
GSH and Zinc Supplementation Attenuate Cadmium-Induced Cellular Stress and Stimulation of Choline Uptake in Cultured Neonatal Rat Choroid Plexus Epithelia



Abstract
:1. Introduction
2. Results
2.1. Cultured Choroid Plexus Epitheial Cells (CPECs) Accumulate Cadmium (Cd) and Zinc (Zn)
2.2. Cadmium Alters Intracellular Glutathione (GSH) and Glutathione Sulfide (GSSG) in CPECs
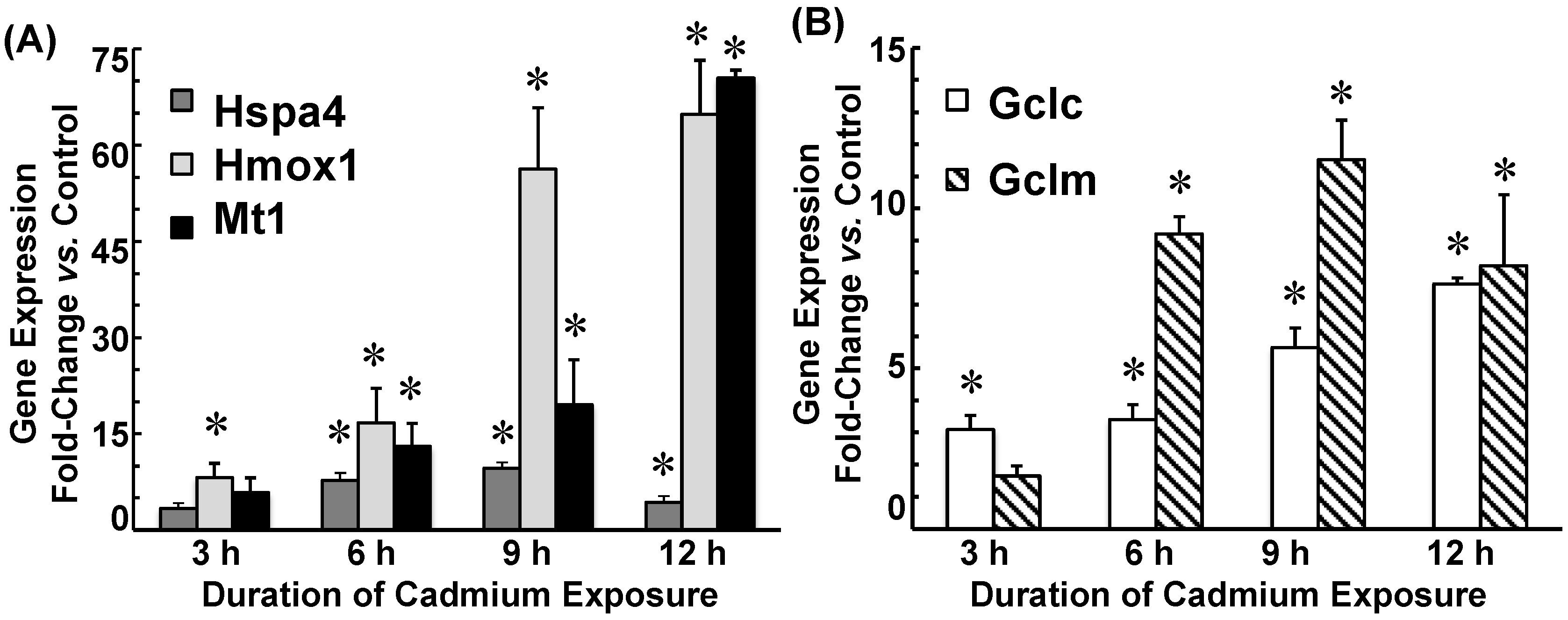
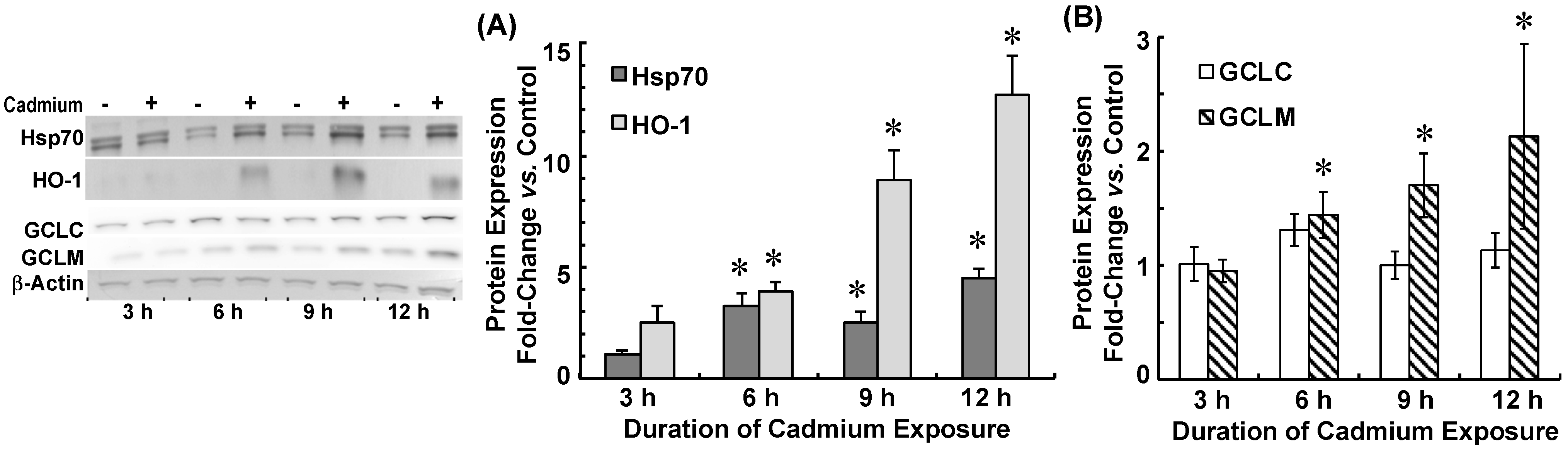
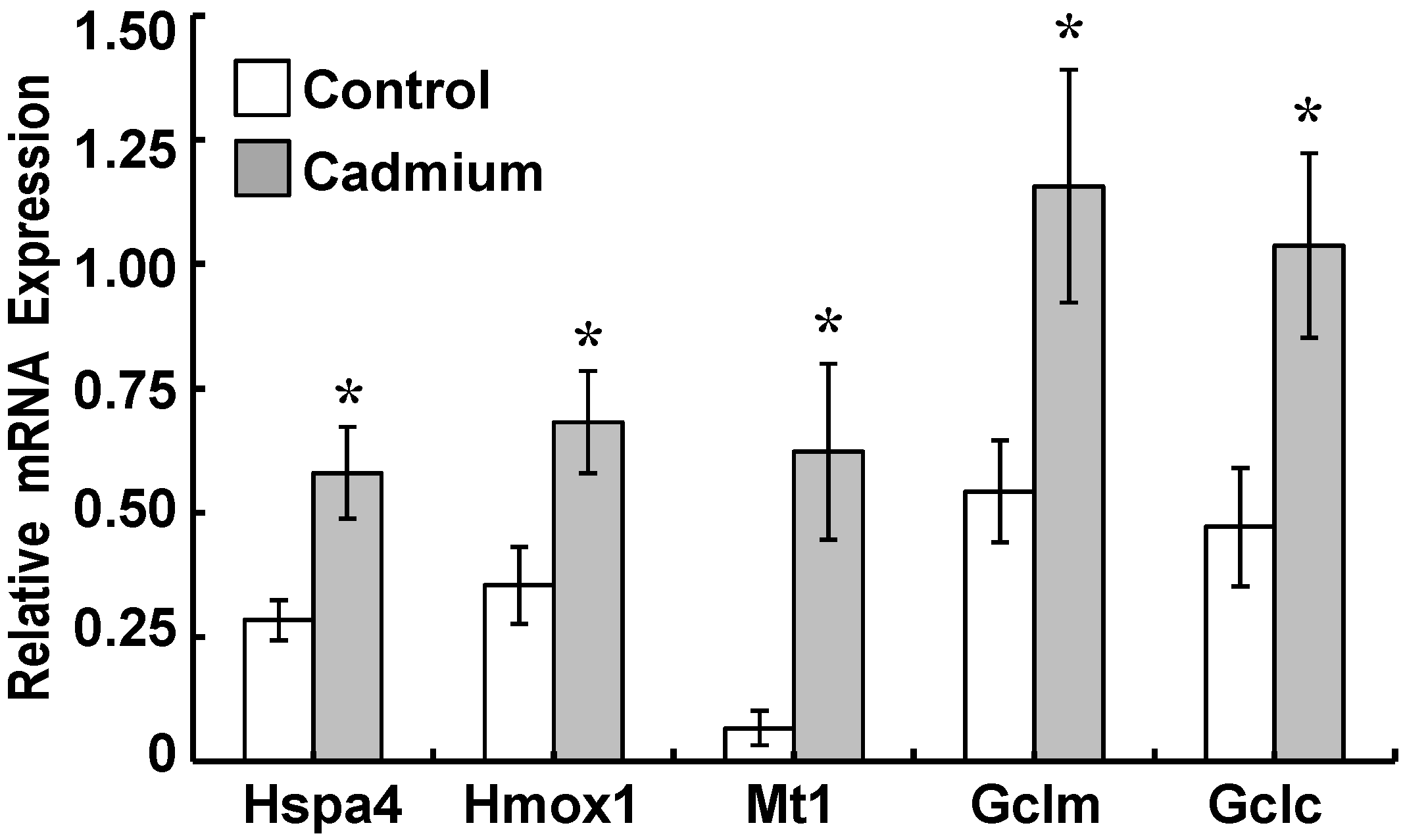
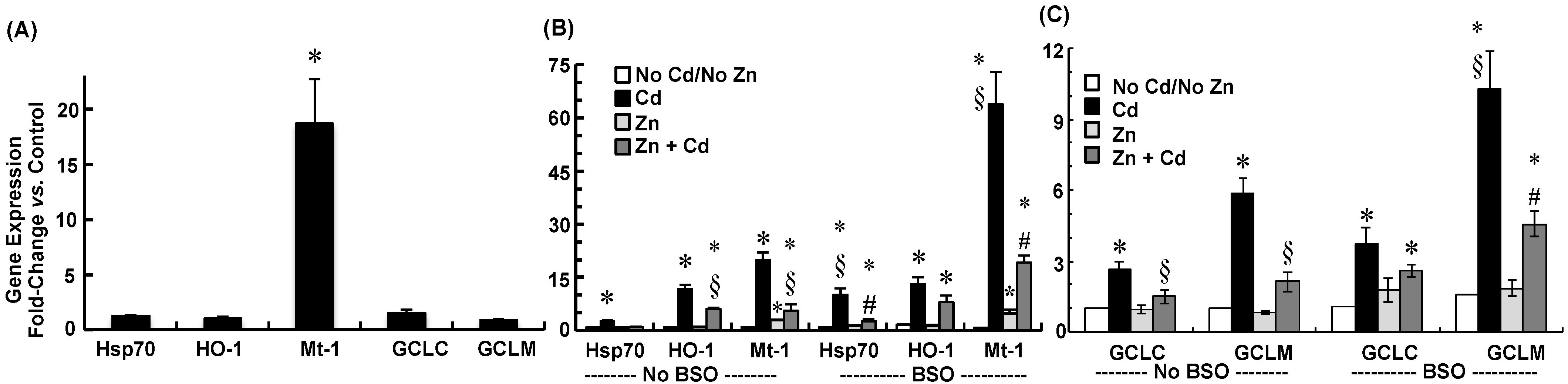
2.3. Cadmium Induced Glutamate Cysteine Ligase (GCL) Subunits and Stress Proteins in CPECs
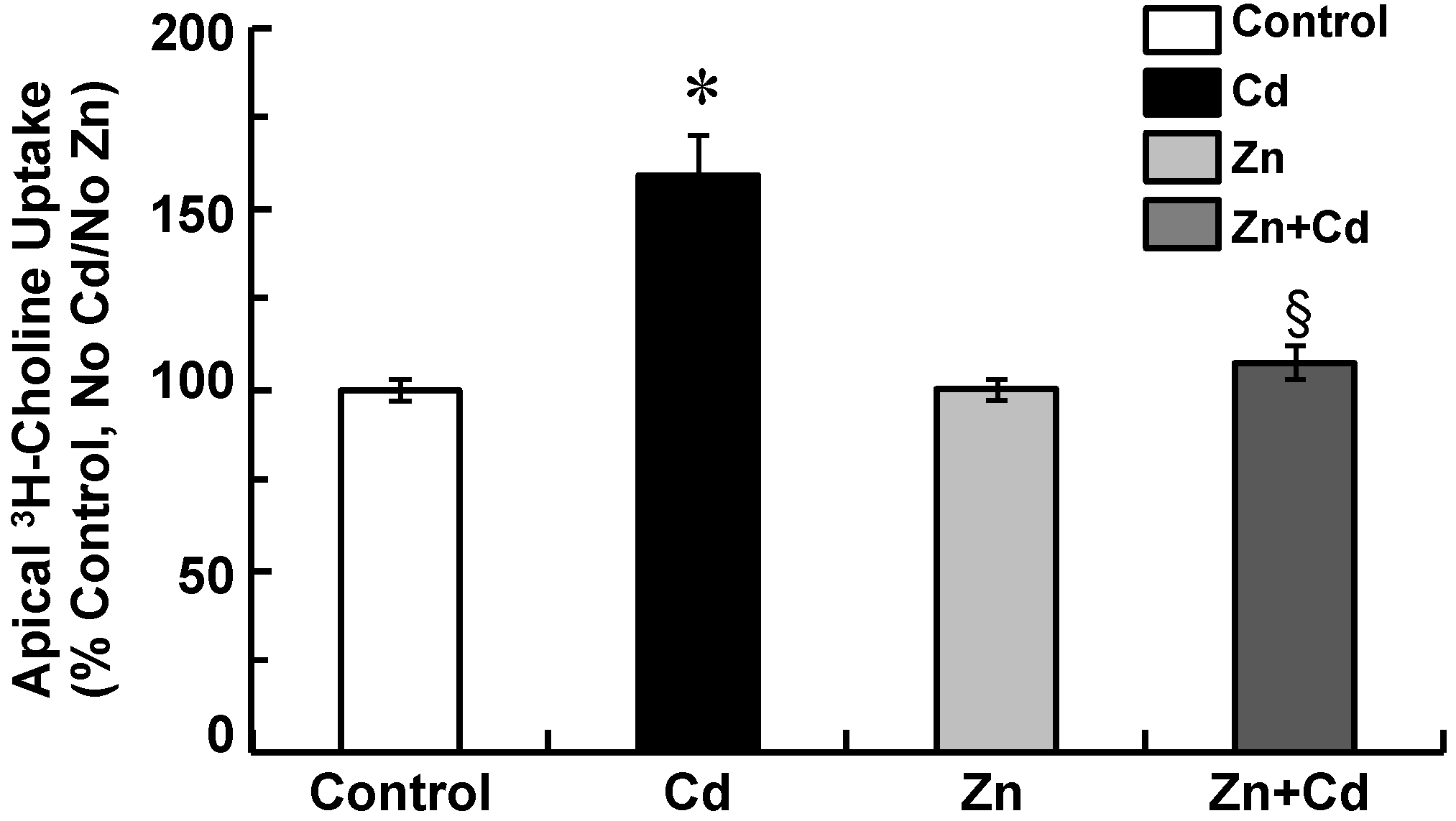
2.4. Zinc Attenuates Increases in Apical Choline Uptake in Cadmium-Exposed CPECs
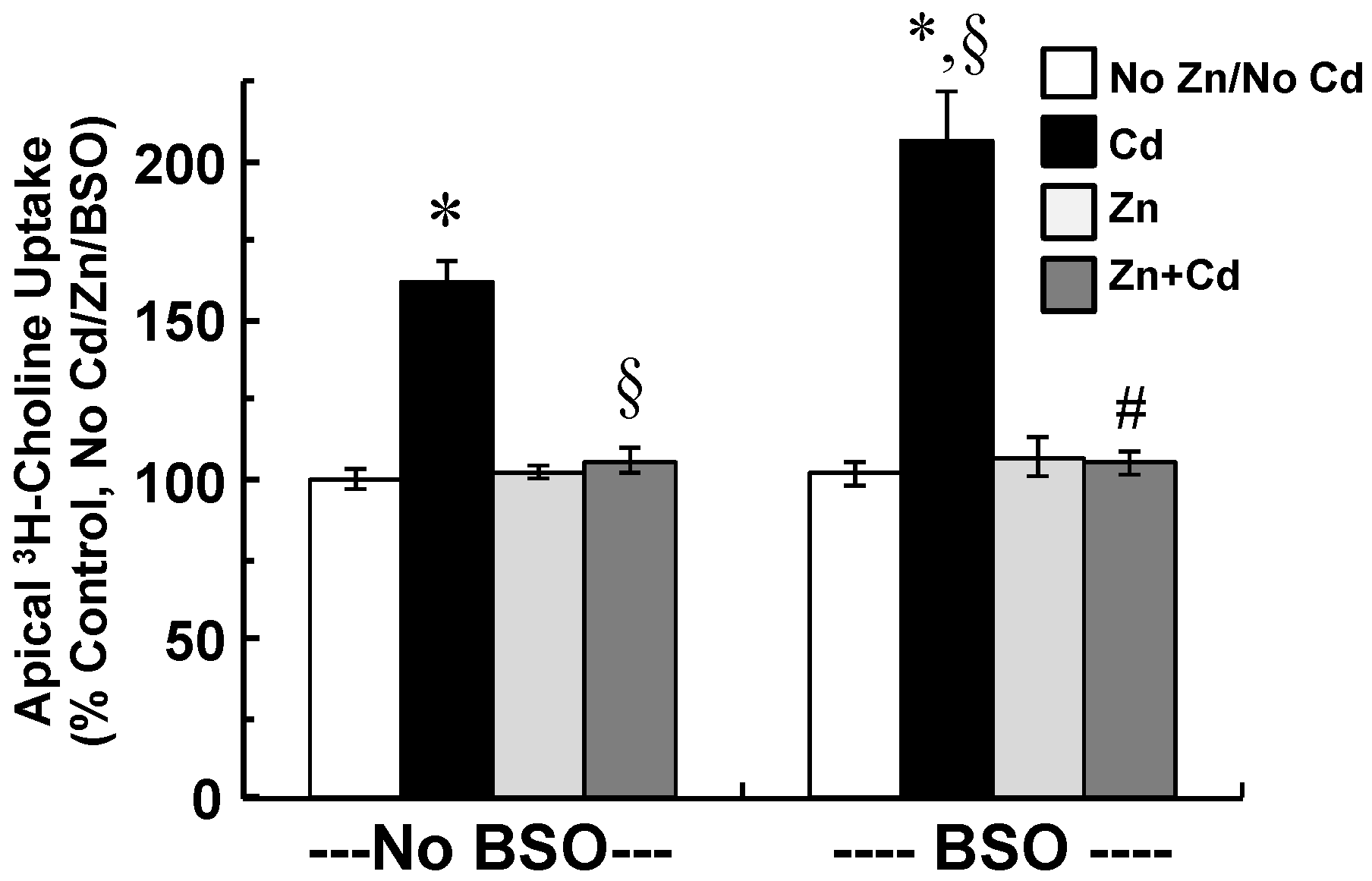
2.5. Zinc Supplementation and Inhibition of GSH Synthesis Modify Cellular Stress Responses and Apical Choline Uptake in Cadmium-Exposed CPECs
3. Discussion
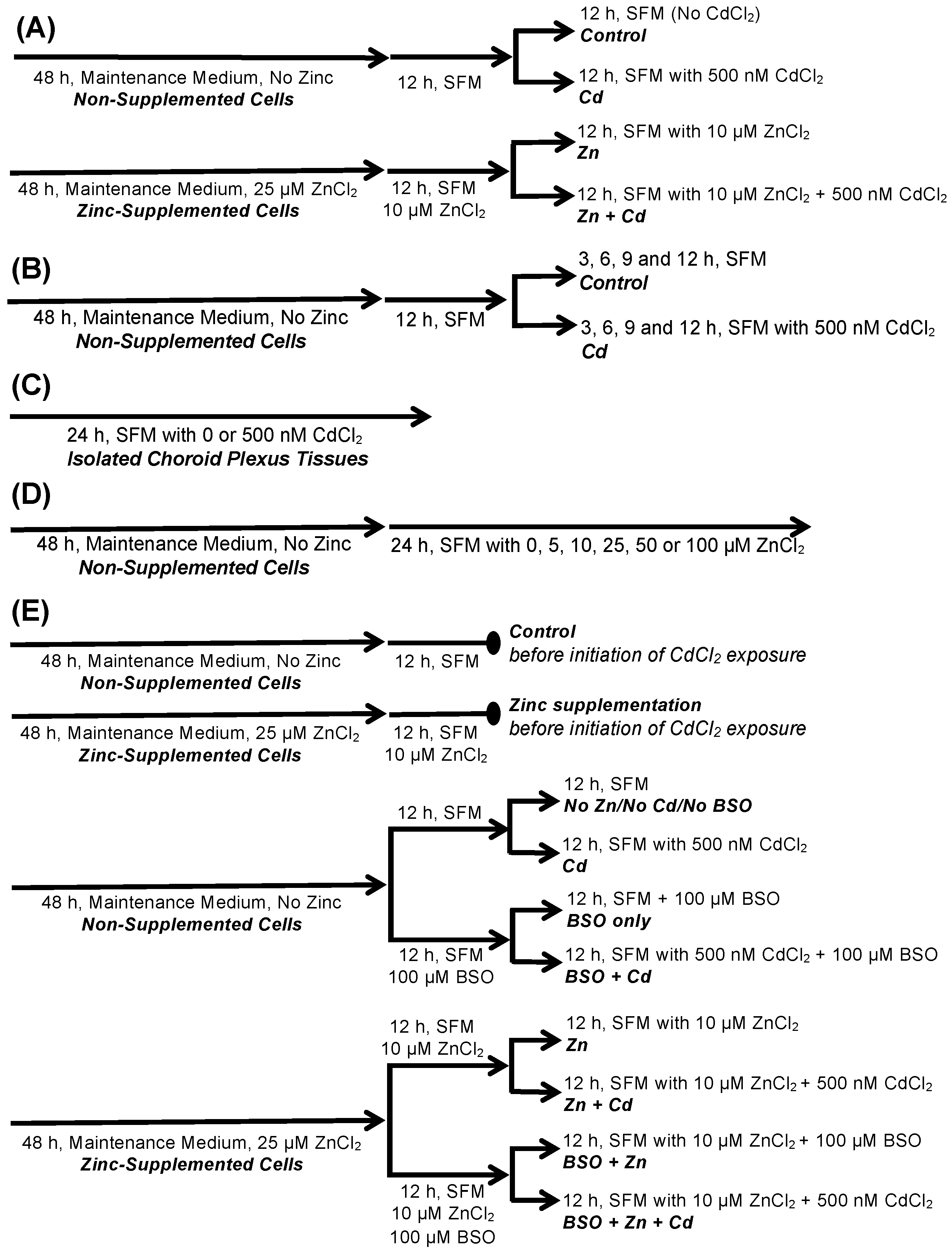
4. Materials and Methods
4.1. Animal Use and Tissue Harvest
4.2. Chemicals, Reagents, and Solutions
4.3. Isolation and Primary Culture of Choroid Plexus Epithelial Cells (CPECs)
4.4. Treatment of CPECs and Isolated CP Tissues with Cadmium, Zinc, and BSO
4.5. Analysis of Cellular Accumulation of Zinc and Cadmium
4.6. Intracellular Reduced Glutathione and Oxidized Glutathione Assay
4.7. Analysis of Gene Expression
4.8. Immunoblot Analysis
4.9. Radiotracer Assay of Apical Choline Transport in CPECs
4.10. Extracellular Lactate Dehydrogenase Assay
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbott, N.J. Dynamics of CNS barriers: Evolution, differentiation, and modulation. Cell. Mol. Neurobiol. 2005, 25, 5–23. [Google Scholar] [CrossRef]
- Praetorius, J.; Damkier, H.H. Transport across the choroid plexus epithelium. Am. J. Physiol.-Cell Physiol. 2017, 312, C673–C686. [Google Scholar] [CrossRef]
- Redzic, Z.B.; Preston, J.E.; Duncan, J.A.; Chodobski, A.; Szmydynger-Chodobska, J. The choroid plexus-cerebrospinal fluid system: From development to aging. Curr. Top. Dev. Biol. 2005, 71, 1–52. [Google Scholar] [CrossRef]
- Strazielle, N.; Ghersi-Egea, J.F. Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol. Pharm. 2013, 10, 1473–1491. [Google Scholar] [CrossRef] [PubMed]
- Friedheim, E.; Corvi, C.; Graziano, J.; Donnelli, T.; Breslin, D. Choroid plexus as a protective sink for heavy metals? Lancet 1983, 1, 981–982. [Google Scholar] [CrossRef]
- Zheng, W. Toxicology of choroid plexus: Special reference to metal-induced neurotoxicities. Microsc. Res. Tech. 2001, 52, 89–103. [Google Scholar] [CrossRef]
