
Role of Neuroinflammation and Blood-Brain Barrier Permutability on Migraine

 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Summary of Search
3. Clinical Evidence of Neuroinflammation
3.1. Serial Analysis of Cytokines
3.2. Cerebral Spinal Fluid (CSF)
3.3. Genetic Analysis
4. Experimental Evidence of Neuroinflammation
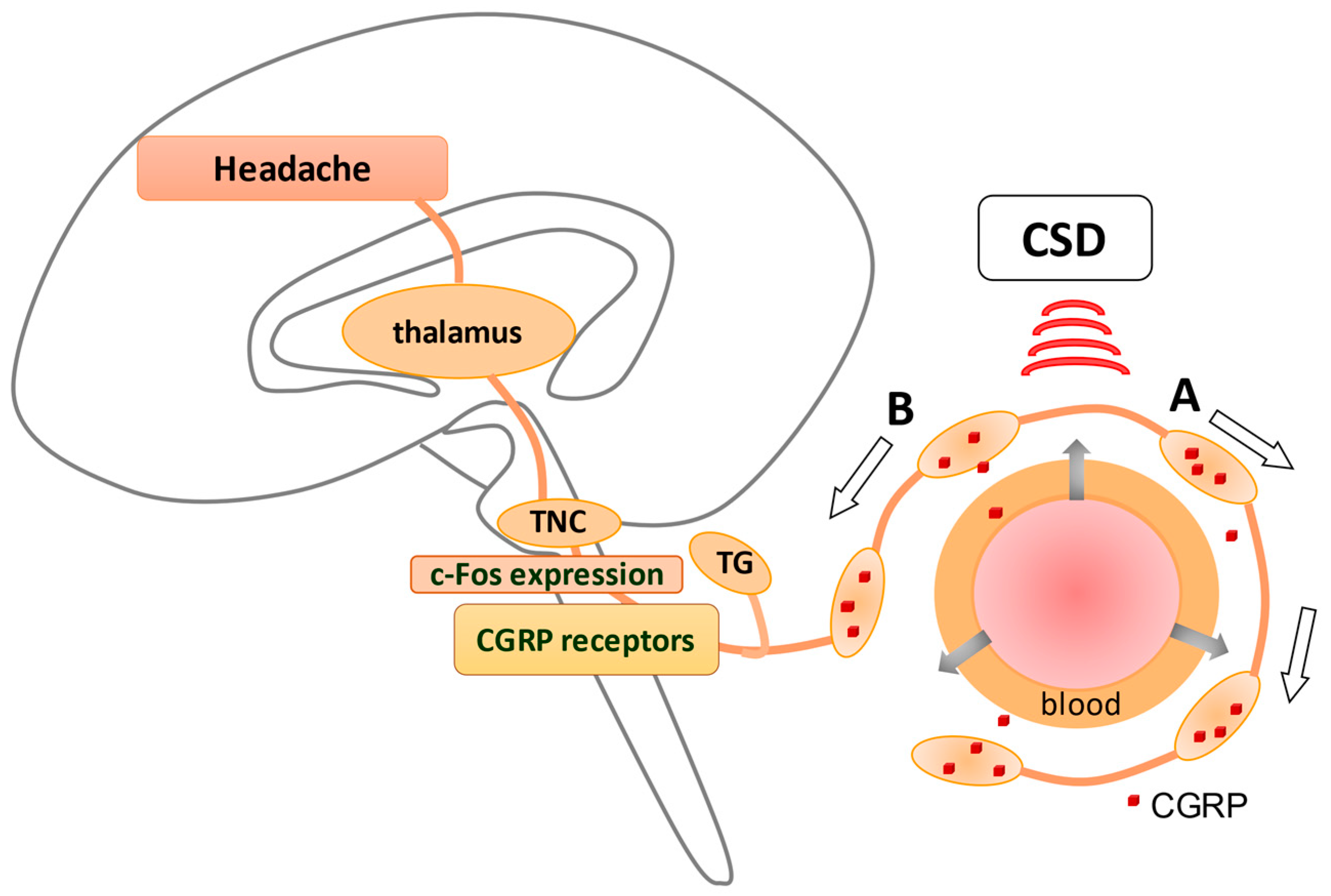
4.1. Inflammation Response Provoked by CSD
4.2. Spontaneous Migraine-like Mouse Model Using NTG
5. Presence of Blood-Brain Barrier (BBB) Permutability in Migraine
5.1. Experimental Research of BBB Permutability
5.2. Clinical Studies of BBB Permutability
6. Participation of Pericytes in Migraine
7. Prospect of Therapeutic Benefit via the IL-1β/IL-1R1 Axis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BBB | blood-brain barrier |
| IL | interleukin |
| MMP-9 | metalloproteinase-9 |
| MRI | magnetic resonance image |
| TNF-α | tumor necrosis factor-α |
| ESM-1 | endothelial cell-specific molecule-1 |
| TG | trigeminal ganglia |
| ROS | reactive oxygen species |
| NO | nitric oxide |
| CGRP | calcitonin gene-related peptide |
| CNS | central nervous system |
| ICAM-1 | intercellular adhesion molecule-1 |
| CSF | cerebral spinal fluid |
| CSD | cortical spreading depolarization |
| NTG | nitroglycerin |
| NLRP3 | leucine-rich repeat pyrin containing protein-3 |
| 5-HT | 5-hydroxytryptamine |
| MIP-1α | macrophage inflammatory protein-1α |
| CM | chronic migraine |
| MCP-1 | monocyte chemoattractant protein-1 |
| CCL5 | C–C chemokine ligand 5 |
| IL-1R1 | IL-1 receptor type 1 |
References
- Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [CrossRef] [Green Version]
- Feigin, V.L.; Vos, T.; Alahdab, F.; Amit, A.M.L.; Bärnighausen, T.W.; Beghi, E.; Beheshti, M.; Chavan, P.P.; Criqui, M.H.; Desai, R.; et al. Burden of Neurological Disorders Across the US From 1990–2017: A Global Burden of Disease Study. JAMA Neurol. 2021, 78, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.P.; Feniuk, W.; Perren, M.J.; Beresford, I.J.; Skingle, M.; Whalley, E.T. Serotonin and migraine. Ann. N. Y. Acad. Sci. 1990, 600, 587–598; discussion 598–600. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L.; Uddman, R. Neurobiology in primary headaches. Brain Res. Brain Res. Rev. 2005, 48, 438–456. [Google Scholar] [CrossRef] [Green Version]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K.; Krause, D.N. CGRP as the target of new migraine therapies—successful translation from bench to clinic. Nat. Rev. Neurol. 2018, 14, 338–350. [Google Scholar] [CrossRef]
- Raddant, A.C.; Russo, A.F. Calcitonin gene-related peptide in migraine: Intersection of peripheral inflammation and central modulation. Expert Rev. Mol. Med. 2011, 13, e36. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, R. Neurogenic inflammation and its role in migraine. Semin. Immunopathol. 2018, 40, 301–314. [Google Scholar] [CrossRef]
- Malhotra, R. Understanding migraine: Potential role of neurogenic inflammation. Ann. Indian Acad. Neurol. 2016, 19, 175–182. [Google Scholar] [CrossRef]
- Gao, Y.J.; Ji, R.R. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol. Ther. 2010, 126, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K. Does inflammation have a role in migraine? Nat. Rev. Neurol. 2019, 15, 483–490. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, G.; Takata, F.; Kataoka, Y.; Kanou, K.; Morichi, S.; Dohgu, S.; Kawashima, H. The Neuroinflammatory Role of Pericytes in Epilepsy. Biomedicines 2021, 9, 759. [Google Scholar] [CrossRef] [PubMed]
- Covelli, V.; Munno, I.; Pellegrino, N.M.; Di Venere, A.; Jirillo, E.; Buscaino, G.A. Exaggerated spontaneous release of tumor necrosis factor-alpha/cachectin in patients with migraine without aura. Acta Neurol. 1990, 12, 257–263. [Google Scholar]
- Perini, F.; D’Andrea, G.; Galloni, E.; Pignatelli, F.; Billo, G.; Alba, S.; Bussone, G.; Toso, V. Plasma Cytokine Levels in Migraineurs and Controls. Headache J. Head Face Pain 2005, 45, 926–931. [Google Scholar] [CrossRef]
