | 276,091 | 525 | 325 | 168 | 17,225 | 34,755 (12.6%) | 17,259 (6.3%) | 89,843,229 | |
| 51 | 337,219 | 373 | 278 | 160 | 17,210 | 33,450 (9.9%) | 15,902 (4.7%) | 93,877,046 | |
| 63 | 273,737 | 397 | 307 | 181 | 17,292 | 31,027 (11.3%) | 14,829 (5.4%) | 84,133,522 | |
| 71 | 209,952 | 484 | 345 | 199 | 16,558 | 28,293 (13.5%) | 14,006 (6.7%) | 72,338,185 | |
| 91 | 46,669 | 1062 | 585 | 294 | 11,896 | 15,185 (32.5%) | 8137 (17.4%) | 27,278,317 | |
| CLC_Trinity_SOAPdenovo (kmer31) _CD-HIT-EST | N.A. | 226,250 | 1396 | 837 | 469 | 17,202 | 106,994 (47.3%) | 58,421 (25.8%) | 189,317,381 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rai, M.; Rai, A.; Mori, T.; Nakabayashi, R.; Yamamoto, M.; Nakamura, M.; Suzuki, H.; Saito, K.; Yamazaki, M. Gene-Metabolite Network Analysis Revealed Tissue-Specific Accumulation of Therapeutic Metabolites in Mallotus japonicus. Int. J. Mol. Sci. 2021, 22, 8835. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168835
Rai M, Rai A, Mori T, Nakabayashi R, Yamamoto M, Nakamura M, Suzuki H, Saito K, Yamazaki M. Gene-Metabolite Network Analysis Revealed Tissue-Specific Accumulation of Therapeutic Metabolites in Mallotus japonicus. International Journal of Molecular Sciences. 2021; 22(16):8835. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168835
Chicago/Turabian StyleRai, Megha, Amit Rai, Tetsuya Mori, Ryo Nakabayashi, Manami Yamamoto, Michimi Nakamura, Hideyuki Suzuki, Kazuki Saito, and Mami Yamazaki. 2021. "Gene-Metabolite Network Analysis Revealed Tissue-Specific Accumulation of Therapeutic Metabolites in Mallotus japonicus" International Journal of Molecular Sciences 22, no. 16: 8835. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168835