- Zheng, W.; Perry, D.F.; Nelson, D.L.; Aposhian, H.V. Choroid plexus protects cerebrospinal fluid against toxic metals. FASEB J. 1991, 5, 2188–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvidson, B.; Tjälve, H. Distribution of 109Cd in the nervous system of rats after intravenous injection. Acta Neuropathol. 1986, 69, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Takefuta, S.; Ijiro, H.; Okada, S.; Oku, N. 109Cd transport in rat brain. Brain Res. Bull. 1999, 49, 453–457. [Google Scholar] [CrossRef]
- Valois, A.A.; Webster, W.S. The choroid plexus as a target site for cadmium toxicity following chronic exposure in the adult mouse: An ultrastructural study. Toxicology 1989, 55, 193–205. [Google Scholar] [CrossRef]
- Agency for Toxic Substances and Disease Registry. Toxicological Profile for Cadmium; Department of Health and Human Services, Public Health Service: Atlanta, GA, USA, 2012. [Google Scholar]
- Satarug, S.; Garrett, S.H.; Sens, M.A.; Sens, D.A. Cadmium, environmental exposure, and health outcomes. Environ. Health Perspect. 2010, 118, 182–190. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef]
- Casalino, E.; Sblano, C.; Landriscina, C. Enzyme activity alteration by cadmium administration to rats: The possibility of iron involvement in lipid peroxidation. Arch. Biochem. Biophys. 1997, 346, 171–179. [Google Scholar] [CrossRef] [PubMed]
- L’hoste, S.; Chargui, A.; Belfodil, R.; Duranton, C.; Rubera, I.; Mograbi, B.; Poujeol, C.; Tauc, M.; Poujeol, P. CFTR mediates cadmium-induced apoptosis through modulation of ROS level in mouse proximal tubule cells. Free Radic. Biol. Med. 2009, 46, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, J.; Leonard, S.S.; Rao, K.M. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic. Biol. Med. 2004, 36, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Young, R.K.; Villalobos, A.R. Stress-induced stimulation of choline transport in cultured choroid plexus epithelium exposed to low concentrations of cadmium. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R291–R303. [Google Scholar] [CrossRef] [Green Version]
- Sweet, D.H.; Miller, D.S.; Pritchard, J.B. Ventricular choline transport: A role for organic cation transporter 2 expressed in choroid plexus. J. Biol. Chem. 2001, 276, 41611–41619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, S.H. Choline: Essential for brain development and function. Adv. Pediatrics 1997, 44, 263–295. [Google Scholar] [CrossRef]
- Zeisel, S.H. Nutritional importance of choline for brain development. J. Am. Coll. Nutr. 2004, 23, 621S–626S. [Google Scholar] [CrossRef]