- Kacinski, M.; Gergont, A.; Kubik, A.; Steczkowska-Klucznik, M. Proinflammatory cytokines in children with migraine with or without aura. Przegl. Lek. 2005, 62, 1276–1280. [Google Scholar]
- Sarchielli, P.; Alberti, A.; Baldi, A.; Coppola, F.; Rossi, C.; Pierguidi, L.; Floridi, A.; Calabresi, P. Proinflammatory cytokines, adhesion molecules, and lymphocyte integrin expression in the internal jugular blood of migraine patients without aura assessed ictally. Headache 2006, 46, 200–207. [Google Scholar] [CrossRef]
- Wang, F.; He, Q.; Ren, Z.; Li, F.; Chen, W.; Lin, X.; Zhang, H.; Tai, G. Association of serum levels of intercellular adhesion molecule-1 and interleukin-6 with migraine. Neurol. Sci. 2015, 36, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Yücel, M.; Kotan, D.; Gurol Çiftçi, G.; Çiftçi, I.H.; Cikriklar, H.I. Serum levels of endocan, claudin-5 and cytokines in migraine. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 930–936. [Google Scholar]
- Van Hilten, J.J.; Ferrari, M.D.; Van der Meer, J.W.M.; Gijsman, H.J.; Looij, B.J., Jr. Plasma interleukin-1, tumour necrosis factor and hypothalamic-pituitary-adrenal axis responses during migraine attacks. Cephalalgia 1991, 11, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Tanure, M.T.; Gomez, R.S.; Hurtado, R.C.; Teixeira, A.L.; Domingues, R.B. Increased serum levels of brain-derived neurotropic factor during migraine attacks: A pilot study. J. Headache Pain 2010, 11, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Martelletti, P.; Stirparo, G.; Morrone, S.; Rinaldi, C.; Giacovazzo, M. Inhibition of intercellular adhesion molecule-1 (ICAM-1), soluble ICAM-1 and interleukin-4 by nitric oxide expression in migraine patients. J. Mol. Med. 1997, 75, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Fidan, I.; Yüksel, S.; Ymir, T.; Irkeç, C.; Aksakal, F.N. The importance of cytokines, chemokines and nitric oxide in pathophysiology of migraine. J. Neuroimmunol. 2006, 171, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Munno, I.; Marinaro, M.; Bassi, A.; Cassiano, M.A.; Causarano, V.; Centonze, V. Immunological aspects in migraine: Increase of IL-10 plasma levels during attack. Headache 2001, 41, 764–767. [Google Scholar] [CrossRef]
- Boćkowski, L.; Sobaniec, W.; Zelazowska-Rutkowska, B. Proinflammatory plasma cytokines in children with migraine. Pediatr. Neurol. 2009, 41, 17–21. [Google Scholar] [CrossRef]
- Uzar, E.; Evliyaoglu, O.; Yucel, Y.; Ugur Cevik, M.; Acar, A.; Guzel, I.; Islamoglu, Y.; Colpan, L.; Tasdemir, N. Serum cytokine and pro-brain natriuretic peptide (BNP) levels in patients with migraine. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 1111–1116. [Google Scholar]
- Oliveira, A.B.; Bachi, A.L.L.; Ribeiro, R.T.; Mello, M.T.; Tufik, S.; Peres, M.F.P. Unbalanced plasma TNF-α and IL-12/IL-10 profile in women with migraine is associated with psychological and physiological outcomes. J. Neuroimmunol. 2017, 313, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, H.; Teixeira, A.L.; Rocha, N.P.; Domingues, R.B. Increased interictal serum levels of CXCL8/IL-8 and CCL3/MIP-1α in migraine. Neurol. Sci. 2015, 36, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Empl, M.; Sostak, P.; Riedel, M.; Schwarz, M.; Müller, N.; Förderreuther, S.; Straube, A. Decreased sTNF-RI in migraine patients? Cephalalgia 2003, 23, 55–58. [Google Scholar] [CrossRef]
- Michalak, S.; Kalinowska-Lyszczarz, A.; Wegrzyn, D.; Niezgoda, A.; Losy, J.; Osztynowicz, K.; Kozubski, W. Increased Serum CD14 Level Is Associated with Depletion of TNF-alpha in Monocytes in Migraine Patients during Interictal Period. Int. J. Mol. Sci. 2017, 18, 398. [Google Scholar] [CrossRef] [Green Version]
