
Vitamin D Reverses Disruption of Gut Epithelial Barrier Function Caused by Campylobacter jejuni

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
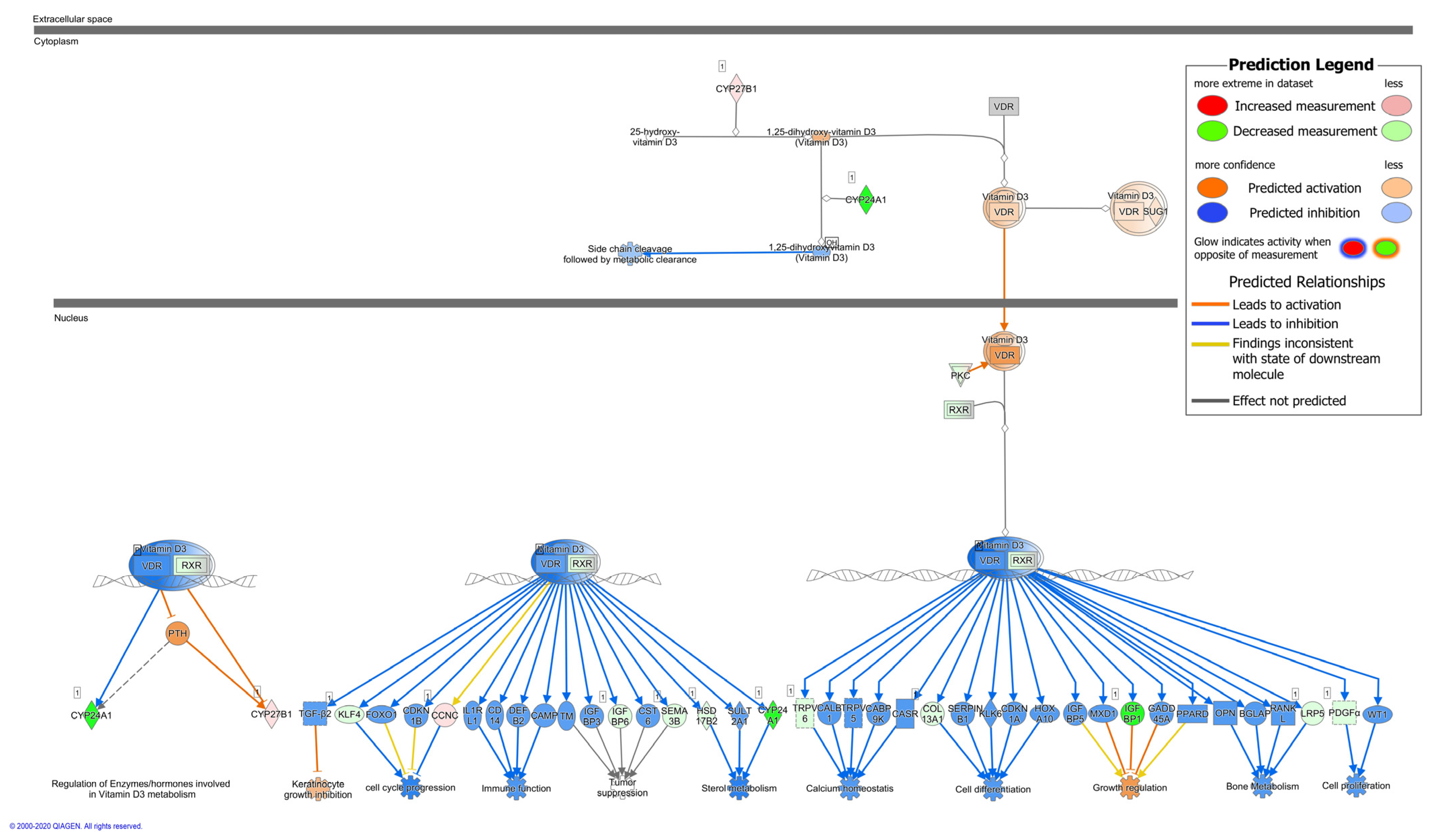
2.1. Upstream Regulator Analysis of C. jejuni—Infected Human Colonic Mucosa and VDR Downstream Targets
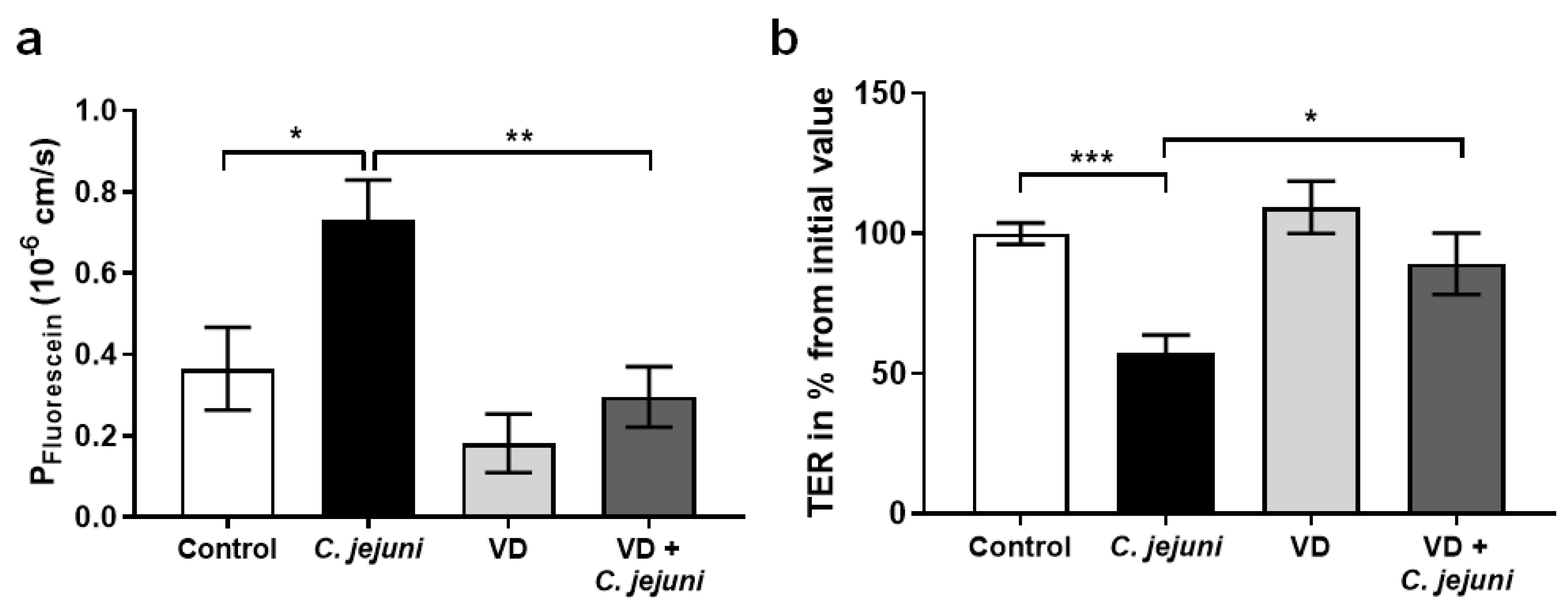
2.2. Epithelial Barrier Dysfunction in C. jejuni Infection Is Reversed by Vitamin D
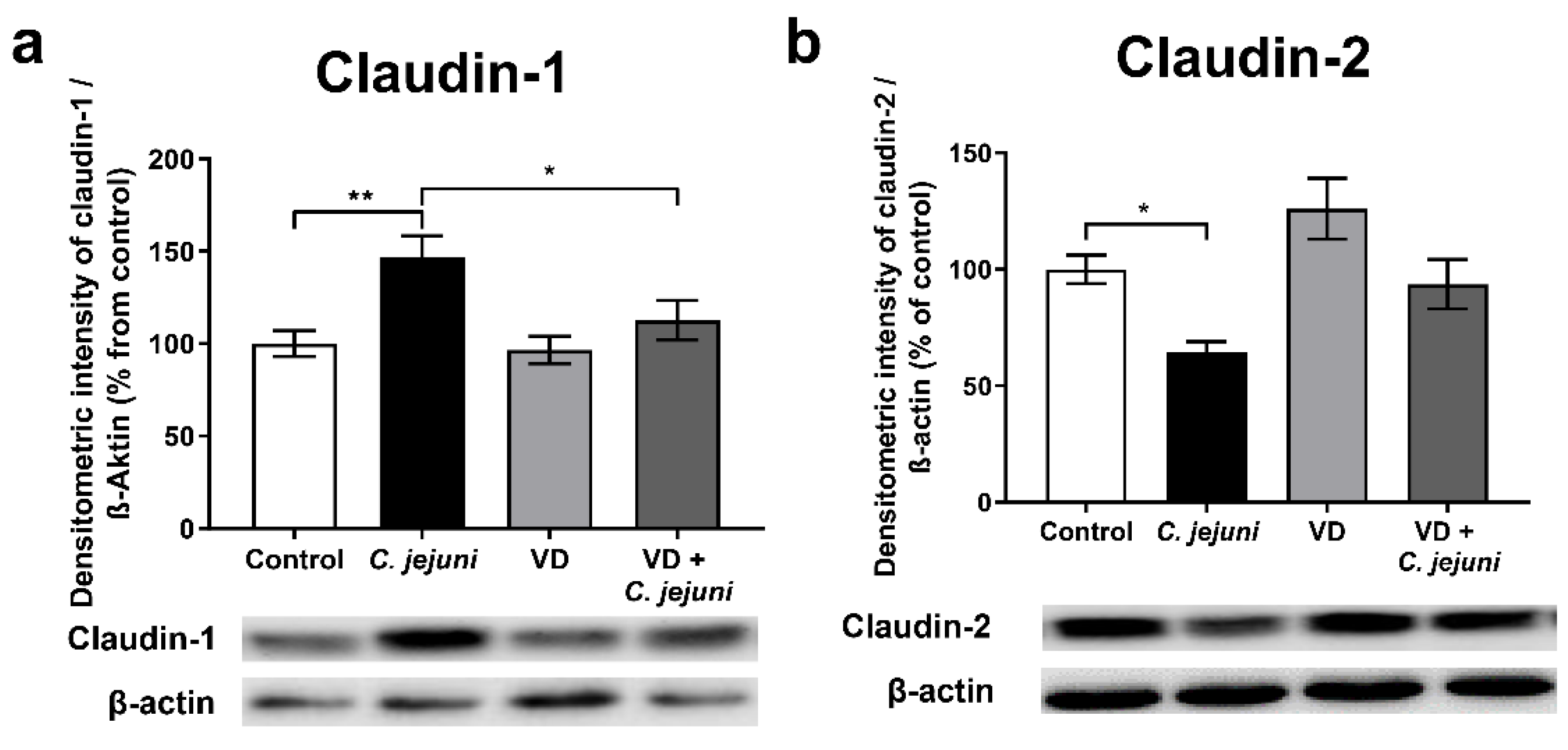
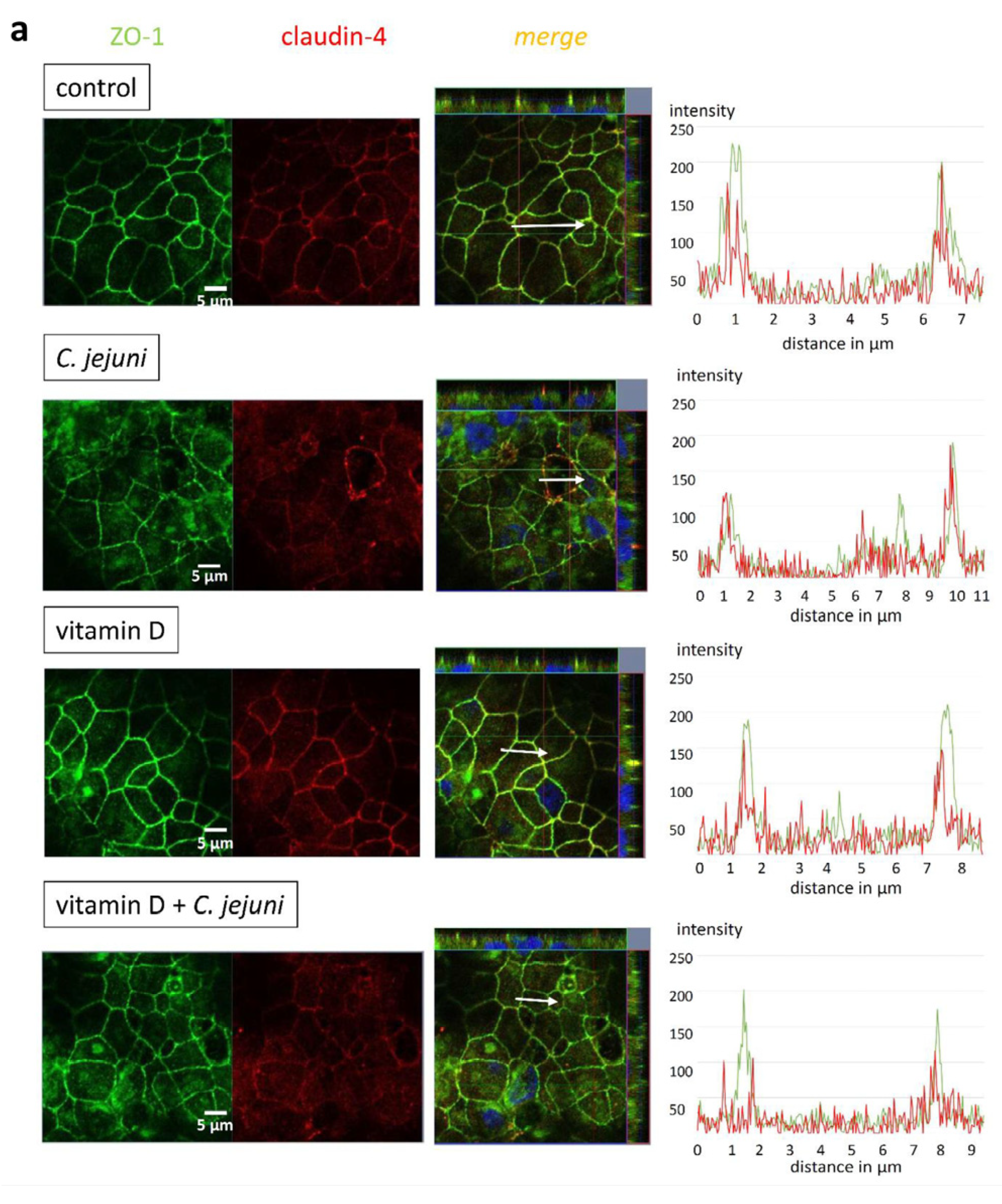
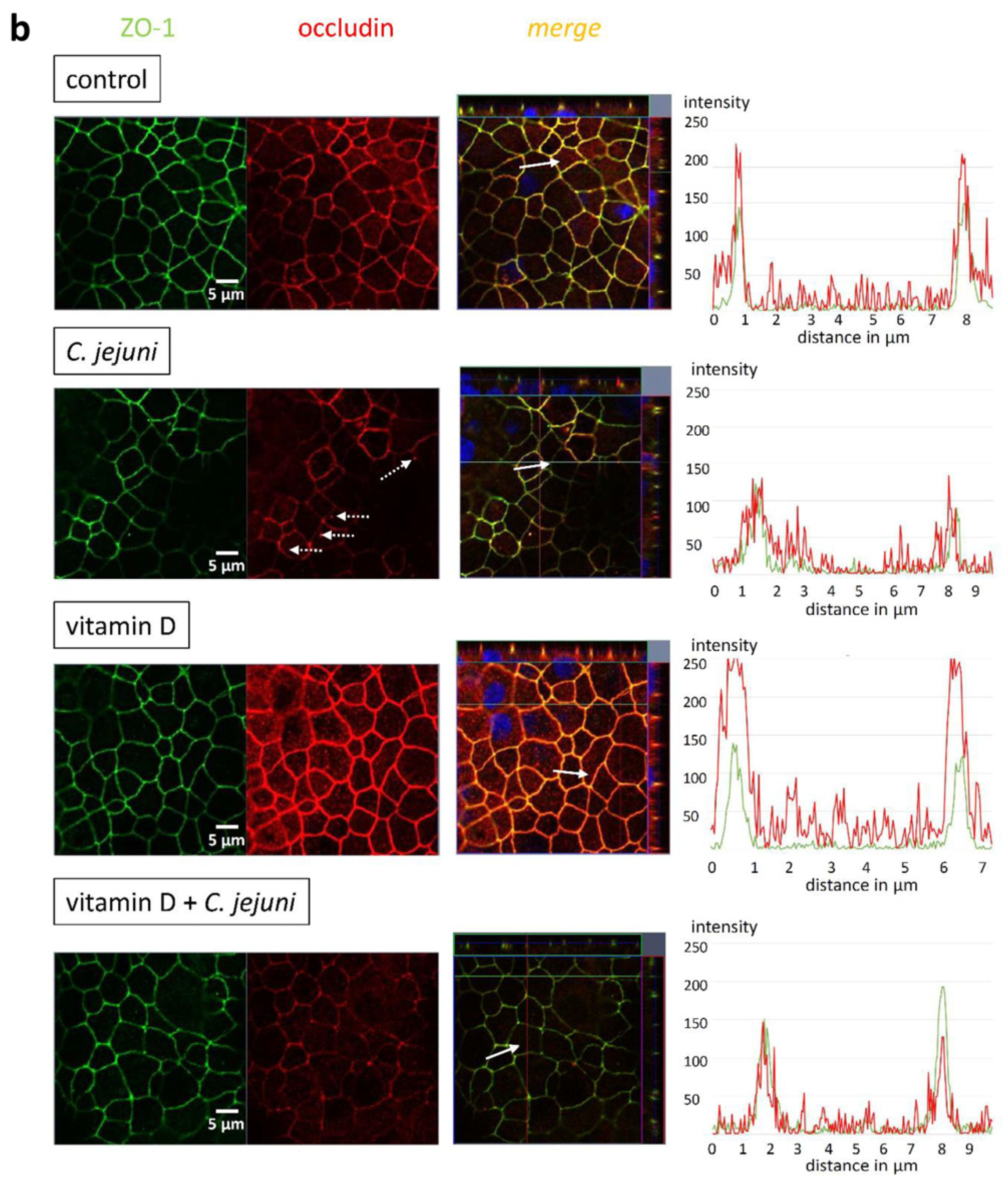
2.3. Influence of C. jejuni and Vitamin D on the Protein Expression and Distribution of Tight Junction Proteins
2.4. Vitamin D Prevents C. jejuni—Induced Epithelial Apoptosis
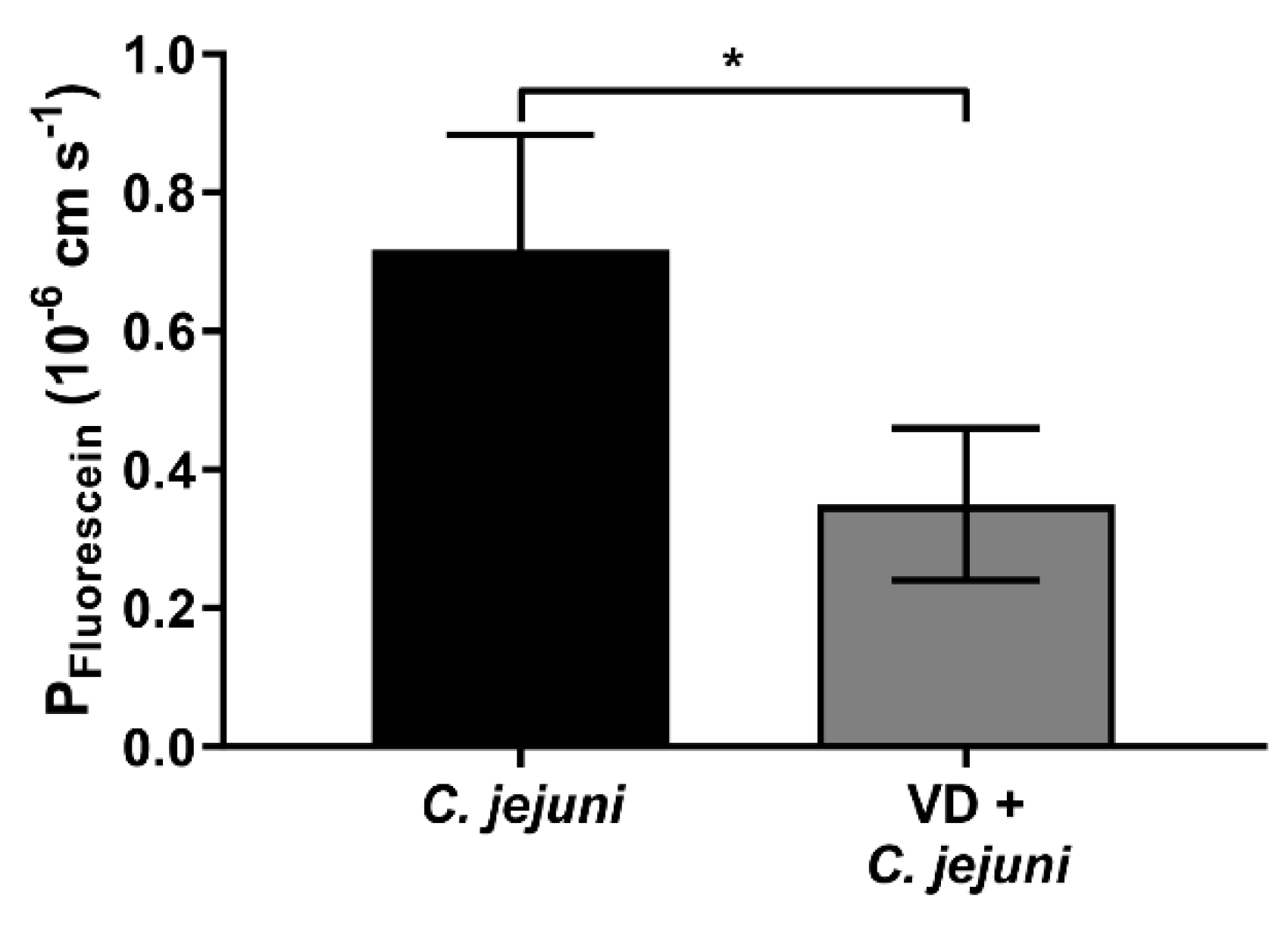
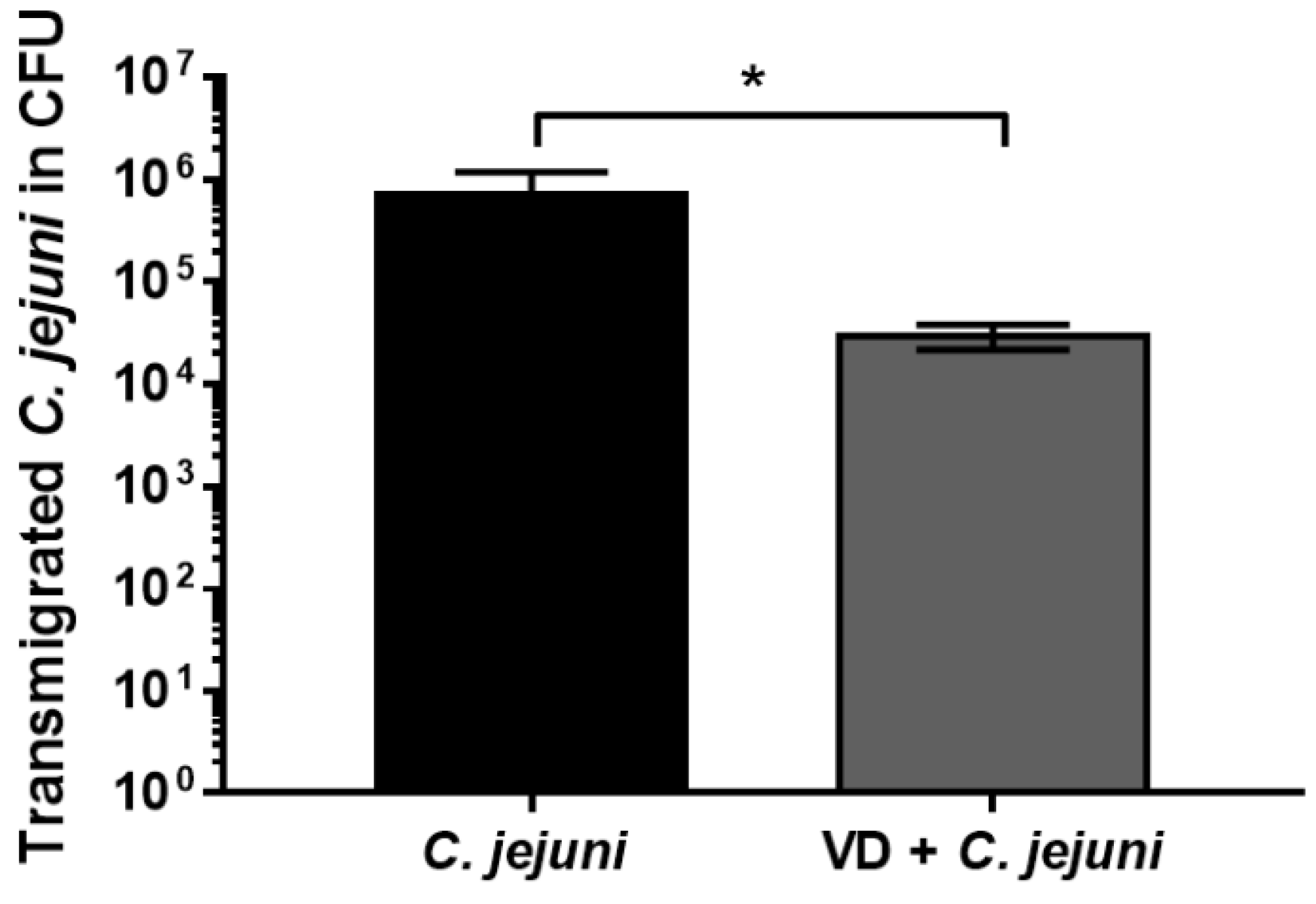
2.5. Vitamin D Supplementation Reduces the Number of Transmigrated C. jejuni
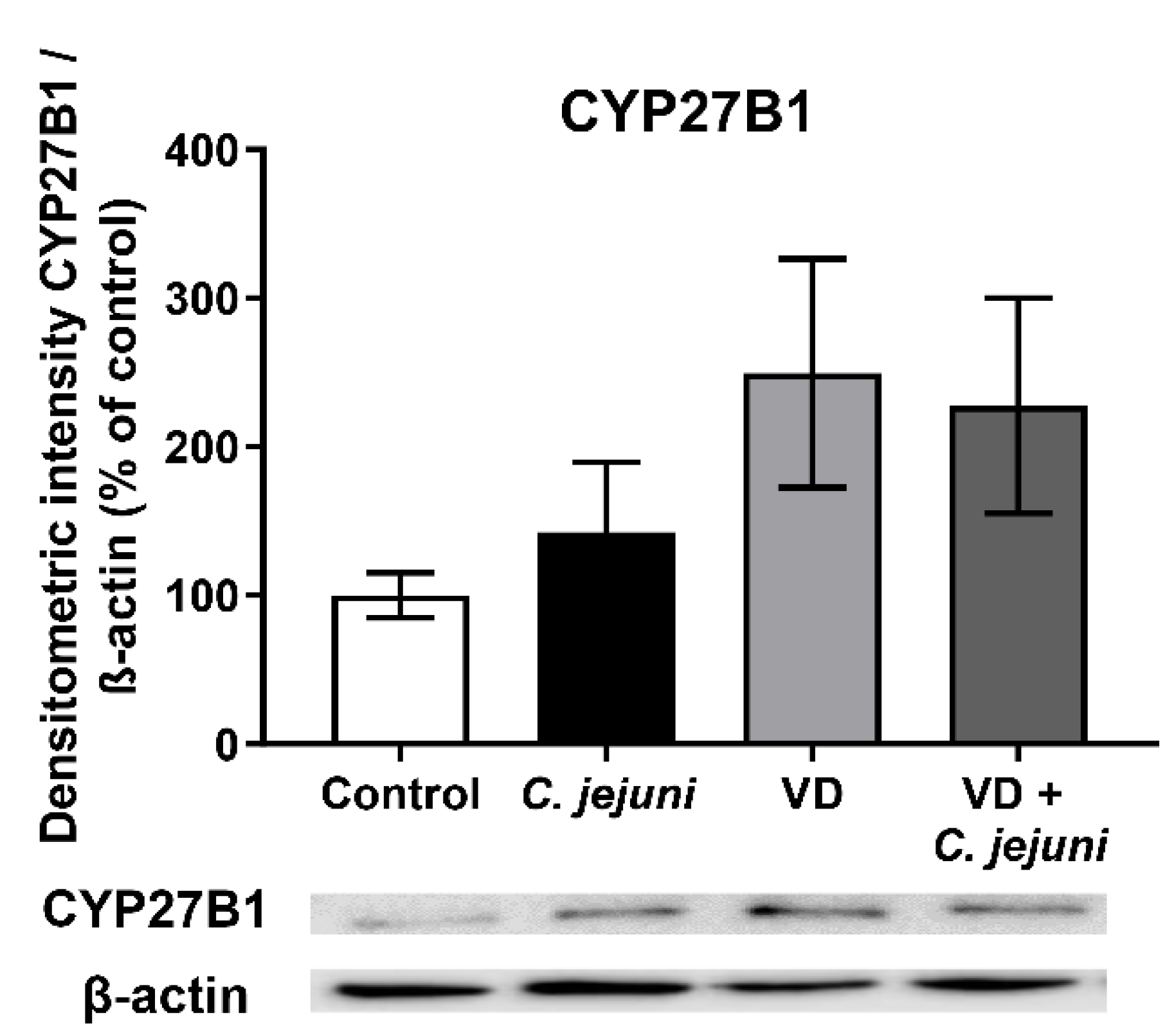
2.6. Influence of C. jejuni on the Vitamin D Pathway and Its Functional Importance
3. Discussion
3.1. Effect of C. jejuni and Vitamin D on Tight Junction Protein Expression, Localization, and Apoptosis Induction
3.2. C. jejuni Interference with the Vitamin D Pathway
4. Materials and Methods
4.1. Cell Culture and Treatment of the Cells
4.2. C. jejuni Cultivation and Experimental Infection In Vitro
4.3. Generation of Secondary Abiotic IL-10−/− Mice, Treatment and Experimental Infection In Vivo
4.4. Electrophysiological Methods
4.5. Epithelial Permeability Measurements
4.6. Transmigration Assay
4.7. Western Blot
4.8. Immunostaining, Apoptosis Assay, and Confocal Microscopy
4.9. RNA Expression Analysis
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CFU | colony forming unit |
| CLSM | confocal laser-scanning microscopy |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DSS | dextran sulfate sodium |
| FCS | fetal calf serum |
| HtrA | high-temperature requirement A |
| IBD | inflammatory bowel disease |
| IBS | irritable bowel syndrome |
| IFN | interferon |
| IPA | Ingenuity Pathway Analysis |