- Derbyshire, E.; Obeid, R. Choline, Neurological development and brain function: A systematic review focusing on the first 1000 days. Nutrients 2020, 12, 1731. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Singhal, R.K.; Anderson, M.E.; Meister, A. Glutathione, a first line of defense against cadmium toxicity. FASEB J. 1987, 1, 220–223. [Google Scholar] [CrossRef]
- Suzuki, C.A.; Cherian, M.G. Renal glutathione depletion and nephrotoxicity of cadmium-metallothionein in rats. Toxicol. Appl. Pharmacol. 1989, 98, 544–552. [Google Scholar] [CrossRef]
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, A.R.; Munters, E.; Artois, T.J.; et al. Cadmium stress: An oxidative challenge. Biometals 2010, 23, 27–40. [Google Scholar] [CrossRef]
- Tate, S.S.; Ross, L.L.; Meister, A. The g-glutamyl cycle in the choroid plexus: Its possible function in amino acid transport. Proc. Natl. Acad. Sci. USA 1973, 70, 1447–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghersi-Egea, J.F.; Strazielle, N.; Murat, A.; Jouvet, A.; Buénerd, A.; Belin, M.F. Brain protection at the blood-cerebrospinal fluid interface involves a glutathione-dependent metabolic barrier mechanism. J. Cereb. Blood Flow Metab. 2006, 26, 1165–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudrais, E.; Strazielle, N.; Ghersi-Egea, J.F. Choroid plexus glutathione peroxidases are instrumental in protecting the brain fluid environment from hydroperoxides during postnatal development. Am. J. Physiol.-Cell Physiol. 2018, 315, C445–C456. [Google Scholar] [CrossRef] [PubMed]
- Kasarskis, E.J. Zinc metabolism in normal and Zinc-deficient rat brain. Exp. Neurol. 1984, 85, 114–127. [Google Scholar] [CrossRef]
- Nitzan, Y.B.; Sekler, I.; Silverman, W.F. Histochemical and histofluorescence tracing of chelatable Zinc in the developing mouse. J. Histochem. Cytochem. 2004, 52, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Y.; Stoltenberg, M.; Jo, S.M.; Huang, L.; Larsen, A.; Dahlström, A.; Danscher, G. Dynamic Zinc pools in mouse choroid plexus. Neuroreport 2004, 15, 1801–1804. [Google Scholar] [CrossRef]
- Damkier, H.H.; Brown, P.D.; Praetorius, J. Cerebrospinal fluid secretion by the choroid plexus. Physiol. Rev. 2013, 93, 1847–1892. [Google Scholar] [CrossRef] [Green Version]
- Vallee, B.L.; Falchuk, K.H. The biochemical basis of Zinc physiology. Physiol. Rev. 1993, 73, 79–118. [Google Scholar] [CrossRef]
- Oteiza, P.I. Zinc and the modulation of redox homeostasis. Free Radic. Biol. Med. 2012, 53, 1748–1759. [Google Scholar] [CrossRef] [Green Version]