- Sarchielli, P.; Alberti, A.; Vaianella, L.; Pierguidi, L.; Floridi, A.; Mazzotta, G.; Floridi, A.; Gallai, V. Chemokine levels in the jugular venous blood of migraine without aura patients during attacks. Headache 2004, 44, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Sarchielli, P.; Floridi, A.; Mancini, M.L.; Rossi, C.; Coppola, F.; Baldi, A.; Pini, L.A.; Calabresi, P. NF-kappaB activity and iNOS expression in monocytes from internal jugular blood of migraine without aura patients during attacks. Cephalalgia 2006, 26, 1071–1079. [Google Scholar] [CrossRef]
- Rozen, T.; Swidan, S.Z. Elevation of CSF Tumor Necrosis Factor α Levels in New Daily Persistent Headache and Treatment Refractory Chronic Migraine. Headache J. Head Face Pain 2007, 47, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Bø, S.H.; Davidsen, E.M.; Gulbrandsen, P.; Dietrichs, E.; Bovim, G.; Stovner, L.J.; White, L.R. Cerebrospinal fluid cytokine levels in migraine, tension-type headache and cervicogenic headache. Cephalalgia 2009, 29, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Gormley, P.; Anttila, V.; Winsvold, B.S.; Palta, P.; Esko, T.; Pers, T.H.; Farh, K.H.; Cuenca-Leon, E.; Muona, M.; Furlotte, N.A.; et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 2016, 48, 856–866. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, I.A.; Ozge, A.; Erdal, M.E.; Edgünlü, T.G.; Cakmak, S.E.; Yalin, O.O. Cytokine polymorphism in patients with migraine: Some suggestive clues of migraine and inflammation. Pain Med. 2010, 11, 492–497. [Google Scholar] [CrossRef] [Green Version]
- Perry, C.J.; Blake, P.; Buettner, C.; Papavassiliou, E.; Schain, A.J.; Bhasin, M.K.; Burstein, R. Upregulation of inflammatory gene transcripts in periosteum of chronic migraineurs: Implications for extracranial origin of headache. Ann. Neurol. 2016, 79, 1000–1013. [Google Scholar] [CrossRef] [Green Version]
- Somjen, G.G.; Aitken, P.G.; Czéh, G.L.; Herreras, O.; Jing, J.; Young, J.N. Mechanism of spreading depression: A review of recent findings and a hypothesis. Can. J. Physiol. Pharmacol. 1992, 70, S248–S254. [Google Scholar] [CrossRef]
- Somjen, G.G. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev. 2001, 81, 1065–1096. [Google Scholar] [CrossRef] [Green Version]
- Leao, A.A. Further observations on the spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1947, 10, 409–414. [Google Scholar] [CrossRef]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; Shuttleworth, C.W.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; Shuttleworth, C.W.; Kirov, S.A.; Ayata, C.; Hinzman, J.M.; Foreman, B.; Andrew, R.D.; Boutelle, M.G.; Brennan, K.C.; Carlson, A.P.; et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leão’s legacy. J. Cereb. Blood Flow Metab. 2017, 37, 1571–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Levy, D.; Kainz, V.; Noseda, R.; Jakubowski, M.; Burstein, R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann. Neurol. 2011, 69, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayata, C.; Jin, H.; Kudo, C.; Dalkara, T.; Moskowitz, M.A. Suppression of cortical spreading depression in migraine prophylaxis. Ann. Neurol. 2006, 59, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Erdener, S.E.; Gursoy-Ozdemir, Y.; Lule, S.; Eren-Koçak, E.; Sen, Z.D.; Dalkara, T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013, 339, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Kraig, R.P.; Mitchell, H.M.; Christie-Pope, B.; Kunkler, P.E.; White, D.M.; Tang, Y.P.; Langan, G. TNF-alpha and Microglial Hormetic Involvement in Neurological Health & Migraine. Dose Response 2010, 8, 389–413. [Google Scholar] [CrossRef]
- Levy, D. Endogenous mechanisms underlying the activation and sensitization of meningeal nociceptors: The role of immuno-vascular interactions and cortical spreading depression. Curr. Pain Headache Rep. 2012, 16, 270–277. [Google Scholar] [CrossRef]