| LPS | lipopolysaccharide |
| MOI | multiplicity of infection |
| OD | optical density |
| RXR | retinoid X receptor |
| TER | transepithelial electrical resistance |
| TJ | tight junction |
| TNBS | 2,4,6-trinitrobenzenesulfonic acid |
| TNF | tumor necrosis factor |
| TUNEL | TdT-mediated dUTP-biotin nick end labeling |
| VD | vitamin D |
| VDR | vitamin D receptor |
| ZO-1 | zonula occludens-1 |
References
- Black, R.E.; Levine, M.M.; Clements, M.L.; Hughes, T.P.; Blaser, M.J. Experimental Campylobacter jejuni Infection in Humans. J. Infect. Dis. 1988, 157, 472–479. [Google Scholar] [CrossRef]
- Bücker, R.; Krug, S.M.; Moos, V.; Bojarski, C.; Schweiger, M.R.; Kerick, M.; Fromm, A.; Janßen, S.; Fromm, M.; Hering, N.A.; et al. Erratum: Campylobacter jejuni impairs sodium transport and epithelial barrier function via cytokine release in human colon. Mucosal Immunol. 2017, 11, 575–577. [Google Scholar] [CrossRef] [Green Version]
- Lobo de Sá, F.D.; Butkevych, E.; Nattramilarasu, P.K.; Fromm, A.; Mousavi, S.; Moos, V.; Golz, J.C.; Stingl, K.; Kittler, S.; Seinige, D.; et al. Curcumin Mitigates Immune-Induced Epithelial Barrier Dysfunction by Campylobacter jejuni. Int. J. Mol. Sci. 2019, 20, 4830. [Google Scholar] [CrossRef] [Green Version]
- Butkevych, E.; Lobo de Sá, F.D.; Nattramilarasu, P.K.; Bücker, R. Contribution of Epithelial Apoptosis and Subepithelial Immune Responses in Campylobacter jejuni-Induced Barrier Disruption. Front. Microbiol. 2020, 11, 344. [Google Scholar] [CrossRef]
- Meeker, S.; Seamons, A.; Maggio-Price, L.; Paik, J. Protective links between vitamin D, inflammatory bowel disease and colon cancer. World J. Gastroenterol. 2016, 22, 933–948. [Google Scholar] [CrossRef] [PubMed]
- Assa, A.; Vong, L.; Pinnell, L.J.; Avitzur, N.; Johnson-Henry, K.C.; Sherman, P.M. Vitamin D Deficiency Promotes Epithelial Barrier Dysfunction and Intestinal Inflammation. J. Infect. Dis. 2014, 210, 1296–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasnezhad, A.; Amani, R.; Hasanvand, A.; Rad, E.Y.; Alipour, M.; Saboori, S.; Choghakhori, R. Association