
Cytostatic Effect of a Novel Mitochondria-Targeted Pyrroline Nitroxide in Human Breast Cancer Lines

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
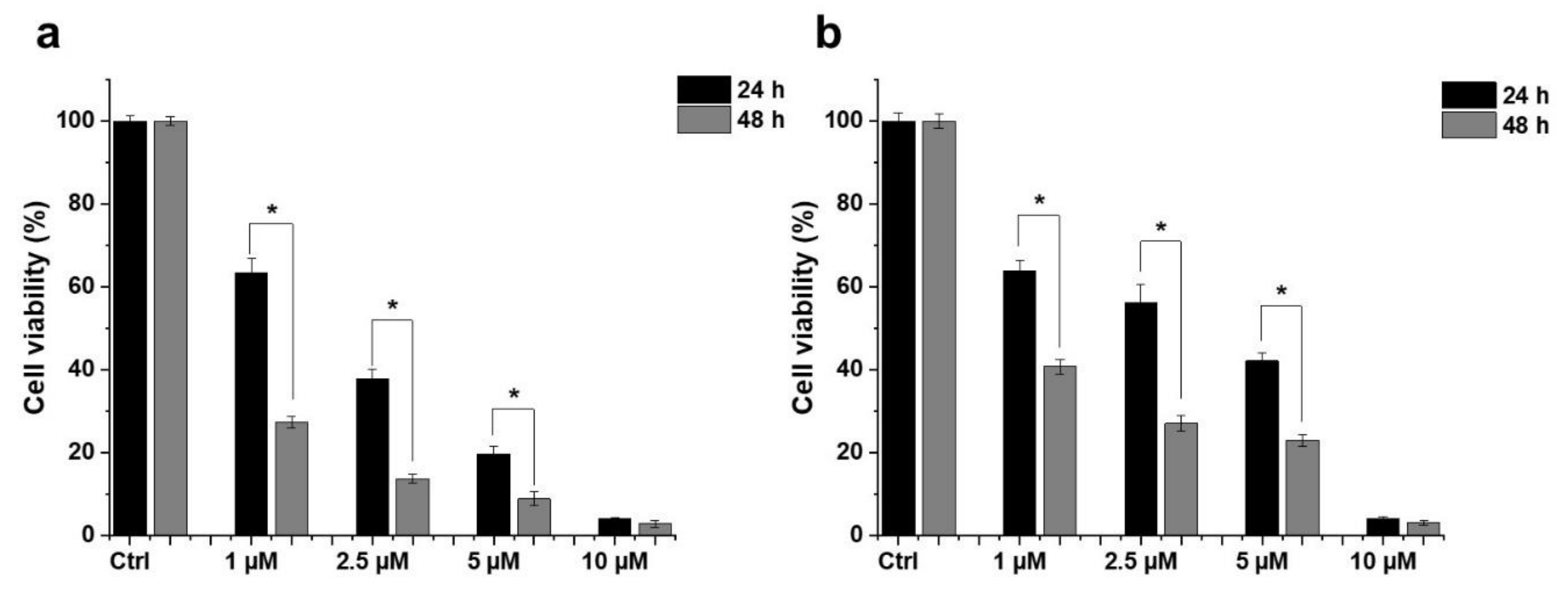
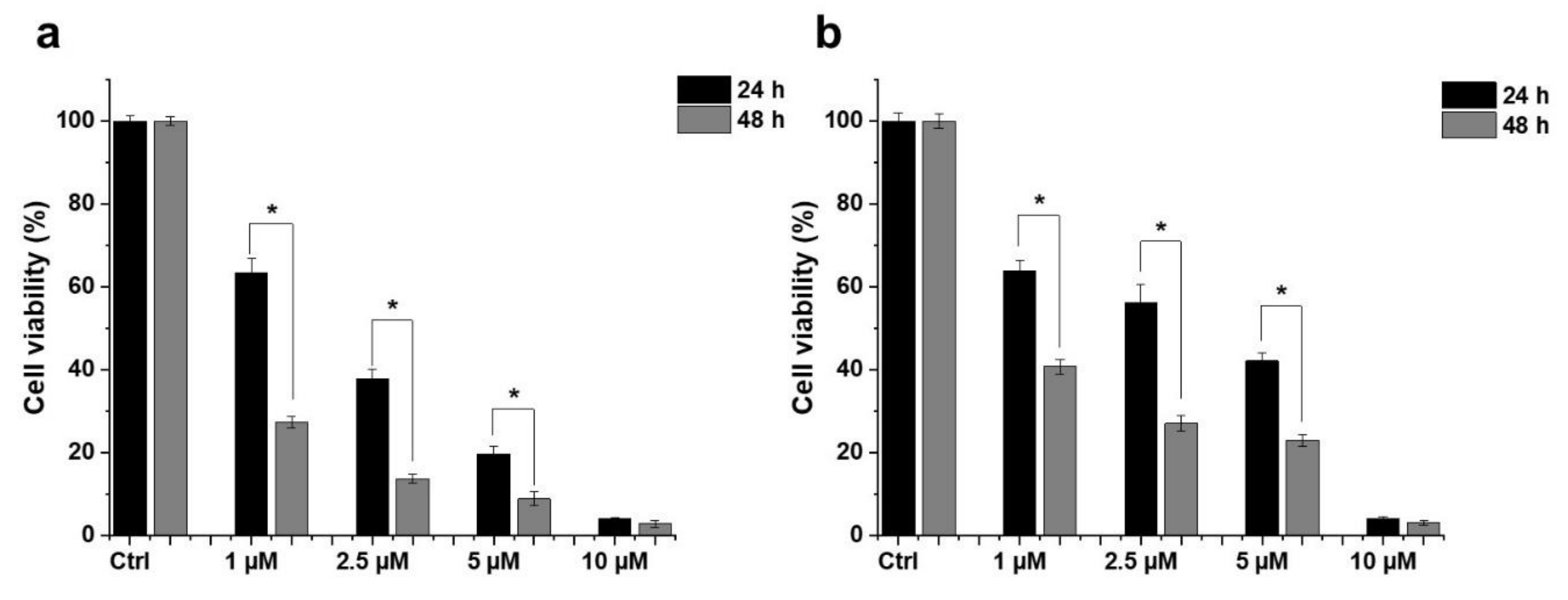
2.1. Effect of HO-5114 on Cell Viability
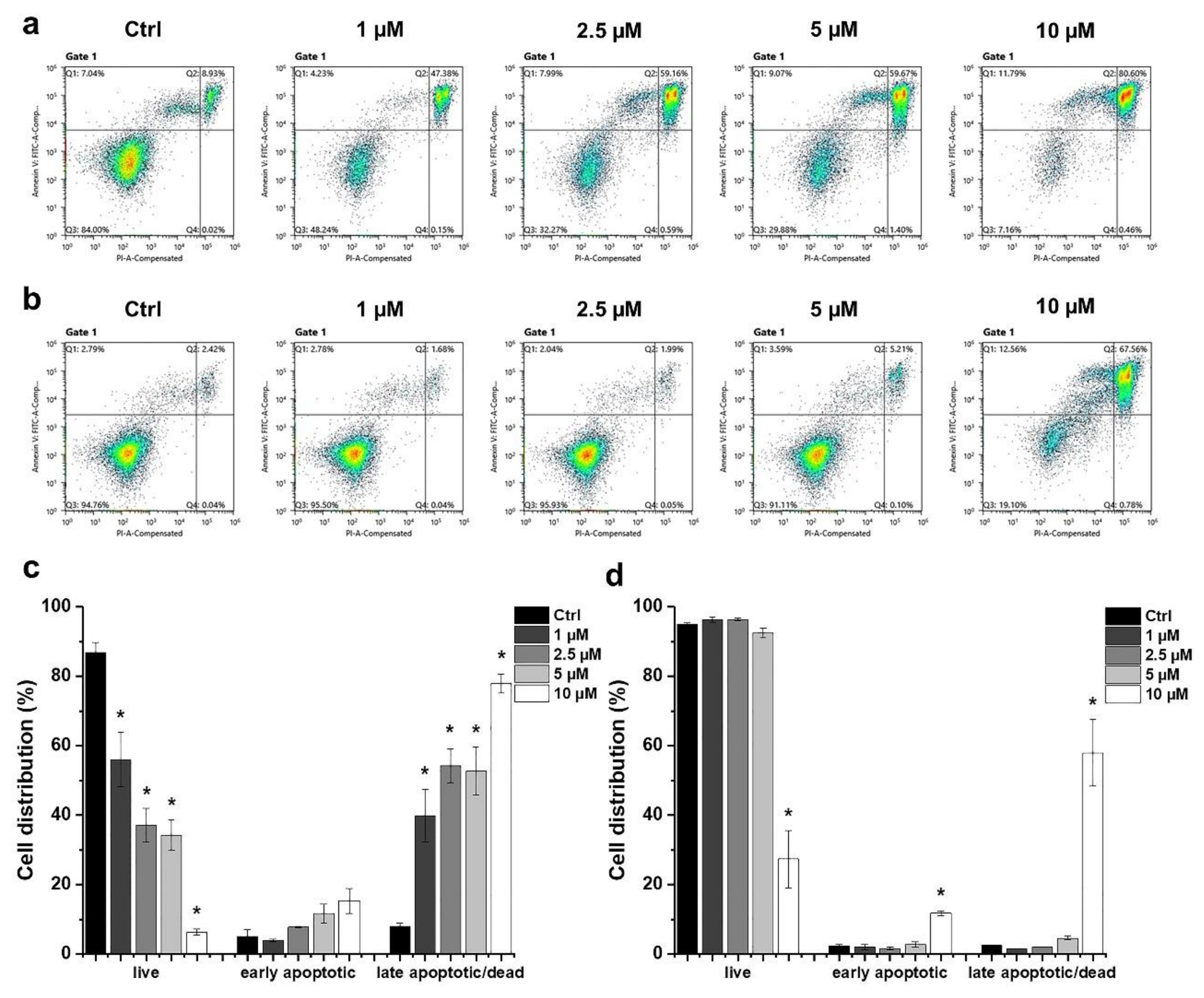
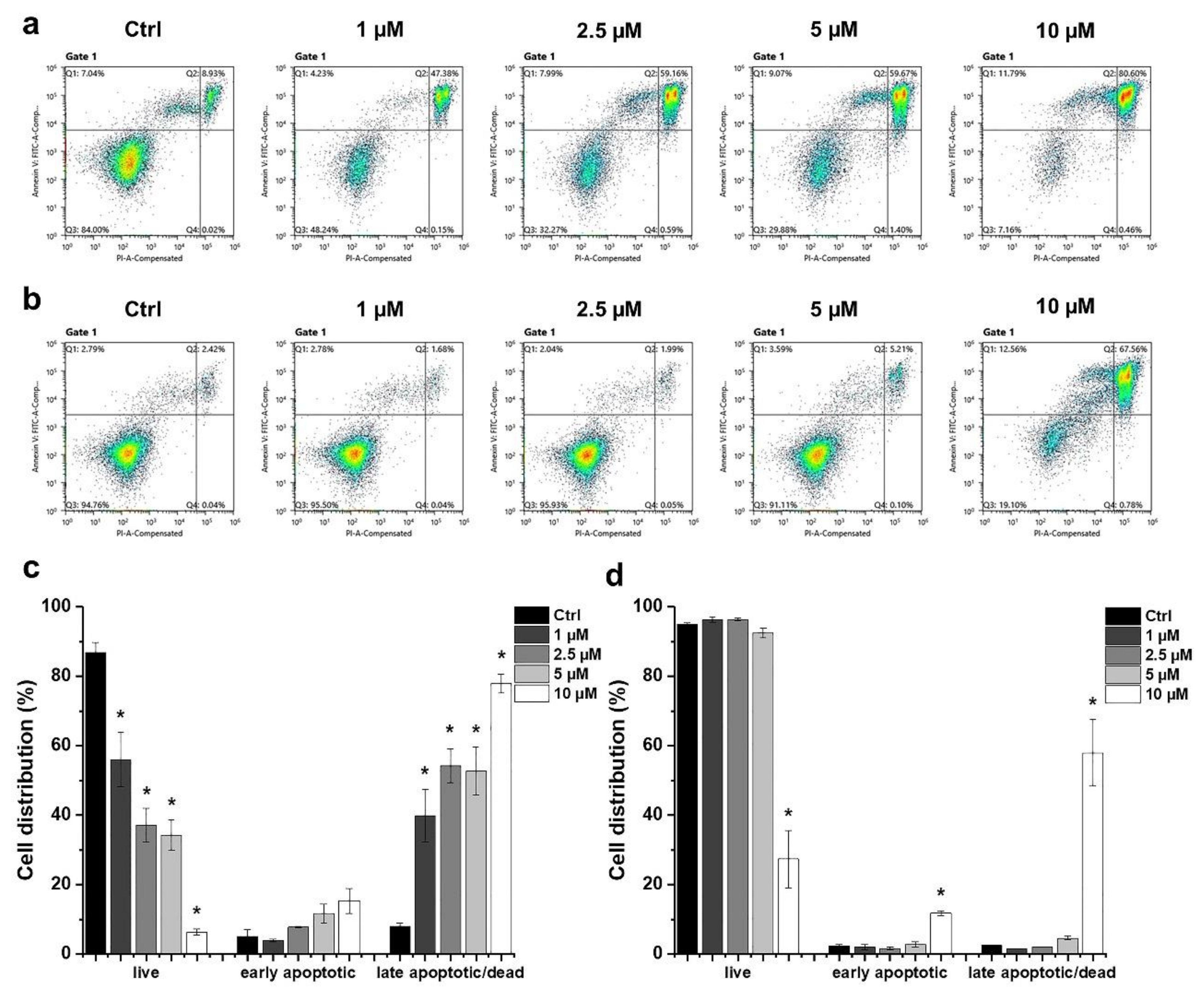
2.2. Determination of the Type of HO-5114-Induced Cell Death
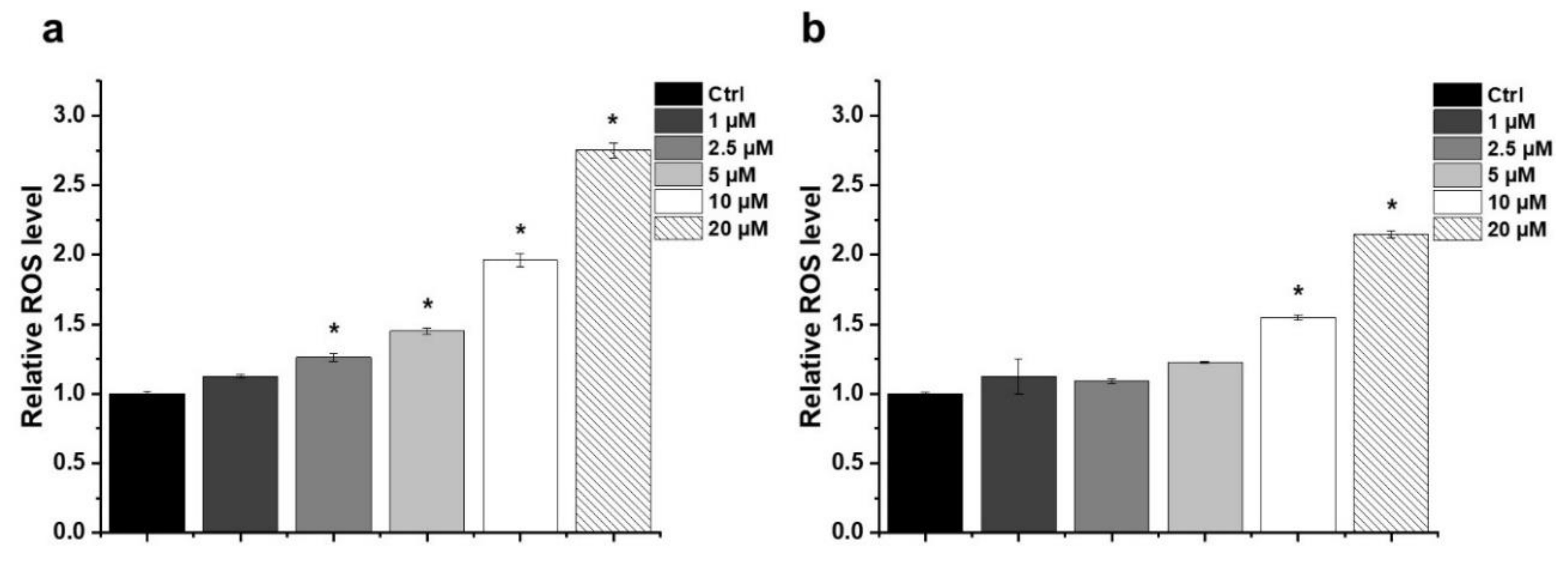
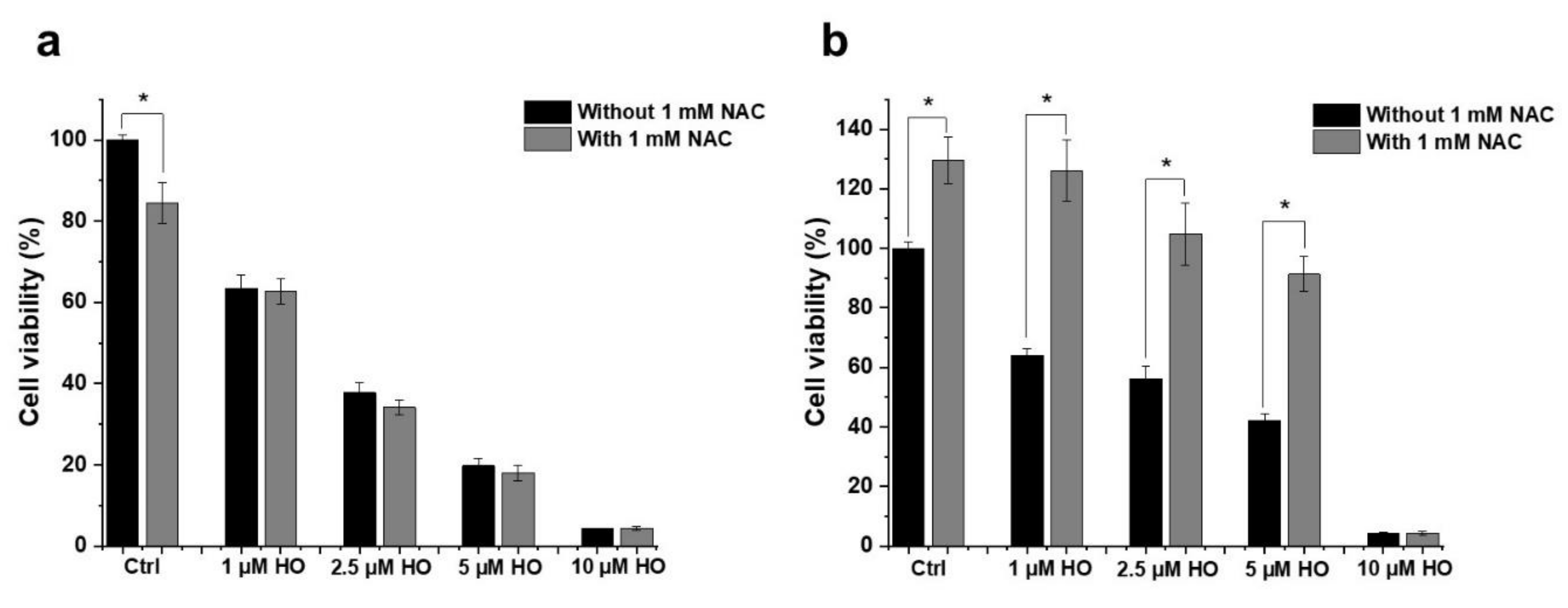
2.3. Effect of HO-5114 on Reactive Oxygen Species (ROS) Generation
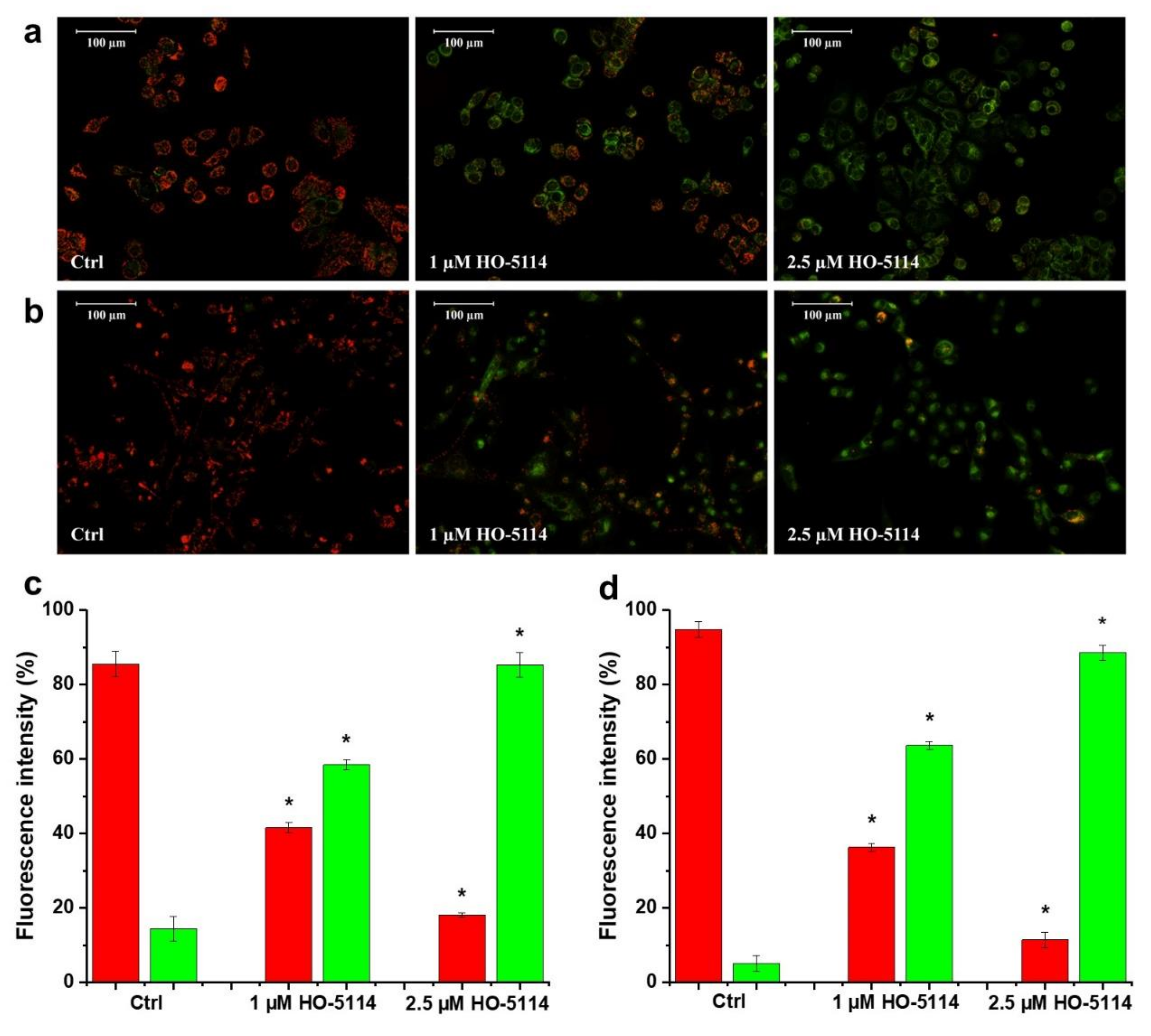
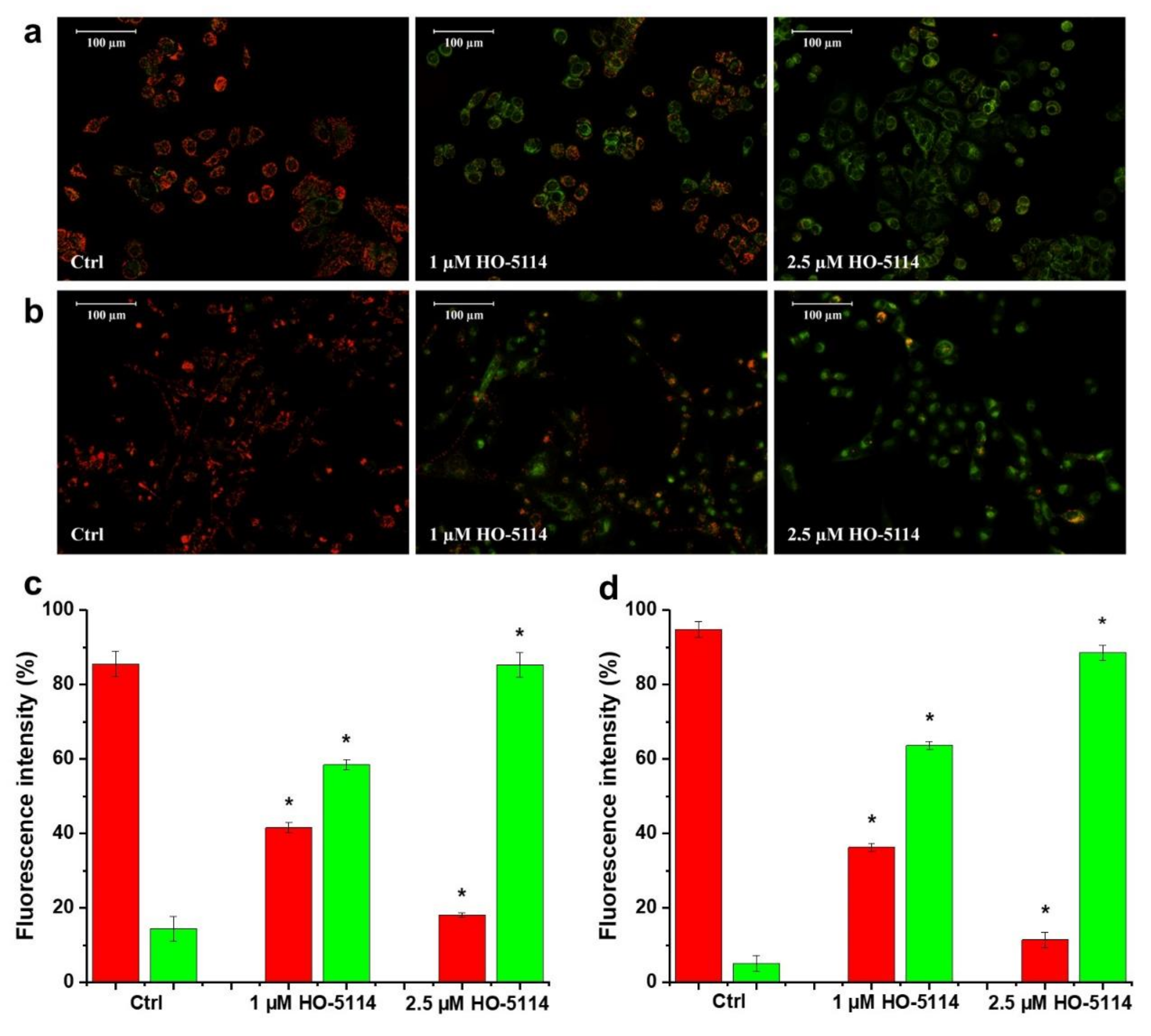
2.4. Effect of HO-5114 on ΔΨm
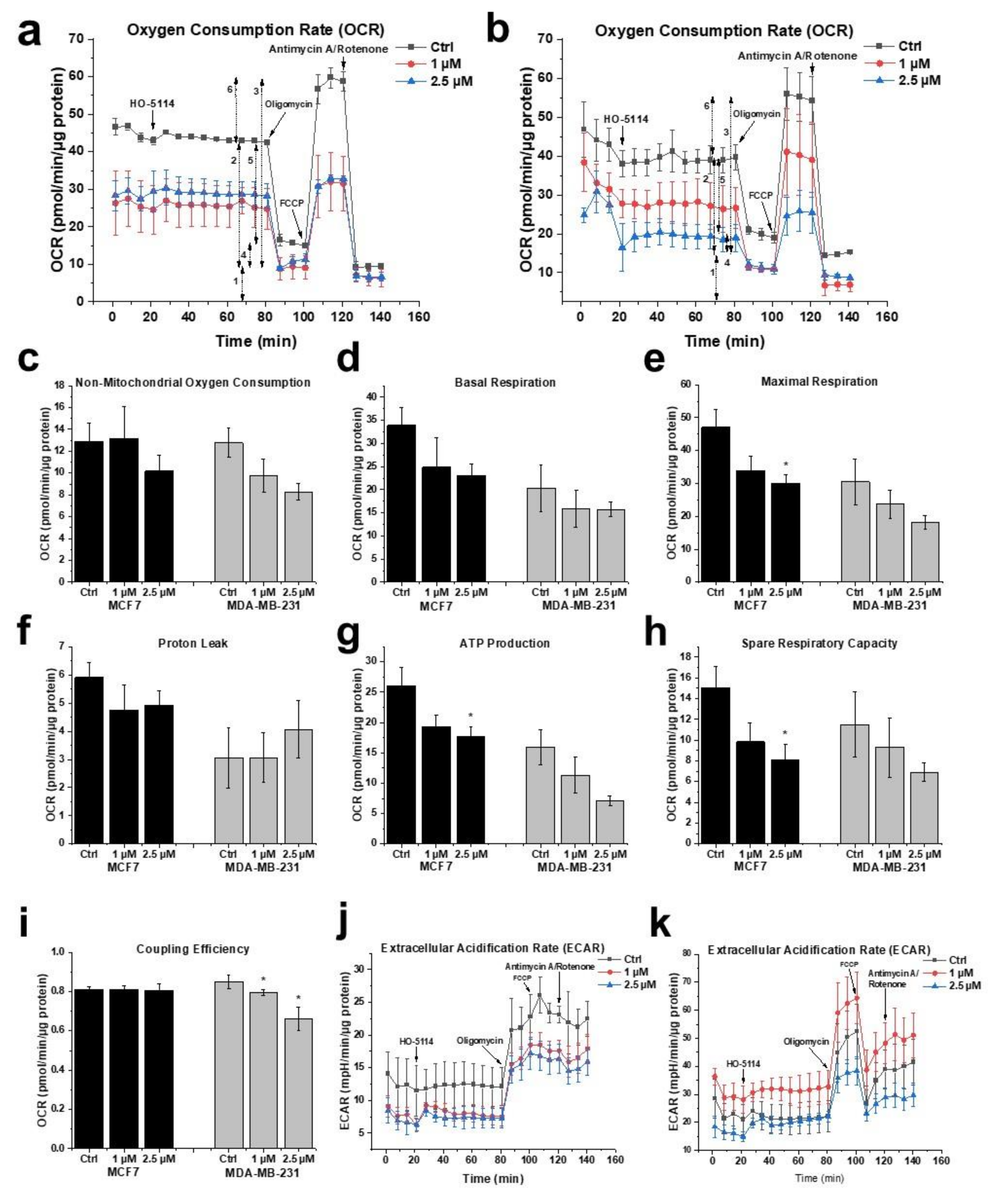
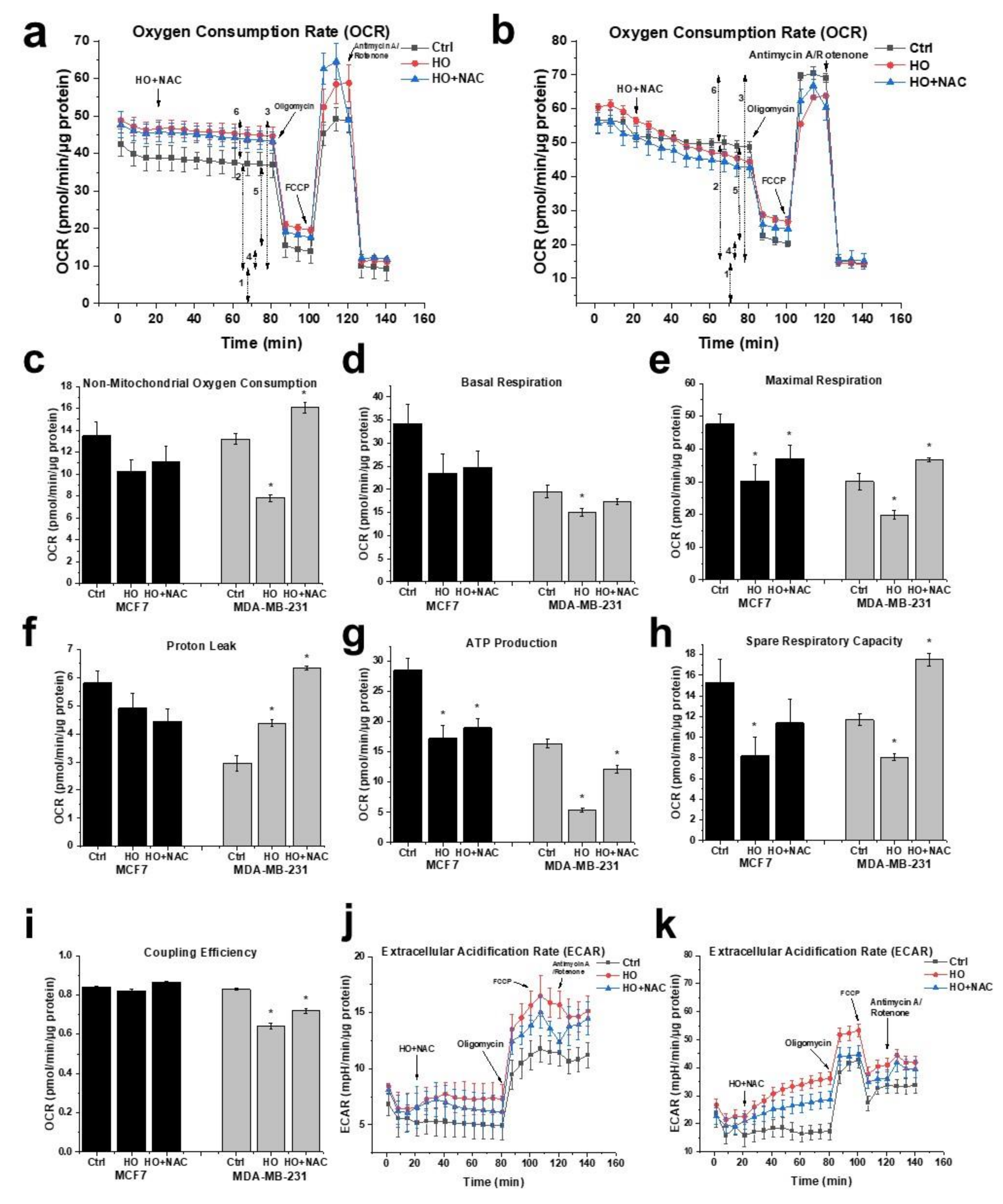
2.5. Effect of HO-5114 on Mitochondrial Energy Production
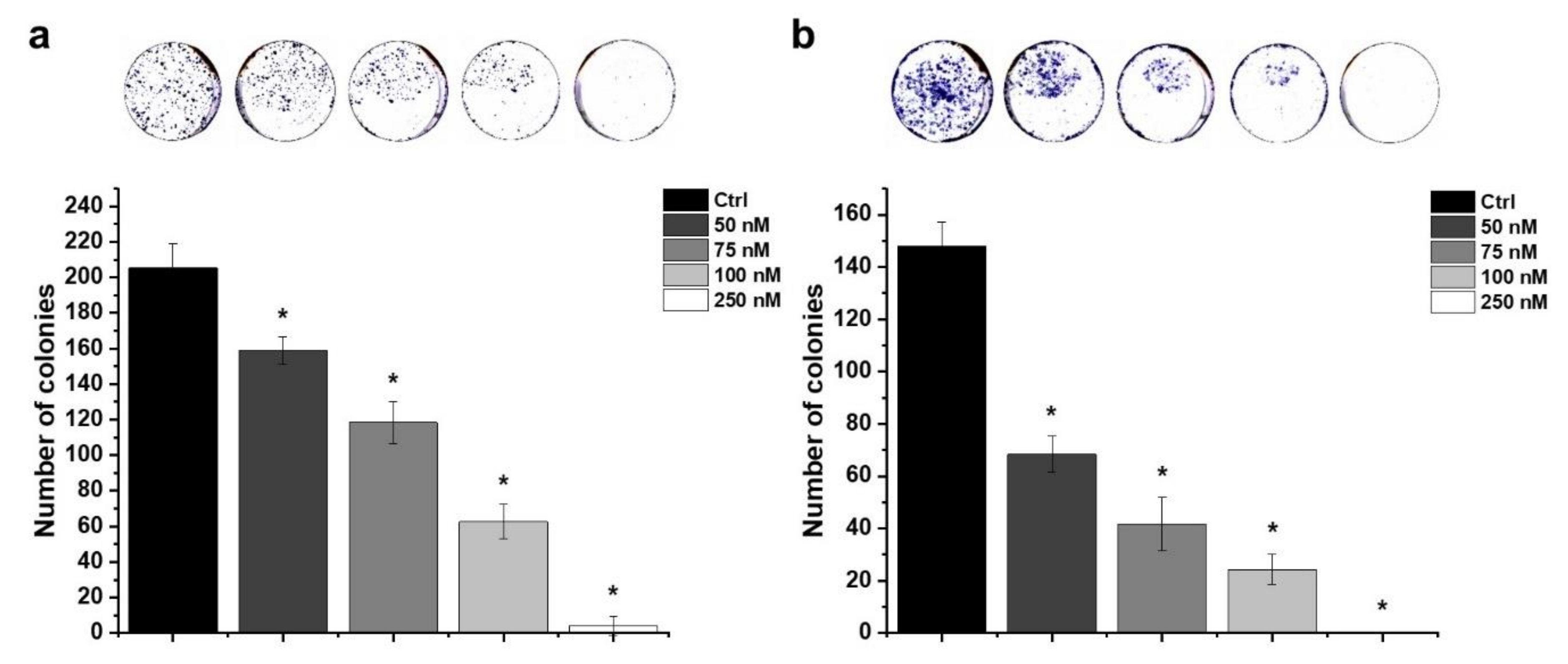
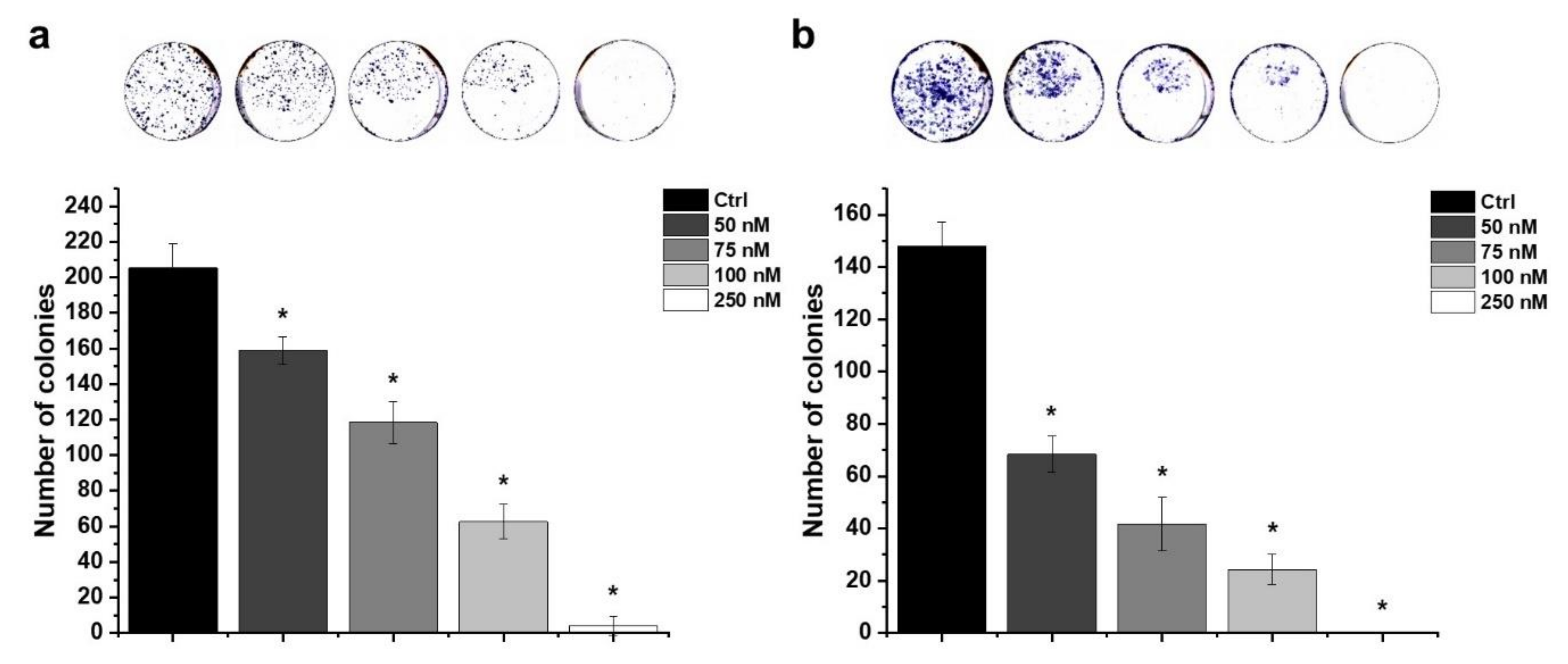
2.6. Effect of HO-5114 on Colony Formation
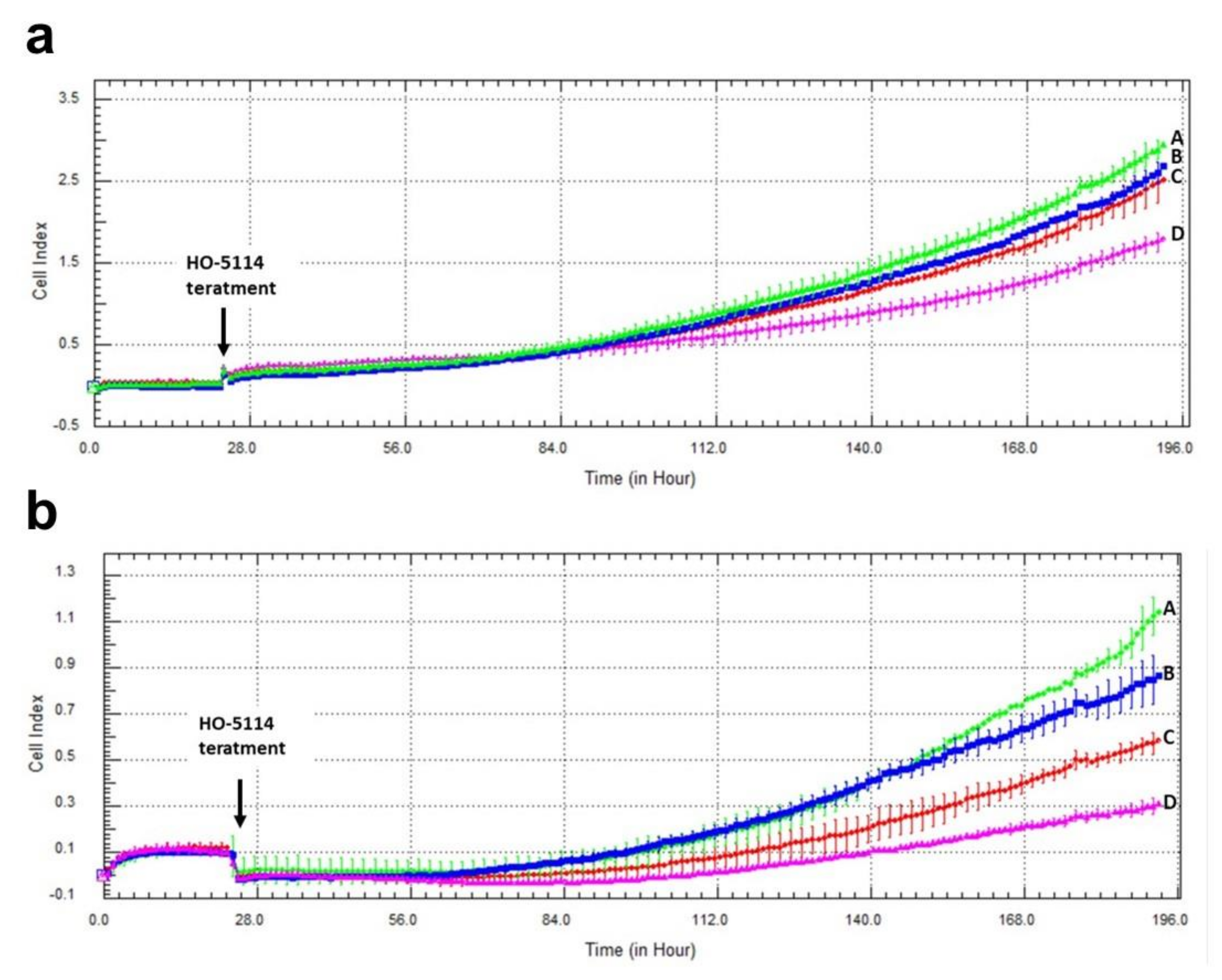
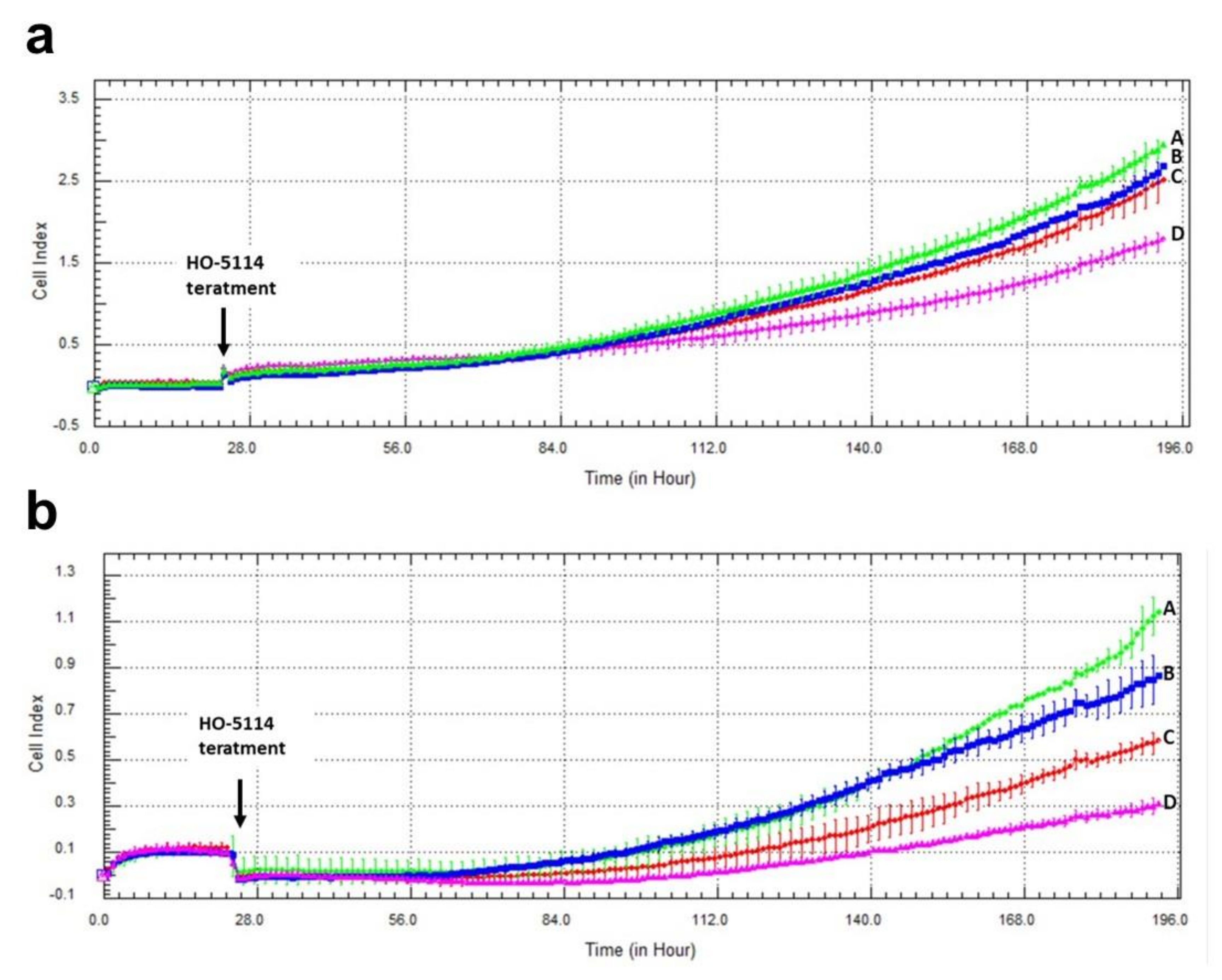
2.7. Effect of HO-5114 on Invasive Growth