- Jander, S.; Schroeter, M.; Peters, O.; Witte, O.W.; Stoll, G. Cortical spreading depression induces proinflammatory cytokine gene expression in the rat brain. J. Cereb. Blood Flow Metab. 2001, 21, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Ghaemi, A.; Sajadian, A.; Khodaie, B.; Lotfinia, A.A.; Lotfinia, M.; Aghabarari, A.; Khaleghi Ghadiri, M.; Meuth, S.; Gorji, A. Immunomodulatory Effect of Toll-Like Receptor-3 Ligand Poly I:C on Cortical Spreading Depression. Mol. Neurobiol. 2016, 53, 143–154. [Google Scholar] [CrossRef]
- Ghaemi, A.; Alizadeh, L.; Babaei, S.; Jafarian, M.; Khaleghi Ghadiri, M.; Meuth, S.G.; Kovac, S.; Gorji, A. Astrocyte-mediated inflammation in cortical spreading depression. Cephalalgia 2018, 38, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Kunkler, P.E.; Hulse, R.E.; Kraig, R.P. Multiplexed cytokine protein expression profiles from spreading depression in hippocampal organotypic cultures. J. Cereb. Blood Flow Metab. 2004, 24, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, C.S.; Hakim, A.M. Cortical spreading depression modifies components of the inflammatory cascade. Mol. Neurobiol. 2005, 32, 51–57. [Google Scholar] [CrossRef]
- Chen, S.P.; Qin, T.; Seidel, J.L.; Zheng, Y.; Eikermann, M.; Ferrari, M.D.; van den Maagdenberg, A.; Moskowitz, M.A.; Ayata, C.; Eikermann-Haerter, K. Inhibition of the P2X7-PANX1 complex suppresses spreading depolarization and neuroinflammation. Brain 2017, 140, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, R.; Cui, L.; Yong, C.; Bowyer, S.; Klein, R.M.; Welch, K.M.; Berman, N.E. Cortical spreading depression and gene regulation: Relevance to migraine. Ann. Neurol. 2002, 51, 499–506. [Google Scholar] [CrossRef]
- Urbach, A.; Bruehl, C.; Witte, O.W. Microarray-based long-term detection of genes differentially expressed after cortical spreading depression. Eur. J. Neurosci. 2006, 24, 841–856. [Google Scholar] [CrossRef]
- Wang, H.; Peca, J.; Matsuzaki, M.; Matsuzaki, K.; Noguchi, J.; Qiu, L.; Wang, D.; Zhang, F.; Boyden, E.; Deisseroth, K.; et al. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc. Natl. Acad. Sci. USA 2007, 104, 8143–8148. [Google Scholar] [CrossRef] [Green Version]
- Arenkiel, B.R.; Peca, J.; Davison, I.G.; Feliciano, C.; Deisseroth, K.; Augustine, G.J.; Ehlers, M.D.; Feng, G. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron 2007, 54, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.Y.; Sadeghian, H.; Qin, T.; Lule, S.; Lee, H.; Karakaya, F.; Goins, S.; Oka, F.; Yaseen, M.A.; Houben, T.; et al. Determinants of Optogenetic Cortical Spreading Depolarizations. Cereb. Cortex 2019, 29, 1150–1161. [Google Scholar] [CrossRef]
- Takizawa, T.; Qin, T.; Lopes de Morais, A.; Sugimoto, K.; Chung, J.Y.; Morsett, L.; Mulder, I.; Fischer, P.; Suzuki, T.; Anzabi, M.; et al. Non-invasively triggered spreading depolarizations induce a rapid pro-inflammatory response in cerebral cortex. J. Cereb. Blood Flow Metab. 2019, 40, 1117–1131. [Google Scholar] [CrossRef]
- Friedman, B.W.; Greenwald, P.; Bania, T.C.; Esses, D.; Hochberg, M.; Solorzano, C.; Corbo, J.; Chu, J.; Chew, E.; Cheung, P.; et al. Randomized trial of IV dexamethasone for acute migraine in the emergency department. Neurology 2007, 69, 2038–2044. [Google Scholar] [CrossRef]
- Rowe, B.H.; Colman, I.; Edmonds, M.L.; Blitz, S.; Walker, A.; Wiens, S. Randomized controlled trial of intravenous dexamethasone to prevent relapse in acute migraine headache. Headache 2008, 48, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.; Sundermann, R.; Jackson, R.; Bastani, A. Intravenous dexamethasone vs placebo as adjunctive therapy to reduce the recurrence rate of acute migraine headaches: A multicenter, double-blinded, placebo-controlled randomized clinical trial. Am. J. Emerg. Med. 2008, 26, 124–130. [Google Scholar] [CrossRef]
- Demartini, C.; Greco, R.; Zanaboni, A.M.; Sances, G.; De Icco, R.; Borsook, D.; Tassorelli, C. Nitroglycerin as a comparative experimental model of migraine pain: From animal to human and back. Prog. Neurobiol. 2019, 177, 15–32. [Google Scholar] [CrossRef]