of Serum Vitamin D Concentration With Clinical Symptoms and Quality of Life in Patients With Irritable Bowel Syndrome. J. Am. Coll. Nutr. 2018, 38, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Mahyar, A.; Ayazi, P.; Saffari Rad, M.; Dalirani, R.; Javadi, A.; Esmaeily, S. The Correlation Between Vitamin D and Bacterial Diarrhea in Children. Arch. Pediatr. Infect. Dis. 2019, 7, e84382. [Google Scholar] [CrossRef] [Green Version]
- Ooi, J.H.; Li, Y.; Rogers, C.J.; Cantorna, M.T. Vitamin D Regulates the Gut Microbiome and Protects Mice from Dextran Sodium Sulfate–Induced Colitis. J. Nutr. 2013, 143, 1679–1686. [Google Scholar] [CrossRef]
- Barbáchano, A.; Fernández-Barral, A.; Ferrer-Mayorga, G.; Costales, A.; Larriba, M.J.; Muñoz, A. The endocrine vitamin D system in the gut. Mol. Cell. Endocrinol. 2017, 453, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Stio, M.; Retico, L.; Annese, V.; Bonanomi, A.G. Vitamin D regulates the tight-junction protein expression in active ulcerative colitis. Scand. J. Gastroenterol. 2016, 51, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-W.; Ma, Y.-Y.; Zhu, J.; Zuo, S.; Zhang, J.-L.; Chen, Z.-Y.; Chen, G.-W.; Wang, X.; Pan, Y.-S.; Liu, Y.-C.; et al. Protective effect of 1,25-dihydroxyvitamin D3 on ethanol-induced intestinal barrier injury both in vitro and in vivo. Toxicol. Lett. 2015, 237, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, J.; Chen, G.; Zuo, S.; Zhang, J.; Chen, Z.; Wang, X.; Li, J.; Liu, Y.; Wang, P. 1,25-Dihydroxyvitamin D3 preserves intestinal epithelial barrier function from TNF-α induced injury via suppression of NF-kB p65 mediated MLCK-P-MLC signaling pathway. Biochem. Biophys. Res. Commun. 2015, 460, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.; Lobo de Sá, F.D.; Schulzke, J.-D.; Bücker, R.; Bereswill, S.; Heimesaat, M.M. Vitamin D in Acute Campylobacteriosis–Results From an Intervention Study Applying a Clinical Campylobacter jejuni Induced Enterocolitis Model. Front. Immunol. 