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Cultures
4.3. Viability Assay
4.4. Flow Cytometric Analysis of Cell Death
4.5. Measurement of ROS Production
4.6. Measurement of Mitochondrial Bioenergetics
4.7. Measurement of Mitochondrial Membrane Potential
4.8. Colony Formation Assay
4.9. Measurement of Invasive Growth
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| ΔΨm | Mitochondrial membrane potential |
| ECAR | Extracellular acidification rate |
| FBS | Fetal bovine serum |
| FCCP | Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone |
| FITC | Fluorescein isothiocyanate |
| HR+BC | Hormone receptor positive breast cancer |
| NAC | N-acetylcysteine |
| OCR | Oxygen consumption rate |
| PI | Propidium iodide |
| R + AmA | Rotenone and antimycin A |
| ROS | Reactive oxygen species |
| SD | Standard deviation |
| SEM | Standard error of the mean |
| SRB | Sulforhodamine B |
| TCA | Trichloroacetic acid |
| TNBC | Triple-negative breast cancer |
| TPP | Tryphenylphosphonium |
References
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans Revisited: Mitochondrial Targeting as Efficient Anti-Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 7941. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Neuzil, J. Targeting mitochondria as an anticancer strategy. Cancer Commun. 2019, 39, 63. [Google Scholar] [CrossRef] [Green Version]
- Battogtokh, G.; Cho, Y.Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondrial-Targeting Anticancer Agent Conjugates and Nanocarrier Systems for Cancer Treatment. Front. Pharmacol. 2018, 9, 922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997, 15, 326–330. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Porteous, C.M.; Logan, A.; Evans, C.; Ledgerwood, E.C.; Menon, D.K.; Aigbirhio, F.; Smith, R.A.; Murphy, M.P. Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: Implications for mitochondria-specific therapies and probes. Biochim. Biophys. Acta 2010, 1800, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef]
- Reda, A.; Refaat, A.; Abd-Rabou, A.A.; Mahmoud, A.M.; Adel, M.; Sabet, S.; Ali, S.S. Role of mitochondria in rescuing glycolytically inhibited subpopulation of triple negative but not hormone-responsive breast cancer