- Reuter, U.; Bolay, H.; Jansen-Olesen, I.; Chiarugi, A.; Sanchez del Rio, M.; Letourneau, R.; Theoharides, T.C.; Waeber, C.; Moskowitz, M.A. Delayed inflammation in rat meninges: Implications for migraine pathophysiology. Brain 2001, 124, 2490–2502. [Google Scholar] [CrossRef] [PubMed]
- Reuter, U.; Chiarugi, A.; Bolay, H.; Moskowitz, M.A. Nuclear factor-kappaB as a molecular target for migraine therapy. Ann. Neurol. 2002, 51, 507–516. [Google Scholar] [CrossRef]
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Redavide, E.; Pampalone, S.; Toldi, J.; Fülöp, F.; Blandini, F.; Nappi, G.; Sandrini, G.; et al. Effects of kynurenic acid analogue 1 (KYNA-A1) in nitroglycerin-induced hyperalgesia: Targets and anti-migraine mechanisms. Cephalalgia 2017, 37, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Demartini, C.; Zanaboni, A.; Casini, I.; De Icco, R.; Reggiani, A.; Misto, A.; Piomelli, D.; Tassorelli, C. Characterization of the peripheral FAAH inhibitor, URB937, in animal models of acute and chronic migraine. Neurobiol. Dis. 2021, 147, 105157. [Google Scholar] [CrossRef]
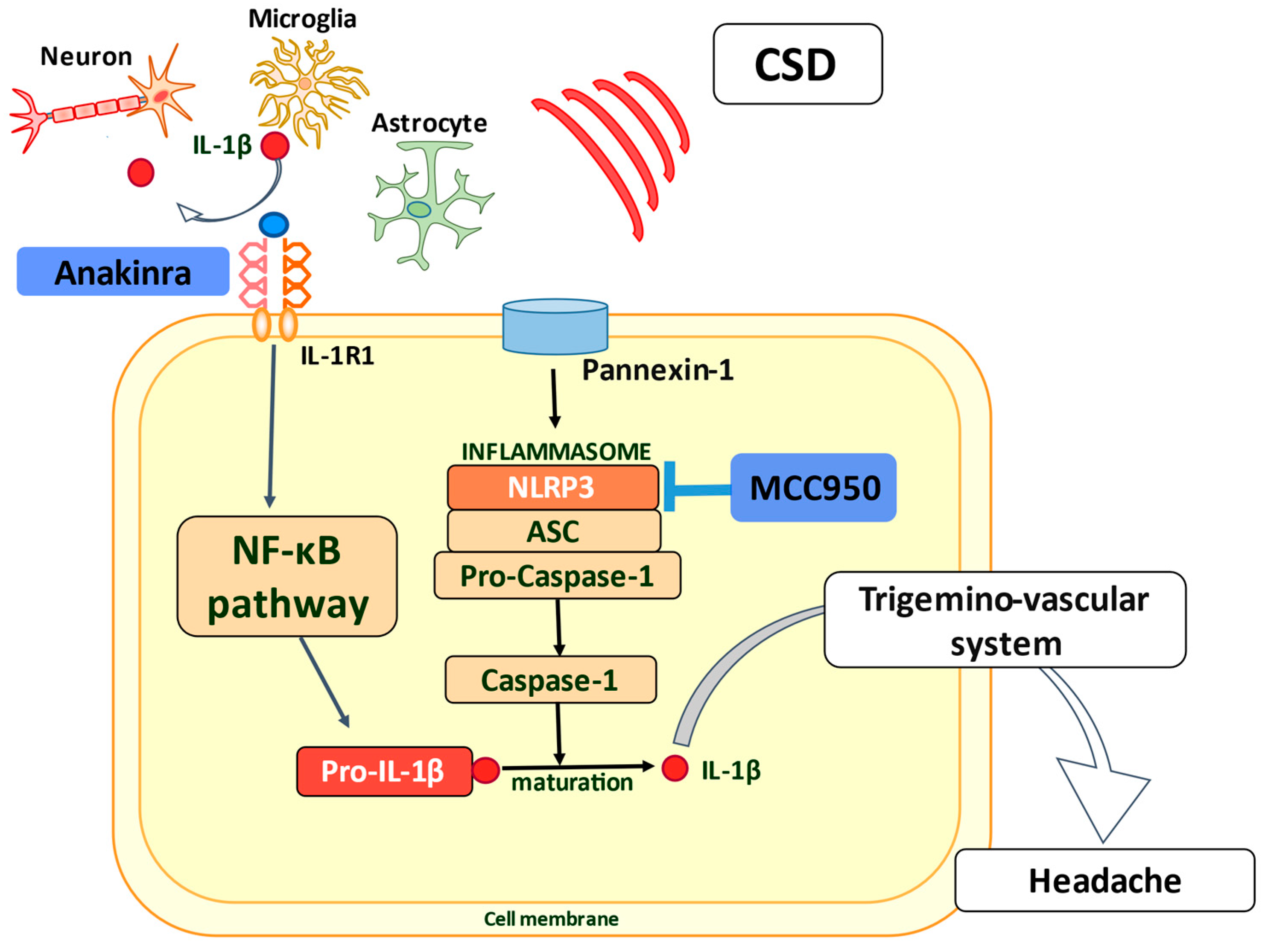
- He, W.; Long, T.; Pan, Q.; Zhang, S.; Zhang, Y.; Zhang, D.; Qin, G.; Chen, L.; Zhou, J. Microglial NLRP3 inflammasome activation mediates IL-1β release and contributes to central sensitization in a recurrent nitroglycerin-induced migraine model. J. Neuroinflammation 2019, 16, 78. [Google Scholar] [CrossRef] [Green Version]
- Sparaco, M.; Feleppa, M.; Lipton, R.B.; Rapoport, A.M.; Bigal, M.E. Mitochondrial dysfunction and migraine: Evidence and hypotheses. Cephalalgia 2006, 26, 361–372. [Google Scholar] [CrossRef]
- Yilmaz, G.; Sürer, H.; Inan, L.E.; Coskun, O.; Yücel, D. Increased nitrosative and oxidative stress in platelets of migraine patients. Tohoku J. Exp. Med. 2007, 211, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Rajamäki, K.; Nordström, T.; Nurmi, K.; Åkerman, K.E.; Kovanen, P.T.; Öörni, K.; Eklund, K.K. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J. Biol. Chem. 2013, 288, 13410–13419. [Google Scholar] [CrossRef] [Green Version]
- Gölöncsér, F.; Sperlágh, B. Effect of genetic deletion and pharmacological antagonism of P2X7 receptors in a mouse animal model of migraine. J. Headache Pain 2014, 15, 24. [Google Scholar] [CrossRef] [Green Version]
- Gursoy-Ozdemir, Y.; Qiu, J.; Matsuoka, N.; Bolay, H.; Bermpohl, D.; Jin, H.; Wang, X.; Rosenberg, G.A.; Lo, E.H.; Moskowitz, M.A. Cortical spreading depression activates and upregulates MMP-9. J. Clin. Investig. 2004, 113, 1447–1455. [Google Scholar] [CrossRef]
- Cottier, K.E.; Galloway, E.A.; Calabrese, E.C.; Tome, M.E.; Liktor-Busa, E.; Kim, J.; Davis, T.P.; Vanderah, T.W.; Largent-Milnes, T.M. Loss of Blood-Brain Barrier Integrity in a KCl-Induced Model of Episodic Headache Enhances CNS Drug Delivery. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Sadeghian, H.; Lacoste, B.; Qin, T.; Toussay, X.; Rosa, R.; Oka, F.; Chung, D.Y.; Takizawa, T.; Gu, C.; Ayata, C. Spreading depolarizations trigger caveolin-1-dependent endothelial transcytosis. Ann. Neurol. 2018, 84, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Harriott, A.M.; Takizawa, T.; Chung, D.Y.; Chen, S.P. Spreading depression as a preclinical model of migraine. J. Headache Pain 2019, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, C.; Haanes, K.A.; Grände, G.; Edvinsson, L. Experimental inflammation following dural application of complete Freund’s adjuvant or inflammatory soup does not alter brain and trigeminal microvascular passage. J. Headache Pain 2015, 16, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edvinsson, L.; Tfelt-Hansen, P. The blood-brain barrier in migraine treatment. Cephalalgia 2008, 28, 1245–1258. [Google Scholar] [CrossRef]