2019, 10, 2094. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-G.; Wu, S.; Lu, R.; Zhou, D.; Zhou, J.; Carmeliet, G.; Petrof, E.; Claud, E.C.; Sun, J. Tight junction CLDN2 gene is a direct target of the vitamin D receptor. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-G.; Lu, R.; Xia, Y.; Zhou, D.; Petrof, E.; Claud, E.C.; Sun, J. Lack of Vitamin D Receptor Leads to Hyperfunction of Claudin-2 in Intestinal Inflammatory Responses. Inflamm. Bowel Dis. 2018, 25, 97–110. [Google Scholar] [CrossRef]
- Meckel, K.; Li, Y.C.; Lim, J.; Kocherginsky, M.; Weber, C.; Almoghrabi, A.; Chen, X.; Kaboff, A.; Sadiq, F.; Hanauer, S.B.; et al. Serum 25-hydroxyvitamin D concentration is inversely associated with mucosal inflammation in patients with ulcerative colitis. Am. J. Clin. Nutr. 2016, 104, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Sikaroudi, M.K.; Mokhtare, M.; Shidfar, F.; Janani, L.; Kashani, A.F.; Masoodi, M.; Agah, S.; Dehnad, A.; Shidfar, S. Effects of vitamin D3 supplementation on clinical symptoms, quality of life, serum serotonin (5-hydroxytryptamine), 5-hydroxy-indole acetic acid, and ratio of 5-HIAA/5-HT in patients with diarrhea-predominant irritable bowel syndrome: A randomized clinical trial. EXCLI J. 2020, 19, 652–667. [Google Scholar]
- Wu, S.; Liao, A.P.; Xia, Y.; Li, Y.C.; Li, J.-D.; Sartor, R.B.; Sun, J. Vitamin D Receptor Negatively Regulates Bacterial-Stimulated NF-κB Activity in Intestine. Am. J. Pathol. 2010, 177, 686–697. [Google Scholar] [CrossRef]
- Prüfer, K.; Racz, A.; Lin, G.C.; Barsony, J. Dimerization with Retinoid X Receptors Promotes Nuclear Localization and Subnuclear Targeting of Vitamin D Receptors. J. Biol. Chem. 2000, 275, 41114–41123. [Google Scholar] [CrossRef] [Green Version]
