
Decreased Interactions between Calmodulin and a Mutant Huntingtin Model Might Reduce the Cytotoxic Level of Intracellular Ca2+: A Molecular Dynamics Study


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Molecular Docking and Binding Free Energy Calculations
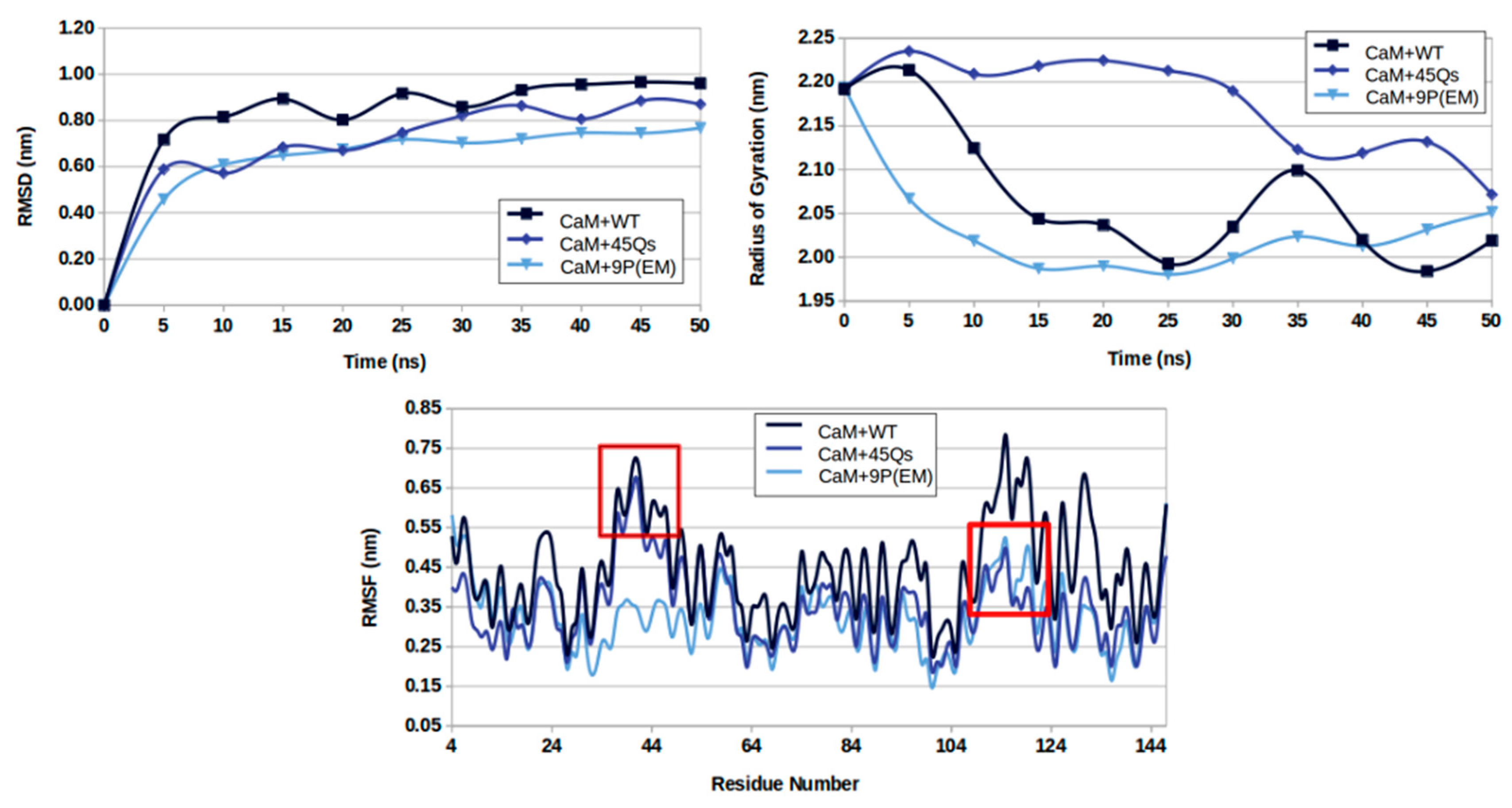
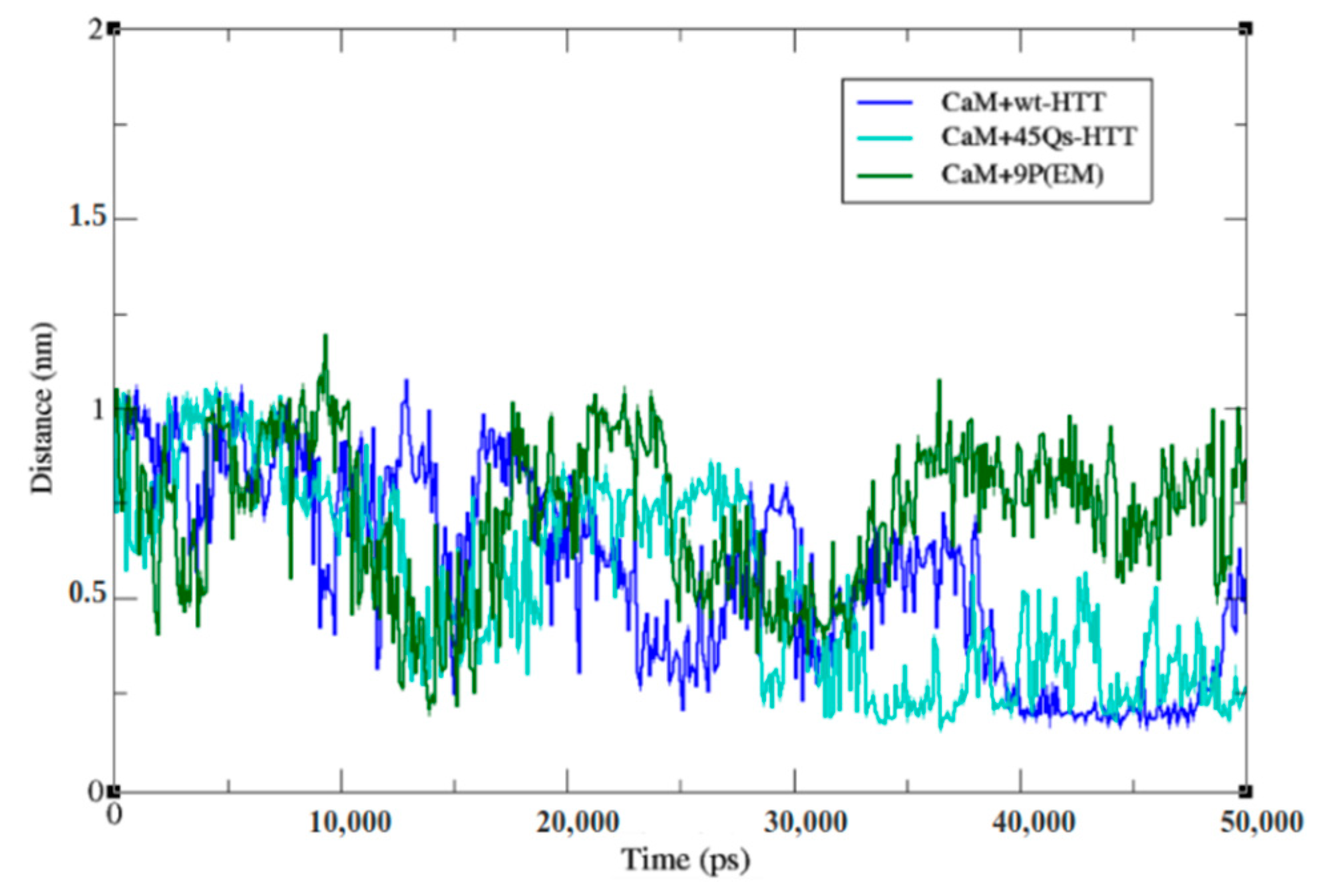
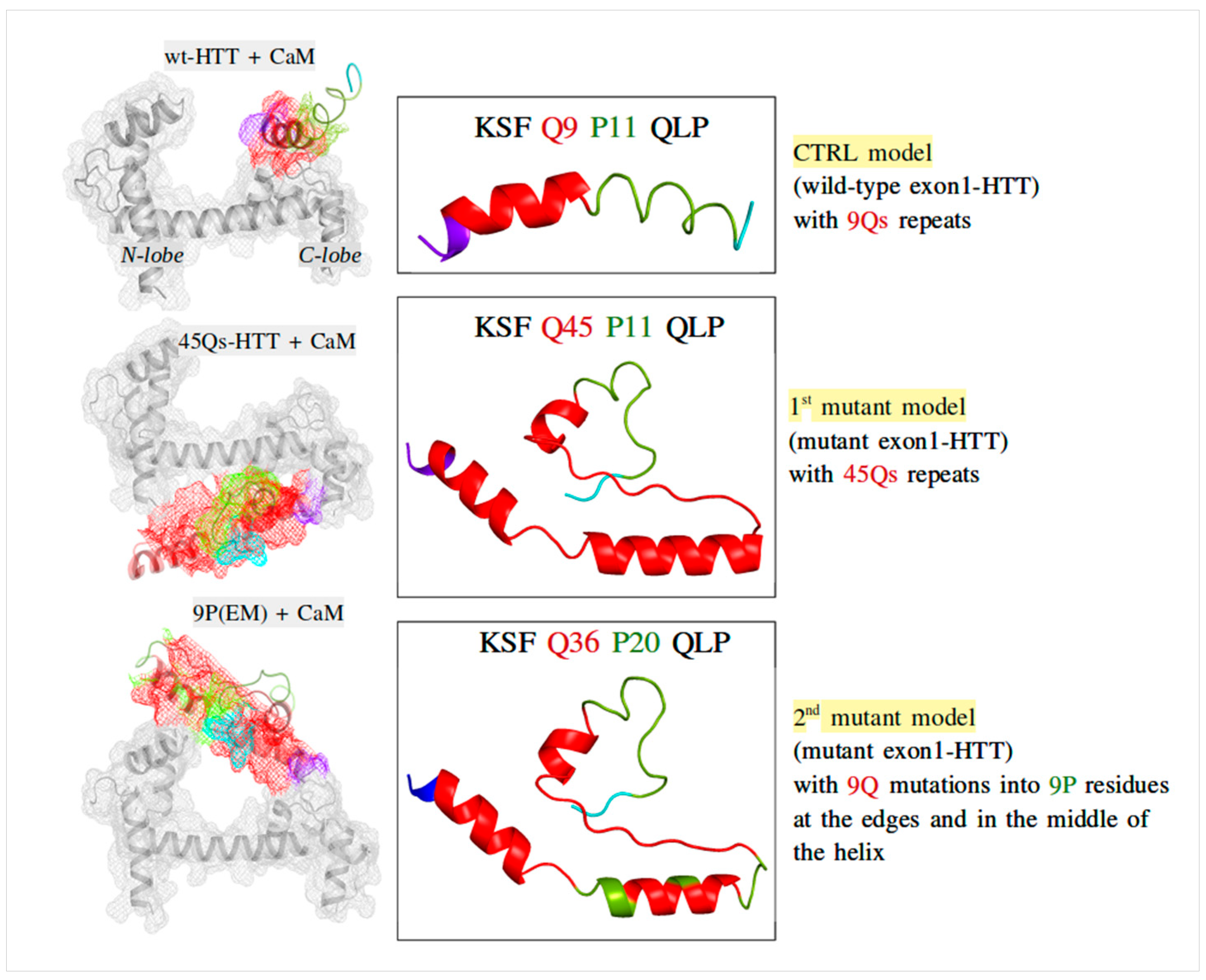
2.2. CaM and wt/m-HTT Interacting Complexes
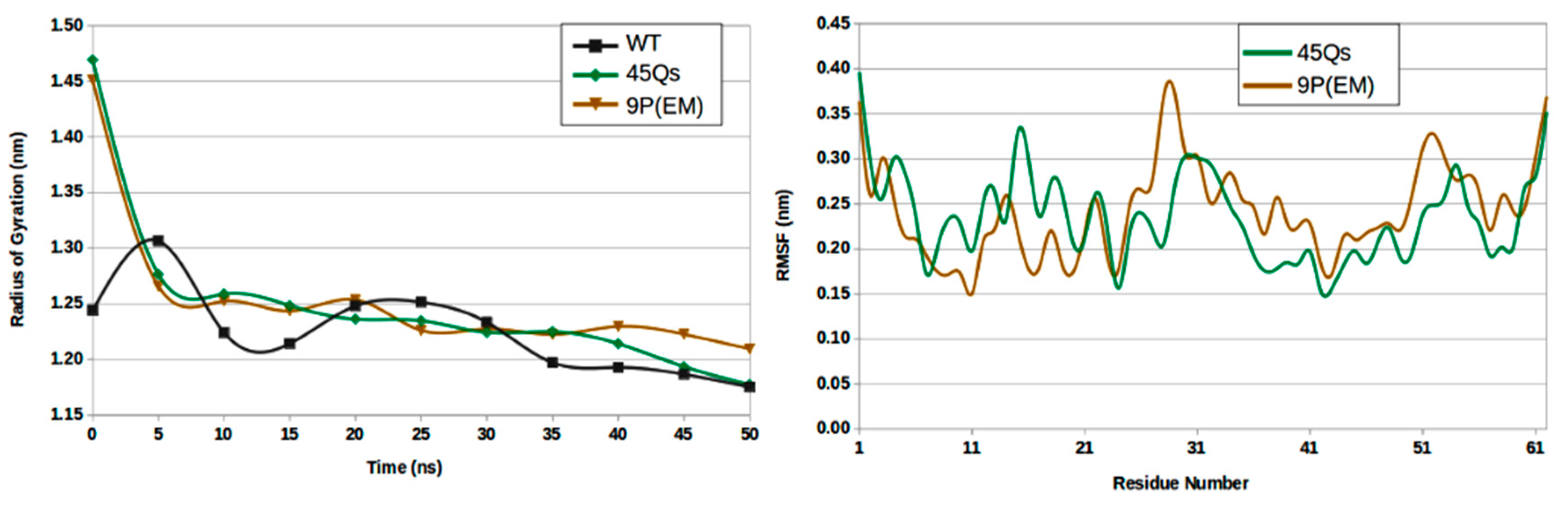