- Imamura, K.; Takeshima, T.; Fusayasu, E.; Nakashima, K. Increased plasma matrix metalloproteinase-9 levels in migraineurs. Headache 2008, 48, 135–139. [Google Scholar] [CrossRef]
- Martins-Oliveira, A.; Gonçalves, F.M.; Speciali, J.G.; Fontana, V.; Izidoro-Toledo, T.C.; Belo, V.A.; Dach, F.; Tanus-Santos, J.E. Specific matrix metalloproteinase 9 (MMP-9) haplotype affect the circulating MMP-9 levels in women with migraine. J. Neuroimmunol. 2012, 252, 89–94. [Google Scholar] [CrossRef]
- Dong, L.; Qiao, H.; Zhang, X.; Zhang, X.; Wang, C.; Wang, L.; Cui, L.; Zhao, J.; Xing, Y.; Li, Y.; et al. Parthenolide is neuroprotective in rat experimental stroke model: Downregulating NF-κB, phospho-p38MAPK, and caspase-1 and ameliorating BBB permeability. Mediat. Inflamm. 2013, 2013, 370804. [Google Scholar] [CrossRef] [Green Version]
- Sarrazin, S.; Adam, E.; Lyon, M.; Depontieu, F.; Motte, V.; Landolfi, C.; Lortat-Jacob, H.; Bechard, D.; Lassalle, P.; Delehedde, M. Endocan or endothelial cell specific molecule-1 (ESM-1): A potential novel endothelial cell marker and a new target for cancer therapy. Biochim. Biophys. Acta 2006, 1765, 25–37. [Google Scholar] [CrossRef]
- Hougaard, A.; Amin, F.M.; Christensen, C.E.; Younis, S.; Wolfram, F.; Cramer, S.P.; Larsson, H.B.W.; Ashina, M. Increased brainstem perfusion, but no blood-brain barrier disruption, during attacks of migraine with aura. Brain 2017, 140, 1633–1642. [Google Scholar] [CrossRef]
- Amin, F.M.; Hougaard, A.; Cramer, S.P.; Christensen, C.E.; Wolfram, F.; Larsson, H.B.W.; Ashina, M. Intact blood-brain barrier during spontaneous attacks of migraine without aura: A 3T DCE-MRI study. Eur. J. Neurol. 2017, 24, 1116–1124. [Google Scholar] [CrossRef]
- Schankin, C.J.; Maniyar, F.H.; Seo, Y.; Kori, S.; Eller, M.; Chou, D.E.; Blecha, J.; Murphy, S.T.; Hawkins, R.A.; Sprenger, T.; et al. Ictal lack of binding to brain parenchyma suggests integrity of the blood-brain barrier for 11C-dihydroergotamine during glyceryl trinitrate-induced migraine. Brain 2016, 139, 1994–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, D.; Rustenhoven, J.; Feng, S.; Hurley, D.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Faull, R.L.; Dragunow, M. A role for human brain pericytes in neuroinflammation. J. Neuroinflamm. 2014, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Khennouf, L.; Gesslein, B.; Brazhe, A.; Octeau, J.C.; Kutuzov, N.; Khakh, B.S.; Lauritzen, M. Active role of capillary pericytes during stimulation-induced activity and spreading depolarization. Brain 2018, 141, 2032–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovac, A.; Erickson, M.A.; Banks, W.A. Brain microvascular pericytes are immunoactive in culture: Cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J. Neuroinflammation 2011, 8, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcendor, D.J.; Charest, A.M.; Zhu, W.Q.; Vigil, H.E.; Knobel, S.M. Infection and upregulation of proinflammatory cytokines in human brain vascular pericytes by human cytomegalovirus. J. Neuroinflammation 2012, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, J.; Takata, F.; Machida, T.; Takahashi, H.; Soejima, Y.; Funakoshi, M.; Futagami, K.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Tumor necrosis factor-α-stimulated brain pericytes possess a unique cytokine and chemokine release profile and enhance microglial activation. Neurosci. Lett. 2014, 578, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, J.; Dohgu, S.; Takata, F.; Machida, T.; Bölükbaşi Hatip, F.F.; Hatip-Al-Khatib, I.; Yamauchi, A.; Kataoka, Y. TNF-α-sensitive brain pericytes activate microglia by releasing IL-6 through cooperation between IκB-NFκB and JAK-STAT3 pathways. Brain Res. 2018, 1692, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Klement, W.; Garbelli, R.; Zub, E.; Rossini, L.; Tassi, L.; Girard, B.; Blaquiere, M.; Bertaso, F.; Perroy, J.; de Bock, F.; et al. Seizure progression and inflammatory mediators promote pericytosis and pericyte-microglia clustering at the cerebrovasculature. Neurobiol. Dis. 2018, 113, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Dziewulska, D.; Kierdaszuk, B. Ultrastructural changes in microvessels in familial hemiplegic migraine with CACNA1A mutation. Clin. Neuropathol. 2018, 37, 283–287. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Andermann, F. Migraine and Epilepsy. Semin. Pediatr. Neurol. 2010, 17, 117–122. [Google Scholar] [CrossRef]