cells. Sci. Rep. 2019, 9, 13748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer 2016, 8, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Dhanasekaran, A.; Kotamraju, S.; Karunakaran, C.; Kalivendi, S.V.; Thomas, S.; Joseph, J.; Kalyanaraman, B. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: Role of mitochondrial superoxide. Free Radic. Biol. Med. 2005, 39, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, J.; Dranka, B.P.; McAllister, D.; Mackinnon, A.C., Jr.; Joseph, J.; Kalyanaraman, B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012, 72, 2634–2644. [Google Scholar] [CrossRef] [Green Version]
- Boyle, K.A.; Van Wickle, J.; Hill, R.B.; Marchese, A.; Kalyanaraman, B.; Dwinell, M.B. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J. Biol. Chem. 2018, 293, 14891–14904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isbera, M.; Bognár, B.; Gallyas, F.; Bényei, A.; Jekő, J.; Kálai, T. Syntheses and Study of a Pyrroline Nitroxide Condensed Phospholene Oxide and a Pyrroline Nitroxide Attached Diphenylphosphine. Molecules 2021, 26, 4366. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Ghoneum, A.; Abdulfattah, A.Y.; Warren, B.O.; Shu, J.; Said, N. Redox Homeostasis and Metabolism in Cancer: A Complex Mechanism and Potential Targeted Therapeutics. Int. J. Mol. Sci. 2020, 21, 3100. [Google Scholar] [CrossRef]
- Dowling, C.M.; Herranz Ors, C.; Kiely, P.A. Using real-time impedance-based assays to monitor the effects of fibroblast-derived media on the adhesion, proliferation, migration and invasion of colon cancer cells. Biosci. Rep. 2014, 34, e00126. [Google Scholar] [CrossRef]
- Masoud, V.; Pages, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat. Commun. 2019, 10, 4862. [Google Scholar] [CrossRef] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Sarmiento-Salinas, F.L.; Delgado-Magallon, A.; Montes-Alvarado, J.B.; Ramirez-Ramirez, D.; Flores-Alonso, J.C.; Cortes-Hernandez, P.; Reyes-Leyva, J.; Herrera-Camacho, I.; Anaya-Ruiz, M.; Pelayo, R.; et al. Breast Cancer Subtypes Present a Differential Production of Reactive Oxygen Species (ROS) and Susceptibility to Antioxidant Treatment. Front. Oncol. 2019, 9, 480. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y. Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast 555 Cancer. Antioxidants 2021, 10, 169. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics—The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, P.; Chen, Y.; Xu, Y.; Swanson, R.A. Mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: A treatable cause of cell death in stroke. Transl. Stroke Res. 2014, 5, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C.; Seyfried, T.N. Mitochondrial Substrate-Level Phosphorylation as Energy Source for Glioblastoma: Review and Hypothesis. ASN Neuro 2018, 10, 1759091418818261. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C. Acute sources of mitochondrial NAD(+) during respiratory chain dysfunction. Exp. Neurol. 2020, 327, 113218. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashley, N.; Poulton, J. Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs. Biochem. Biophys. Res. Commun. 2009, 378, 450–455. [Google Scholar] [CrossRef]
- Bennett, N.K.; Nguyen, M.K.; Darch, M.A.; Nakaoka, H.J.; Cousineau, D.; Ten Hoeve, J.; Graeber, T.G.; Schuelke, M.; Maltepe, E.; Kampmann, M.; et al. Defining the ATPome reveals cross-optimization of metabolic pathways. Nat. Commun. 2020, 11, 4319. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001–1015. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.S.; Liu, L.T.; Ou, L.H.; Pan, S.C.; Lin, C.I.; Wei, Y.H. Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol. Rep. 2018, 39, 316–330. [Google Scholar] [CrossRef] [Green Version]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C.; et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox. Biol. 2019, 25, 101076. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baffy, G. Mitochondrial uncoupling in cancer cells: Liabilities and opportunities. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 655–664. [Google Scholar] [CrossRef]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Karamanou, K.; Franchi, M.; Vynios, D.; Brezillon, S. Epithelial-to-mesenchymal transition and invadopodia markers in breast cancer: Lumican a key regulator. Semin. Cancer Biol. 2020, 62, 125–133. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreidesz, K.; Szabo, A.; Kovacs, D.; Koszegi, B.; Bagone Vantus, V.; Vamos, E.; Isbera, M.; Kalai, T.; Bognar, Z.; Kovacs, K.; et al. Cytostatic Effect of a Novel Mitochondria-Targeted Pyrroline Nitroxide in Human Breast Cancer Lines. Int. J. Mol. Sci. 2021, 22, 9016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22169016
Andreidesz K, Szabo A, Kovacs D, Koszegi B, Bagone Vantus V, Vamos E, Isbera M, Kalai T, Bognar Z, Kovacs K, et al. Cytostatic Effect of a Novel Mitochondria-Targeted Pyrroline Nitroxide in Human Breast Cancer Lines. International Journal of Molecular Sciences. 2021; 22(16):9016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22169016
Chicago/Turabian StyleAndreidesz, Kitti, Aliz Szabo, Dominika Kovacs, Balazs Koszegi, Viola Bagone Vantus, Eszter Vamos, Mostafa Isbera, Tamas Kalai, Zita Bognar, Krisztina Kovacs, and et al. 2021. "Cytostatic Effect of a Novel Mitochondria-Targeted Pyrroline Nitroxide in Human Breast Cancer Lines" International Journal of Molecular Sciences 22, no. 16: 9016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22169016