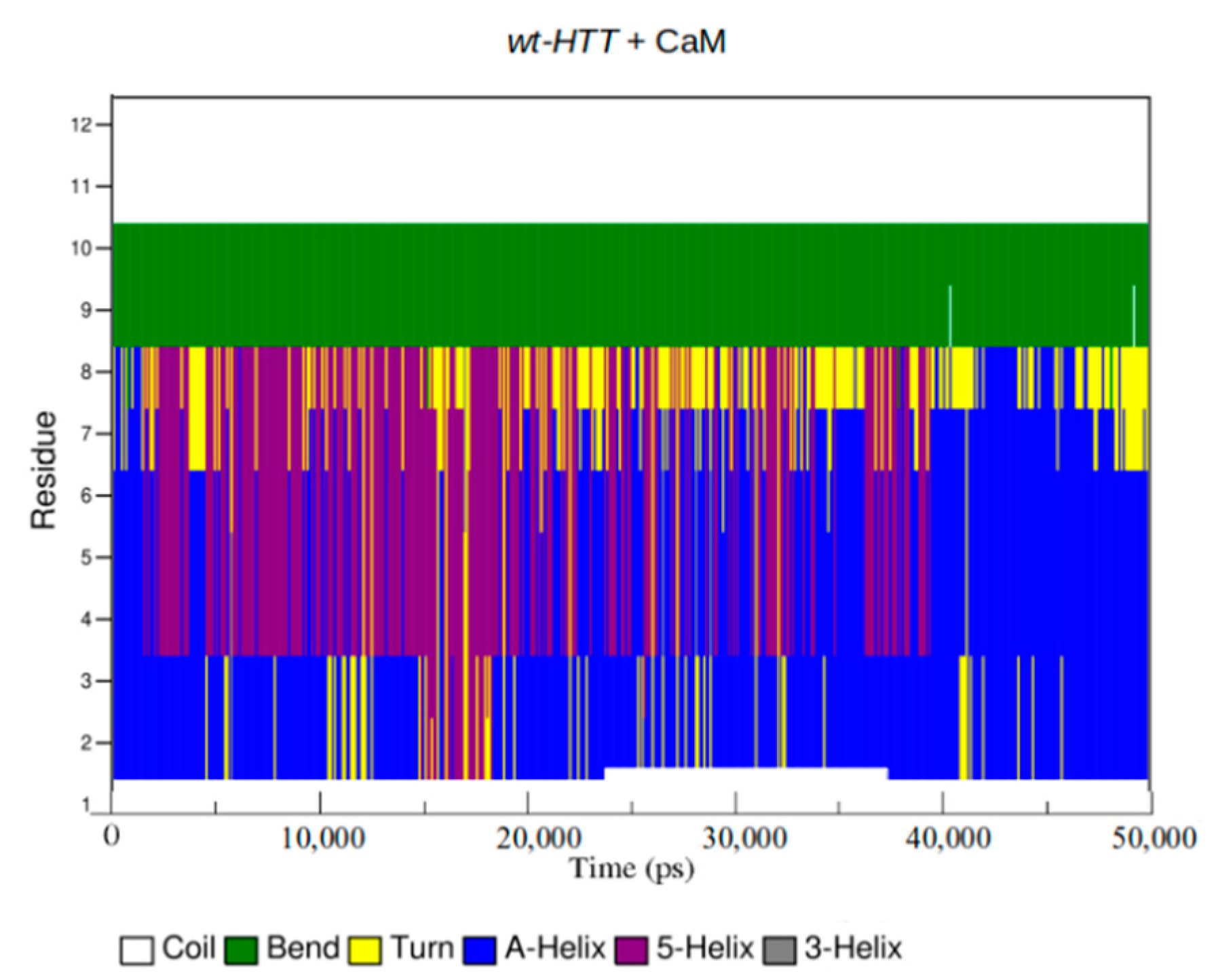
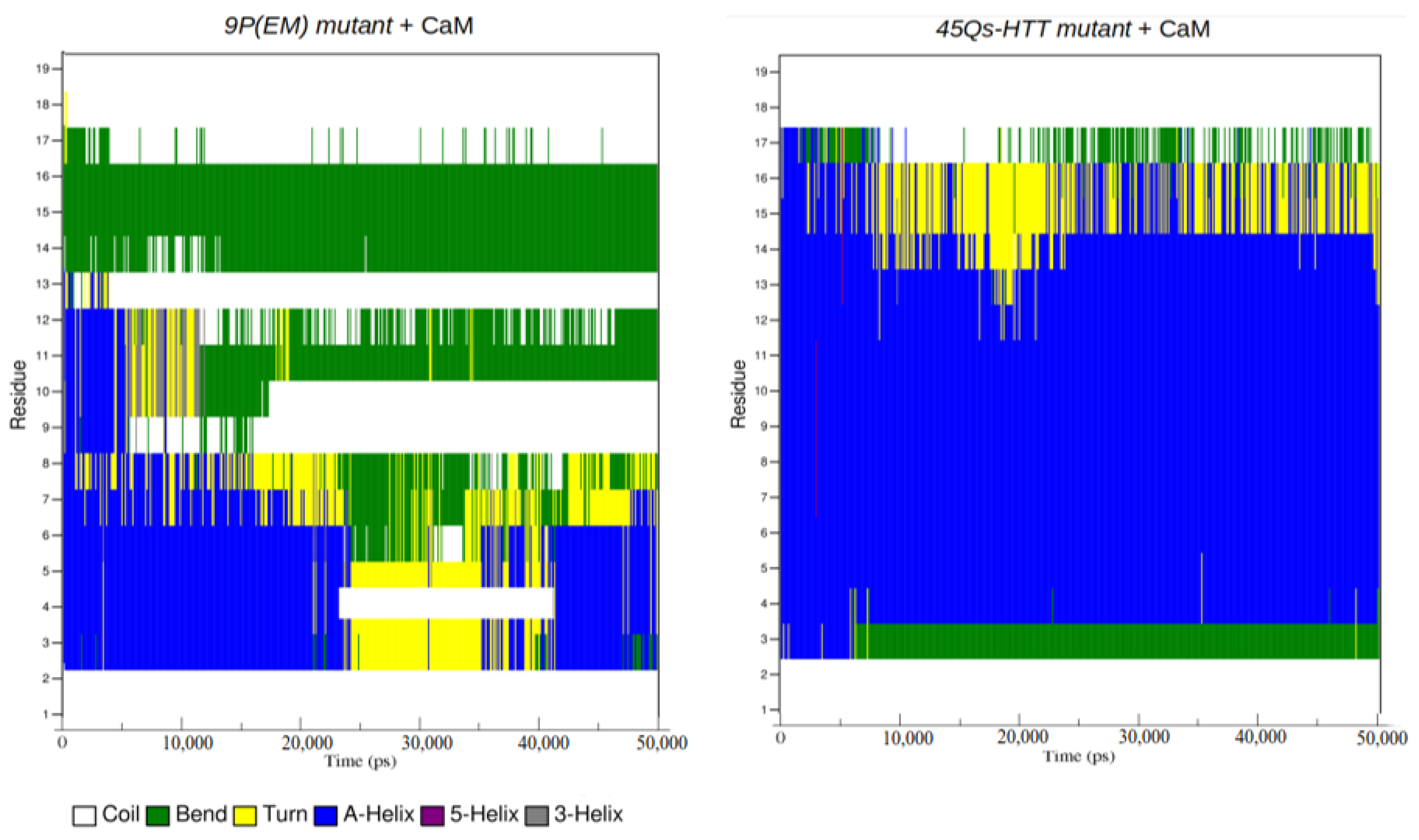
2.3. Structural Changes of wt-HTT, 45Qs-HTT, and 9P(EM) Models
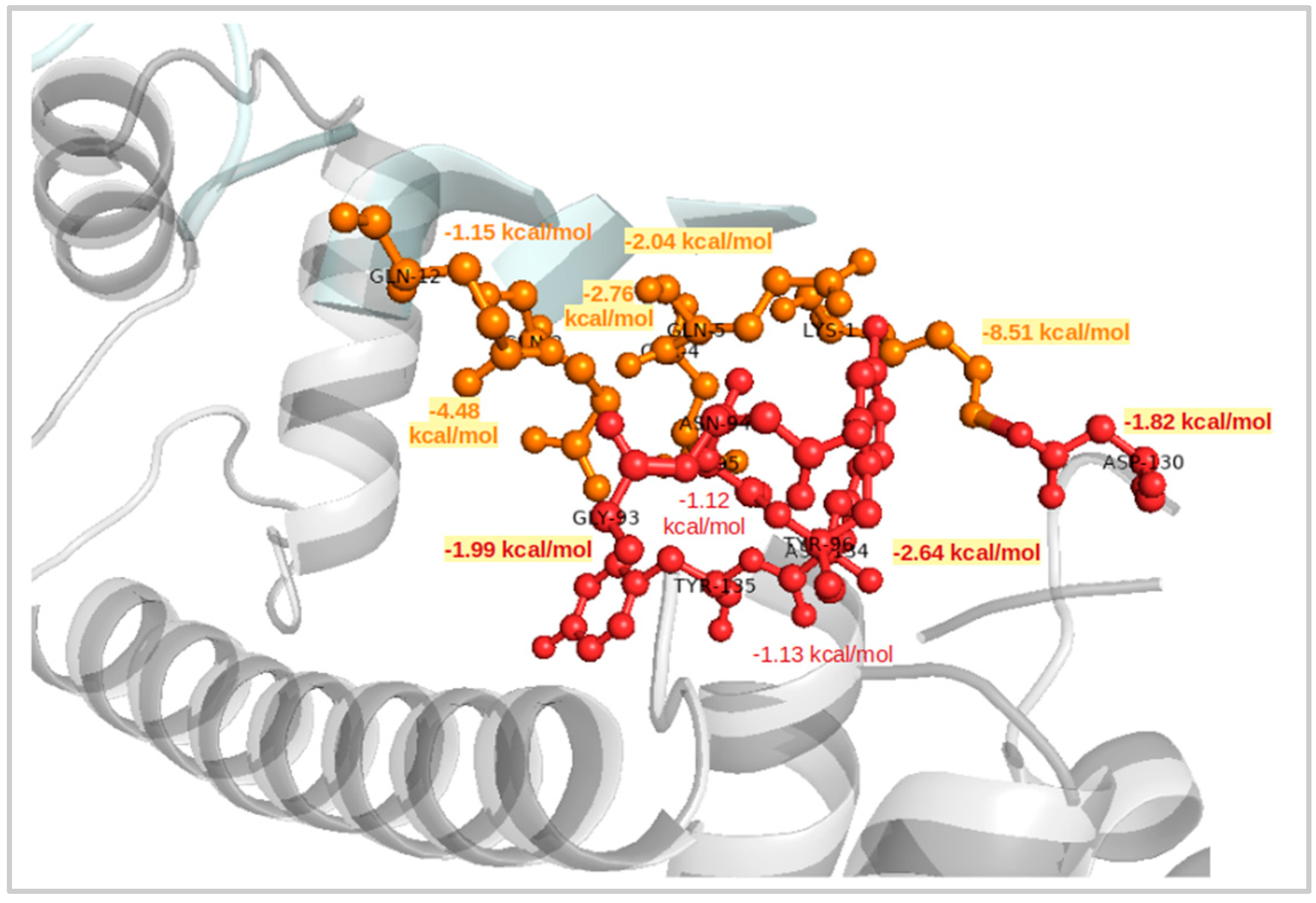
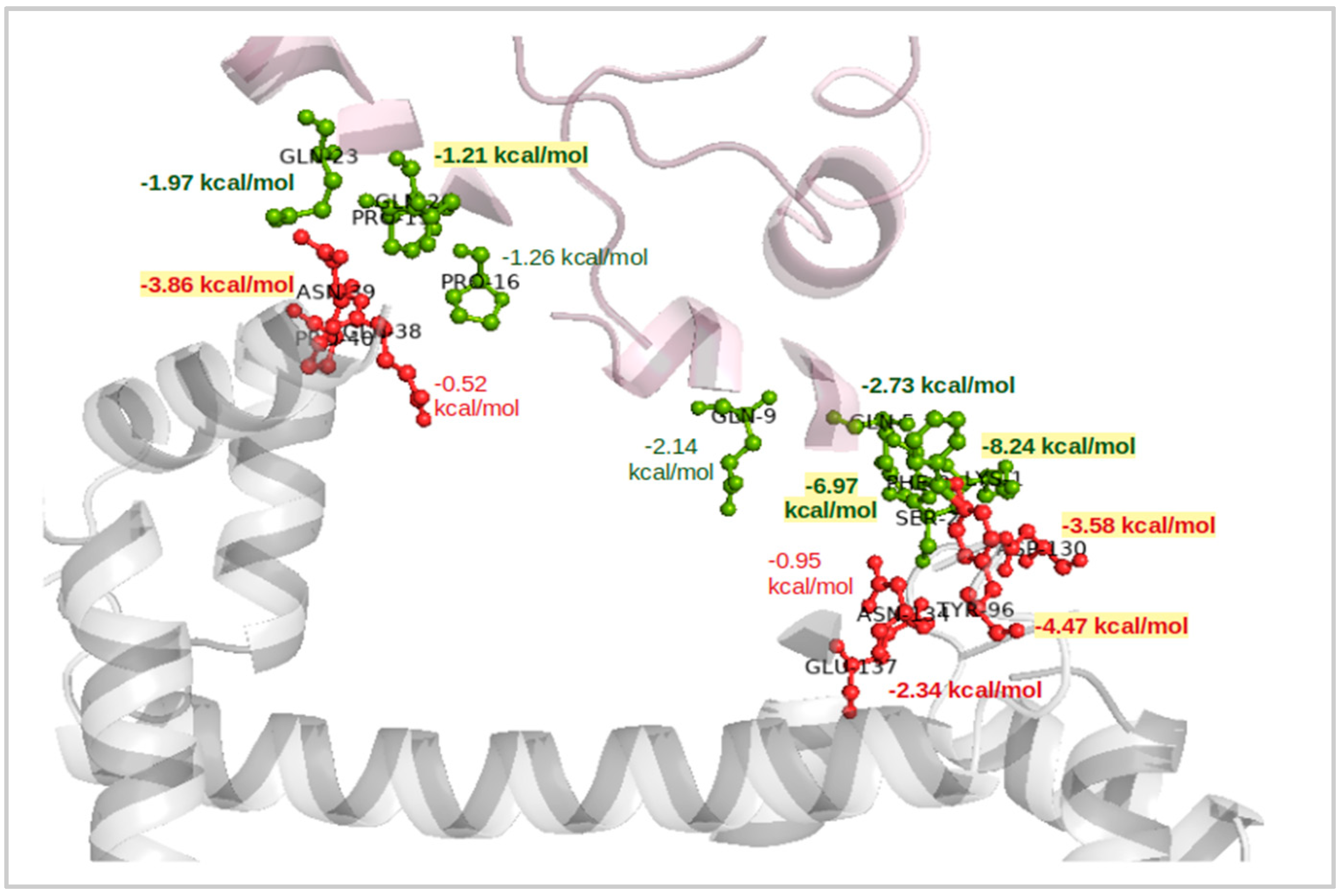
2.4. Interaction Patterns for CaM-HTT Complexes