- Mantegazza, M.; Cestèle, S. Pathophysiological mechanisms of migraine and epilepsy: Similarities and differences. Neurosci. Lett. 2018, 667, 92–102. [Google Scholar] [CrossRef]
- Cheng, J.; Korte, N.; Nortley, R.; Sethi, H.; Tang, Y.; Attwell, D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018, 136, 507–523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Burstein, R.; Levy, D. Local action of the proinflammatory cytokines IL-1β and IL-6 on intracranial meningeal nociceptors. Cephalalgia 2012, 32, 66–72. [Google Scholar] [CrossRef]
- Franceschini, A.; Vilotti, S.; Ferrari, M.D.; van den Maagdenberg, A.M.; Nistri, A.; Fabbretti, E. TNFα levels and macrophages expression reflect an inflammatory potential of trigeminal ganglia in a mouse model of familial hemiplegic migraine. PLoS ONE 2013, 8, e52394. [Google Scholar] [CrossRef]
- Lombardo, S.D.; Mazzon, E.; Basile, M.S.; Cavalli, E.; Bramanti, P.; Nania, R.; Fagone, P.; Nicoletti, F.; Petralia, M.C. Upregulation of IL-1 Receptor Antagonist in a Mouse Model of Migraine. Brain Sci. 2019, 9, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Corato, A.; Lisi, L.; Capuano, A.; Tringali, G.; Tramutola, A.; Navarra, P.; Russo, C.D. Trigeminal satellite cells express functional calcitonin gene-related peptide receptors, whose activation enhances interleukin-1β pro-inflammatory effects. J. Neuroimmunol. 2011, 237, 39–46. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vliet, E.A.; Aronica, E.; Vezzani, A.; Ravizza, T. Review: Neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: Emerging evidence from preclinical and clinical studies. Neuropathol. Appl. Neurobiol. 2018, 44, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Kenney-Jung, D.L.; Vezzani, A.; Kahoud, R.J.; LaFrance-Corey, R.G.; Ho, M.L.; Muskardin, T.W.; Wirrell, E.C.; Howe, C.L.; Payne, E.T. Febrile infection-related epilepsy syndrome treated with anakinra. Ann. Neurol. 2016, 80, 939–945. [Google Scholar] [CrossRef]
- Jyonouchi, H.; Geng, L. Intractable Epilepsy (IE) and Responses to Anakinra, a Human Recombinant IL-1 Receptor Agonist (IL-1ra): Case Reports. J. Clin. Cell. Immunol. 2016, 7, 456. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, G.; Ishida, Y.; Kanou, K.; Suzuki, S.; Watanabe, Y.; Takamatsu, T.; Morichi, S.; Go, S.; Oana, S.; Yamazaki, T.; et al. Towards a Treatment for Neuroinflammation in Epilepsy: Interleukin-1 Receptor Antagonist, Anakinra, as a Potential Treatment in Intractable Epilepsy. Int. J. Mol. Sci. 2021, 22, 6282. [Google Scholar] [CrossRef]
- Smith, C.J.; Hulme, S.; Vail, A.; Heal, C.; Parry-Jones, A.R.; Scarth, S.; Hopkins, K.; Hoadley, M.; Allan, S.M.; Rothwell, N.J.; et al. SCIL-STROKE (Subcutaneous Interleukin-1 Receptor Antagonist in Ischemic Stroke): A Randomized Controlled Phase 2 Trial. Stroke 2018, 49, 1210–1216. [Google Scholar] [CrossRef] [Green Version]
- Galea, J.; Ogungbenro, K.; Hulme, S.; Patel, H.; Scarth, S.; Hoadley, M.; Illingworth, K.; McMahon, C.J.; Tzerakis, N.; King, A.T.; et al. Reduction of inflammation after administration of interleukin-1 receptor antagonist following aneurysmal subarachnoid hemorrhage: Results of the Subcutaneous Interleukin-1Ra in SAH (SCIL-SAH) study. J. Neurosurg. 2018, 128, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Sibley, C.H.; Plass, N.; Snow, J.; Wiggs, E.A.; Brewer, C.C.; King, K.A.; Zalewski, C.; Kim, H.J.; Bishop, R.; Hill, S.; et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: A cohort study to determine three- and five-year outcomes. Arthritis Rheum. 2012, 64, 2375–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullenberg, T.; Löfqvist, M.; Leinonen, M.; Goldbach-Mansky, R.; Olivecrona, H. Long-term safety profile of anakinra in patients with severe cryopyrin-associated periodic syndromes. Rheumatology 2016, 55, 1499–1506. [Google Scholar] [CrossRef] [Green Version]