- Assa, A.; Vong, L.; Pinnell, L.J.; Rautava, J.; Avitzur, N.; Johnson-Henry, K.; Sherman, P. Vitamin D Deficiency Predisposes to Adherent-invasive Escherichia coli-induced Barrier Dysfunction and Experimental Colonic Injury. Inflamm. Bowel Dis. 2015, 21, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.M.S.; Magalhaes, R.S.; Long, K.Z.; Ahmed, T.; Alam, A.; Hossain, I.; Islam, M.; Mahfuz, M.; Mondal, D.; Haque, R.; et al. Association of vitamin D status with incidence of enterotoxigenic, enteropathogenic and enteroaggregative Escherichia coli diarrhoea in children of urban Bangladesh. Trop. Med. Int. Health 2016, 21, 973–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlavaty, T.; Krajcovicova, A.; Payer, J. Vitamin D Therapy in Inflammatory Bowel Diseases: Who, in What Form, and How Much? J. Crohns Colitis 2014, 9, 198–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.; Tinkov, A.; Strand, T.A.; Alehagen, U.; Skalny, A.; Aaseth, J. Early Nutritional Interventions with Zinc, Selenium and Vitamin D for Raising Anti-Viral Resistance Against Progressive COVID-19. Nutrients 2020, 12, 2358. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Chen, Y.; Shi, Y.; Liu, T.; Cao, Y.; Tang, Y.; Ge, X.; Nie, H.; Zheng, C.; Li, Y.C. 1,25-Dihydroxyvitamin D Protects Intestinal Epithelial Barrier by Regulating the Myosin Light Chain Kinase Signaling Pathway. Inflamm. Bowel Dis. 2015, 21, 2495–2506. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Zhang, Z.; Musch, M.W.; Ning, G.; Sun, J.; Hart, J.; Bissonnette, M.; Li, Y.C. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G208–G216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Zhang, H.; Wu, H.; Li, H.; Liu, L.; Guo, J.; Li, C.; Shih, D.Q.; Zhang, X. Protective role of 1,25(OH)2vitamin D3 in the mucosal injury and epithelial barrier disruption in DSS-induced acute colitis in mice. BMC Gastroenterol. 2012, 12, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.I.; McCall, I.C.; Parkos, C.A.; Nusrat, A. Role for Actin Filament Turnover and a Myosin II Motor in Cytoskeleton-driven Disassembly of the Epithelial Apical Junctional Complex. Mol. Biol. Cell 2004, 15, 2639–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bücker, R.; Krug, S.; Rosenthal, R.; Günzel, D.; Fromm, A.; Zeitz, M.; Chakraborty, T.; Fromm, M.; Epple, H.-J.; Schulzke, J.-D. Aerolysin From Aeromonas hydrophila Perturbs Tight Junction Integrity and Cell Lesion Repair in Intestinal Epithelial HT-29/B6 Cells. J. Infect. Dis. 2011, 204, 1283–1292. [Google Scholar] [CrossRef] [Green Version]