- Nzengue, Y.; Candéias, S.M.; Sauvaigo, S.; Douki, T.; Favier, A.; Rachidi, W.; Guiraud, P. The toxicity redox mechanisms of cadmium alone or together with copper and Zinc homeostasis alteration: Its redox biomarkers. J. Trace Elem. Med. Biol. 2011, 25, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Kojima-Yuasa, A.; Ohkita, T.; Yukami, K.; Ichikawa, H.; Takami, N.; Nakatani, T.; Opare Kennedy, D.; Nishiguchi, S.; Matsui-Yuasa, I. Involvement of intracellular glutathione in Zinc deficiency-induced activation of hepatic stellate cells. Chem. Biol. Interact. 2003, 146, 89–99. [Google Scholar] [CrossRef]
- Omata, Y.; Salvador, G.A.; Supasai, S.; Keenan, A.H.; Oteiza, P.I. Decreased Zinc availability affects glutathione metabolism in neuronal cells and in the developing brain. Toxicol. Sci. 2013, 133, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Bernotiene, R.; Ivanoviene, L.; Sadauskiene, I.; Liekis, A.; Ivanov, L. The effects of cadmium chloride and sodium selenite on protein synthesis in mouse liver. Environ. Toxicol. Pharmacol. 2013, 36, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Redox biochemistry of mammalian metallothioneins. J. Biol. Inorg. Chem. 2011, 16, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Sadovic, S.; Shaikh, Z.A. Nephrotoxicity of cadmium-metallothionein: Protection by Zinc and role of glutathione. Toxicol. Appl. Pharmacol. 1998, 151, 276–282. [Google Scholar] [CrossRef]
- Wessells, K.R.; Brown, K.H. Estimating the global prevalence of Zinc deficiency: Results based on Zinc availability in national food supplies and the prevalence of stunting. PLoS ONE 2012, 7, e50568. [Google Scholar] [CrossRef] [Green Version]
- Wessells, K.R.; Singh, G.M.; Brown, K.H. Estimating the global prevalence of inadequate Zinc intake from national food balance sheets: Effects of methodological assumptions. PLoS ONE 2012, 7, e50565. [Google Scholar] [CrossRef]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.E.; Underwood, M.; Bridges, R.J.; Meister, A.M. Glutathione metabolism at the blood-cerebrospinal fluid barrier. FASEB J. 1989, 3, 2527–2531. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, X.; Wu, W.; Wang, J.; Xie, H.; Wu, Z. Regeneration of glutathione by α-lipoic acid via Nrf2/ARE signaling pathway alleviates cadmium-induced HepG2 cell toxicity. Environ. Toxicol. Pharmacol. 2017, 51, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Chin, T.A.; Templeton, D.M. Protective elevations of glutathione and metallothionein in cadmium-exposed mesangial cells. Toxicology 1993, 77, 145–156. [Google Scholar] [CrossRef]