- Oby, E.; Janigro, D. The blood-brain barrier and epilepsy. Epilepsia 2006, 47, 1761–1774. [Google Scholar] [CrossRef]
- Yamanaka, G.; Morichi, S.; Takamatsu, T.; Watanabe, Y.; Suzuki, S.; Ishida, Y.; Oana, S.; Yamazaki, T.; Takata, F.; Kawashima, H. Links between Immune Cells from the Periphery and the Brain in the Pathogenesis of Epilepsy: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 4395. [Google Scholar] [CrossRef]
- Landmann, E.C.; Walker, U.A. Pharmacological treatment options for cryopyrin-associated periodic syndromes. Expert Rev. Clin. Pharmacol. 2017, 10, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, N.P.; Teixeira-Coelho, M.; Saraiva, M.J. Protective role of anakinra against transthyretin-mediated axonal loss and cell death in a mouse model of familial amyloidotic polyneuropathy. J. Neuropathol. Exp. Neurol. 2015, 74, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, N.P.; Vieira, P.; Saraiva, M.J. Interleukin-1 signaling pathway as a therapeutic target in transthyretin amyloidosis. Amyloid 2014, 21, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.W.; Lee, K.S.; Kim, C.W. Curcumin attenuates the expression of IL-1beta, IL-6, and TNF-alpha as well as cyclin E in TNF-alpha-treated HaCaT cells; NF-kappaB and MAPKs as potential upstream targets. Int. J. Mol. Med. 2007, 19, 469–474. [Google Scholar]
- Ferreira, N.; Gonçalves, N.P.; Saraiva, M.J.; Almeida, M.R. Curcumin: A multi-target disease-modifying agent for late-stage transthyretin amyloidosis. Sci. Rep. 2016, 6, 26623. [Google Scholar] [CrossRef]
- Fan, C.; Song, Q.; Wang, P.; Li, Y.; Yang, M.; Yu, S.Y. Neuroprotective Effects of Curcumin on IL-1β-Induced Neuronal Apoptosis and Depression-Like Behaviors Caused by Chronic Stress in Rats. Front. Cell. Neurosci. 2018, 12, 516. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Uncovering the Neuroprotective Mechanisms of Curcumin on Transthyretin Amyloidosis. Int. J. Mol. Sci. 2019, 20, 1287. [Google Scholar] [CrossRef] [Green Version]
- Bulboacă, A.E.; Bolboacă, S.D.; Stănescu, I.C.; Sfrângeu, C.A.; Bulboacă, A.C. Preemptive Analgesic and Antioxidative Effect of Curcumin for Experimental Migraine. Biomed. Res. Int. 2017, 2017, 4754701. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Huang, L.; Wu, Q.; Chen, L.; Wan, Q. Chemical stimulation of the intracranial dura activates NALP3 inflammasome in trigeminal ganglia neurons. Brain Res. 2014, 1566, 1–11. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamanaka, G.; Suzuki, S.; Morishita, N.; Takeshita, M.; Kanou, K.; Takamatsu, T.; Suzuki, S.; Morichi, S.; Watanabe, Y.; Ishida, Y.; et al. Role of Neuroinflammation and Blood-Brain Barrier Permutability on Migraine. Int. J. Mol. Sci. 2021, 22, 8929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168929
Yamanaka G, Suzuki S, Morishita N, Takeshita M, Kanou K, Takamatsu T, Suzuki S, Morichi S, Watanabe Y, Ishida Y, et al. Role of Neuroinflammation and Blood-Brain Barrier Permutability on Migraine. International Journal of Molecular Sciences. 2021; 22(16):8929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168929
Chicago/Turabian StyleYamanaka, Gaku, Shinji Suzuki, Natsumi Morishita, Mika Takeshita, Kanako Kanou, Tomoko Takamatsu, Shunsuke Suzuki, Shinichiro Morichi, Yusuke Watanabe, Yu Ishida, and et al. 2021. "Role of Neuroinflammation and Blood-Brain Barrier Permutability on Migraine" International Journal of Molecular Sciences 22, no. 16: 8929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168929