- Marchiando, A.M.; Shen, L.; Graham, W.; Weber, C.; Schwarz, B.T.; Austin, J.R.; Raleigh, D.R.; Guan, Y.; Watson, A.J.; Montrose, M.H.; et al. Caveolin-1–dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 2010, 189, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-G.; Wu, S.; Xia, Y.; Sun, J. Salmonella Infection Upregulates the Leaky Protein Claudin-2 in Intestinal Epithelial Cells. PLoS ONE 2013, 8, e58606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domazetovic, V.; Iantomasi, T.; Bonanomi, A.G.; Stio, M. Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling. Int. J. Colorectal Dis. 2020, 35, 1231–1242. [Google Scholar] [CrossRef]
- Johnson, C.S.; Muindi, J.R.; Hershberger, P.A.; Trump, D.L. The antitumor efficacy of calcitriol: Preclinical studies. Anticancer Res. 2006, 26, 2543–2549. [Google Scholar] [PubMed]
- Backert, S.; Boehm, M.; Wessler, S.; Tegtmeyer, N. Transmigration route of Campylobacter jejuni across polarized intestinal epithelial cells: Paracellular, transcellular or both? Cell Commun. Signal. 2013, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- Boehm, M.; Lind, J.; Backert, S.; Tegtmeyer, N. Campylobacter jejuni serine protease HtrA plays an important role in heat tolerance, oxygen resistance, host cell adhesion, invasion, and transmigration. Eur. J. Microbiol. Immunol. 2015, 5, 68–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, M.; Hoy, B.; Rohde, M.; Tegtmeyer, N.; Bæk, K.T.; Oyarzabal, O.A.; Brøndsted, L.; Wessler, S.; Backert, S. Rapid paracellular transmigration of Campylobacter jejuni across polarized epithelial cells without affecting TER: Role of proteolytic-active HtrA cleaving E-cadherin but not fibronectin. Gut Pathog. 2012, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Harrer, A.; Bücker, R.; Boehm, M.; Zarzecka, U.; Tegtmeyer, N.; Sticht, H.; Schulzke, J.-D.; Backert, S. Campylobacter jejuni enters gut epithelial cells and impairs intestinal barrier function through cleavage of occludin by serine protease HtrA. Gut Pathog. 2019, 11, 1–16. [Google Scholar] [CrossRef]