- Sauvageau, J.A.; Jumarie, C. Different mechanisms for metal-induced adaptation to cadmium in the human lung cell lines A549 and H441. Cell Biol. Toxicol. 2013, 29, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Solis, W.A.; Dalton, T.P.; Dieter, M.Z.; Freshwater, S.; Harrer, J.M.; He, L.; Shertzer, H.G.; Nebert, D.W. Glutamate-cysteine ligase modifier subunit: Mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem. Pharmacol. 2002, 63, 1739–1754. [Google Scholar] [CrossRef]
- Djukić-Cosić, D.; Ninković, M.; Malicević, Z.; Matović, V.; Soldatović, D. Effect of magnesium pretreatment on reduced glutathione levels in tissues of mice exposed to acute and subacute cadmium intoxication: A time course study. Magnes Res. 2007, 20, 177–186. [Google Scholar] [CrossRef]
- Hahn, H.; Huck, C.W.; Rainer, M.; Najam-ul-Haq, M.; Bakry, R.; Abberger, T.; Jennings, P.; Pfaller, W.; Bonn, G.K. Analysis of glutathione in supernatants and lysates of a human proximal tubular cell line from perfusion culture upon intoxication with cadmium chloride by HPLC and LC-ESI-MS. Anal. Bioanal. Chem. 2007, 388, 1763–1769. [Google Scholar] [CrossRef]
- Li, W.; Zhao, Y.; Chou, I.N. Alterations in cytoskeletal protein sulfhydryls and cellular glutathione in cultured cells exposed to cadmium and nickel ions. Toxicology 1993, 77, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Shukla, G.S.; Shukla, A.; Potts, R.J.; Osier, M.; Hart, B.A.; Chiu, J.F. Cadmium-mediated oxidative stress in alveolar epithelial cells induces the expression of gamma-glutamylcysteine synthetase catalytic subunit and glutathione S-transferase alpha and pi isoforms: Potential role of activator protein-1. Cell Biol. Toxicol. 2000, 16, 347–362. [Google Scholar] [CrossRef]
- McConnachie, L.A.; Botta, D.; White, C.C.; Weldy, C.S.; Wilkerson, H.W.; Yu, J.; Dills, R.; Yu, X.; Griffith, W.C.; Faustman, E.M.; et al. The glutathione synthesis gene Gclm modulates amphiphilic polymer-coated CdSe/ZnS quantum dot-induced lung inflammation in mice. PLoS ONE 2013, 8, e64165. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Gong, P.; Suzuki, K.; Murata, M. Cadmium-responsive element of the human heme oxygenase-1 gene mediates heat shock factor 1-dependent transcriptional activation. J. Biol. Chem. 2007, 282, 8715–8723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, H.M.; Cherian, M.G. Protective roles of metallothionein and glutathione in hepatotoxicity of cadmium. Toxicology 1992, 72, 281–290. [Google Scholar] [CrossRef]
- Dahlgaard, J.; Loeschcke, V.; Michalak, P.; Justesen, J. Induced thermotolerance and associated expression of heat-shock protein Hsp70 in adult Drosophila melanogaster. Funct. Ecol. 1998, 12, 786–793. [Google Scholar] [CrossRef]
- DiDomenico, B.J.; Bugaisky, G.E.; Lindquist, S. The heat shock response is self-regulated at both the transcriptional and posttranscriptional levels. Cell 1982, 31, 593–603. [Google Scholar] [CrossRef]
- Waalkes, M.P.; Perantoni, A. In vitro assessment of target cell specificity in cadmium carcinogenesis: Interactions of cadmium and Zinc with isolated interstitial cells of the rat testes. In Vitro Cell. Dev. Biol. 1988, 24, 558–565. [Google Scholar] [CrossRef]
- Napolitano, J.R.; Liu, M.J.; Bao, S.; Crawford, M.; Nana-Sinkam, P.; Cormet-Boyaka, E.; Knoell, D.L. Cadmium-mediated toxicity of lung epithelia is enhanced through NF-κB-mediated transcriptional activation of the human Zinc transporter ZIP8. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L909–L918. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tashiro, S.; Sun, J.; Doi, H.; Satomi, S.; Igarashi, K. Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J. Biol. Chem. 2003, 278, 49246–49253. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.A.; Upender, R.P.; Hightower, L.E.; Renfro, J.L. Thermoprotection of a functional epithelium: Heat stress effects on transepithelial transport by flounder renal tubule in primary monolayer culture. Proc. Natl. Acad. Sci. USA 1992, 89, 3246–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renfro, J.L.; Brown, M.A.; Parker, S.L.; Hightower, L.E. Relationship of thermal and chemical tolerance to transepithelial transport by cultured flounder renal epithelium. J. Pharmacol. Exp. Ther. 1993, 265, 992–1000. [Google Scholar]
- Sussman, C.R.; Renfro, J.L. Heat shock-induced protection and enhancement of Na+-glucose cotransport by LLC-PK1 monolayers. Am. J. Physiol. 1997, 273, F530–F5307. [Google Scholar] [CrossRef]
- Liu, J.; Squibb, K.S.; Akkerman, M.; Nordberg, G.F.; Lipsky, M.; Fowler, B.A. Cytotoxicity, Zinc protection, and stress protein induction in rat proximal tubule cells exposed to cadmium chloride in primary cell culture. Ren. Fail. 1996, 18, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, A.R.; Renfro, J.L. Trimethylamine oxide suppresses stress-induced alteration of organic anion transport in choroid plexus. J. Exp. Biol. 2007, 210, 541–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jihen, E.H.; Sonia, S.; Fatima, H.; Mohamed Tahar, S.; Abdelhamid, K. Interrelationships between cadmium, Zinc and antioxidants in the liver of the rat exposed orally to relatively high doses of cadmium and Zinc. Ecotoxicol. Environ. Saf. 2011, 74, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, K.A.; Chen, Y.; Cai, J.; Sternberg, P. Modulation of Nrf2-dependent antioxidant functions in the RPE by Zip2, a Zinc transporter protein. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1665–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.S.; Tanaka, N.; Horie, T.; Kawano, H.; Tetsuchikawahara, N.; Onosaka, S. Metallothionein-enriched hepatocytes are resistant to ferric nitriloacetate toxicity during conditions of glutathione depletion. Toxicol. Lett. 2005, 158, 108–115. [Google Scholar] [CrossRef]