- Zilbauer, M.; Dorrell, N.; Boughan, P.K.; Harris, A.; Wren, B.W.; Klein, N.J.; Bajaj-Elliott, M. Intestinal Innate Immunity to Campylobacter jejuni Results in Induction of Bactericidal Human Beta-Defensins 2 and 3. Infect. Immun. 2005, 73, 7281–7289. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-Like Receptor Triggering of a Vitamin D-Mediated Human Antimicrobial Response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Guo, C.; Gombart, A.F. The antibiotic effects of vitamin D. Endocr. Metab. Immune Disord. Drug Targets 2014, 14, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lagishetty, V.; Chun, R.; Liu, N.Q.; Lisse, T.S.; Adams, J.; Hewison, M. 1α-Hydroxylase and innate immune responses to 25-hydroxyvitamin D in colonic cell lines. J. Steroid Biochem. Mol. Biol. 2010, 121, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.; Hewison, M. Extrarenal expression of the 25-hydroxyvitamin D-1-hydroxylase. Arch. Biochem. Biophys. 2012, 523, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Stoffels, K.; Overbergh, L.; Giulietti, A.; Verlinden, L.; Bouillon, R.; Mathieu, C. Immune Regulation of 25-Hydroxyvitamin-D3-1α-Hydroxylase in Human Monocytes. J. Bone Miner. Res. 2005, 21, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Krutzik, S.R.; Hewison, M.; Liu, P.T.; Robles, J.A.; Stenger, S.; Adams, J.S.; Modlin, R.L. IL-15 Links TLR2/1-Induced Macrophage Differentiation to the Vitamin D-Dependent Antimicrobial Pathway. J. Immunol. 2008, 181, 7115–7120. [Google Scholar] [CrossRef]
- Lobo de Sá, F.D.; Heimesaat, M.M.; Bereswill, S.; Nattramilarasu, P.K.; Schulzke, J.-D.; Bücker, R. Resveratrol Prevents Campylobacter jejuni-Induced Leaky gut by Restoring Occludin and Claudin-5 in the Paracellular Leak Pathway. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Lobo de Sá, F.D.; Schulzke, J.-D.; Bücker, R. Diarrheal Mechanisms and the Role of Intestinal Barrier Dysfunction in Campylobacter Infections. Curr. Top. Microbiol. Immunol. 2021, 431, 203–231. [Google Scholar] [CrossRef]
- Bergann, T.; Plöger, S.; Fromm, A.; Zeissig, S.; Borden, S.A.; Fromm, M.; Schulzke, J.-D. A colonic mineralocorticoid receptor cell model expressing epithelial Na+ channels. Biochem. Biophys. Res. Commun. 2009, 382, 280–285. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
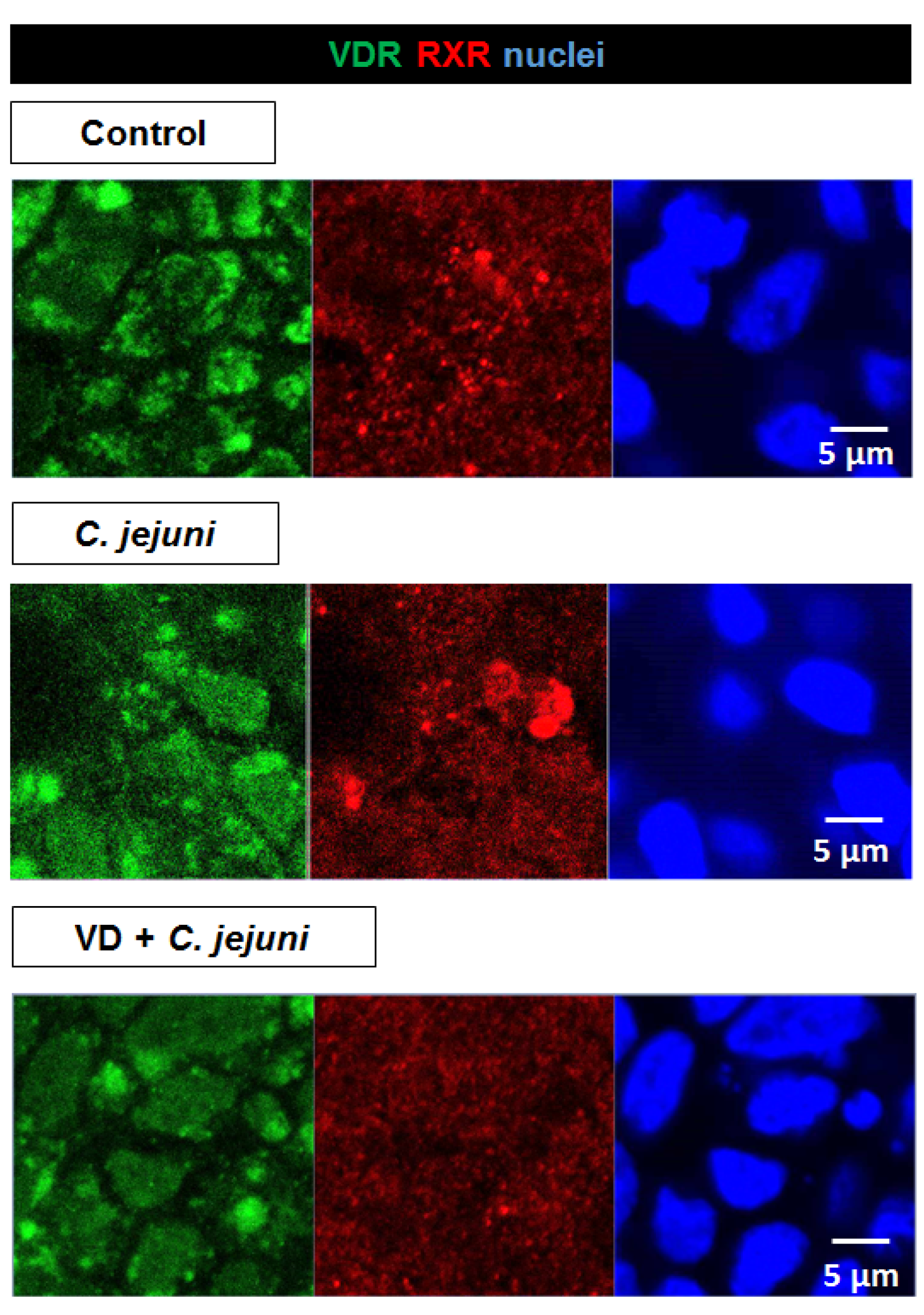{kind=link}
| TJ Protein | C. jejuni | Vitamin D | Vitamin D + C. jejuni |
|---|---|---|---|
| claudin-4 | 98 ± 9% | 95 ± 12% | 90 ± 10% |
| claudin-5 | 85 ± 13% | 93 ± 27% | 86 ± 12% |
| claudin-7 | 89 ± 15% | 101 ± 11% | 100 ± 14% |
| claudin-8 | 92 ± 15% | 93 ± 17% | 110 ± 17% |
| occludin | 103 ± 18% | 109 ± 16% | 99 ± 16% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobo de Sá, F.D.; Backert, S.; Nattramilarasu, P.K.; Mousavi, S.; Sandle, G.I.; Bereswill, S.; Heimesaat, M.M.; Schulzke, J.-D.; Bücker, R. Vitamin D Reverses Disruption of Gut Epithelial Barrier Function Caused by Campylobacter jejuni. Int. J. Mol. Sci. 2021, 22, 8872. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168872
Lobo de Sá FD, Backert S, Nattramilarasu PK, Mousavi S, Sandle GI, Bereswill S, Heimesaat MM, Schulzke J-D, Bücker R. Vitamin D Reverses Disruption of Gut Epithelial Barrier Function Caused by Campylobacter jejuni. International Journal of Molecular Sciences. 2021; 22(16):8872. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168872
Chicago/Turabian StyleLobo de Sá, Fábia D., Steffen Backert, Praveen K. Nattramilarasu, Soraya Mousavi, Geoffrey I. Sandle, Stefan Bereswill, Markus M. Heimesaat, Jörg-Dieter Schulzke, and Roland Bücker. 2021. "Vitamin D Reverses Disruption of Gut Epithelial Barrier Function Caused by Campylobacter jejuni" International Journal of Molecular Sciences 22, no. 16: 8872. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168872