- Chimienti, F.; Jourdan, E.; Favier, A.; Seve, M. Zinc resistance impairs sensitivity to oxidative stress in HeLa cells: Protection through metallothioneins expression. Free Radic. Biol. Med. 2001, 31, 1179–1190. [Google Scholar] [CrossRef]
- Saito, C.; Yan, H.M.; Artigues, A.; Villar, M.T.; Farhood, A.; Jaeschke, H. Mechanism of protection by metallothionein against acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2010, 242, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Lichten, L.A.; Cousins, R.J. Mammalian Zinc transporters: Nutritional and physiologic regulation. Annu. Rev. Nutr. 2009, 29, 153–176. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Vasák, M. Possible role for metallothionein in protection against radiation-induced oxidative stress. Kinetics and mechanism of its reaction with superoxide and hydroxyl radicals. Biochim. Biophys. Acta 1985, 827, 36–44. [Google Scholar] [CrossRef]
- Villalobos, A.R.; Parmelee, J.T.; Renfro, J.L. Choline uptake across the ventricular membrane of neonate rat choroid plexus. Am. J. Physiol. 1999, 276, C1288–C1296. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, A.R.; Parmelee, J.T.; Pritchard, J.B. Functional characterization of choroid plexus epithelial cells in primary culture. J. Pharmacol. Exp. Ther. 1997, 282, 1109–1116. [Google Scholar] [PubMed]
- Lazzaro, V.A.; Walker, R.J.; Duggin, G.G.; Phippard, A.; Horvath, J.S.; Tiller, D.J. Inhibition of fibroblast proliferation in L-valine reduced selective media. Res. Commun. Chem. Pathol. Pharmacol. 1992, 7, 39–48. [Google Scholar]
- Suthanthiran, M.; Anderson, M.E.; Sharma, V.K.; Meister, A. Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proc. Natl. Acad. Sci. USA 1990, 87, 3343–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Zinc, ng/mg Protein | Cadmium, ng/mg Protein |
|---|---|---|
| Control | 333.82 ± 34.04 | 1.02 ± 0.47 |
| Cd | 348.99 ± 42.94 | 265.79 ± 11.79 * |
| Zn | 556.61 ± 53.44 * | 1.14 ± 0.05 |
| Zn + Cd | 463.26 ± 15.82 * | 243.72 ± 8.62 * |
| Condition | GSH, µM | GSSG, µM | GSH:GSSG Ratio |
|---|---|---|---|
| Control | 5.20 ± 0.50 | 0.029 ± 0.010 | 318.4 ± 102.99 |
| Cd | 9.18 ± 0.27 * | 0.207 ± 0.023 * | 43.28 ± 5.89 * |
| Zn | 5.25 ± 0.46 | 0.035 ± 0.010 | 273.73 ± 99.76 |
| Zn + Cd | 7.84 ± 0.87 * | 0.121 ± 0.022 * | 71.94 ± 14.97 * |
| Zinc Concentration, µM | [3H]Choline Uptake, pmol/mg Protein |
|---|---|
| 0 (Control) | 3274.27 ± 287.13 |
| 5 | 3373.58 ± 227.32 |
| 10 | 3288.23 ± 201.05 |
| 25 | 3343.04 ± 269.76 |
| 50 | 3339.53 ± 175.93 |
| 100 | 304.07 ± 96.00 * |
| Condition | GSH, µM | GSSG, µM | GSH:GSSG Ratio |
|---|---|---|---|
| Control | 4.39 ± 0.727 | 0.050 ± 0.017 | 231.76 ± 121.66 |
| Cd | 9.35 ± 1.38 * | 0.167 ± 0.012 * | 46.90 ± 5.68 * |
| Zn | 4.57 ± 0.70 | 0.045 ± 0.025 | 241.33 ± 130.55 |
| Zn + Cd | 8.11 ± 1.12 * | 0.135 ± 0.030 * | 54.09 ± 8.14 * |
| BSO | 0.45 ± 0.50 * | 0.073 ± 0.031 | 8.75 ± 3.25 * |
| BSO + Cd | 0.23 ± 0.02 *,§ | 0.077 ± 0.023 | 9.37 ± 3.29 * |
| BSO + Zn | 0.51 ± 0.07 * | 0.074 ± 0.021 | 6.86 ± 2.26 * |
| BSO + Zn + Cd | 0.33 ± 0.02 *,§ | 0.078 ± 0.025 | 8.09 ± 3.09 * |
| Gene | GeneBank ID | Forward Primer (5–3′) | Reverse Primer (3′–5′) |
|---|---|---|---|
| Actb | NM_031144 | ATGGTGGGTATGGGTCAG | TACTTCAGGGTCAGGATGC |
| Gapdh | NM_017008 | ATGACTCTACCCACGGC | ACTCAGCACCAGCATCA |
| Gclc | NM_012815 | GCTTTCTCCTACCTGTTTCTTG | TGGCAGAGTTCAGTTCCG |
| Gclm | NM_017305 | TGTGATGCCACCAGATTTGA | TGGAAACTTGCCTCAGAGAG |
| Hmox1 | NM_012580 | ACCCCACAAGTTCAAACAG | CCTCTGGCGAAGAACTCTG |
| Hspa4 | NM_153629 | ATGGGGGACAAGTCGGA | GTGGGGATGGTGGAGTT |
| Mt1 | NM_138826 | CACCGTTGCTCCAGATTCA | CAGCAGCACTGTTCGTCA |
| Condition | LDH Release (% Maximal Release) |
|---|---|
| Control | 11.39 ± 0.76 |
| Cd | 14.09 ± 1.54 |
| Zn | 10.34 ± 0.20 |
| Zn + Cd | 10.49 ± 0.22 |
| BSO | 10.89 ± 0.34 |
| BSO + Cd | 12.85 ± 1.01 |
| BSO + Zn | 10.55 ± 0.85 |
| BSO + Zn + Cd | 10.61 ± 0.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francis Stuart, S.D.; Villalobos, A.R. GSH and Zinc Supplementation Attenuate Cadmium-Induced Cellular Stress and Stimulation of Choline Uptake in Cultured Neonatal Rat Choroid Plexus Epithelia. Int. J. Mol. Sci. 2021, 22, 8857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168857
Francis Stuart SD, Villalobos AR. GSH and Zinc Supplementation Attenuate Cadmium-Induced Cellular Stress and Stimulation of Choline Uptake in Cultured Neonatal Rat Choroid Plexus Epithelia. International Journal of Molecular Sciences. 2021; 22(16):8857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168857
Chicago/Turabian StyleFrancis Stuart, Samantha D., and Alice R. Villalobos. 2021. "GSH and Zinc Supplementation Attenuate Cadmium-Induced Cellular Stress and Stimulation of Choline Uptake in Cultured Neonatal Rat Choroid Plexus Epithelia" International Journal of Molecular Sciences 22, no. 16: 8857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168857