3. Materials and Methods
3.1. Preparation of CaM, HTT, and CaM–HTT Complexes
3.2. Molecular Dynamics of CaM–HTT Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vijayvargia, R.; Epand, R.; Leitner, A.; Jung, T.Y.; Shin, B.; Jung, R.; Lloret, A.; Atwal, R.S.; Lee, H.; Lee, J.-M.; et al. Huntingtin’s spherical solenoid structure enables polyglutamine tract-dependent modulation of its structure and function. eLife 2016, 5, e11184. [Google Scholar] [CrossRef] [PubMed]
- Harjes, P.; Wanker, E.E. The hunt for huntingtin function: Interaction partners tell many different stories. Trends Biochem. Sci. 2003, 28, 425–433. [Google Scholar] [CrossRef]
- Ananbeh, H.; Vodicka, P.; Skalnikova, H.K. Emerging Roles of Exosomes in Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 4085. [Google Scholar] [CrossRef]
- De Luca, A.; Morella, A.; Consoli, F.; Fanelli, S.; Thibert, J.R.; Statt, S.; Latham, G.J.; Squitieri, F. A Novel Triplet-Primed PCR Assay to Detect the Full Range of Trinucleotide CAG Repeats in the Huntingtin Gene (HTT). Int. J. Mol. Sci. 2021, 22, 1689. [Google Scholar] [CrossRef]
- Lee, J.-M.; Wheeler, V.C.; Chao, M.J.; Vonsattel, J.P.G.; Pinto, R.M.; Lucente, D.; Abu-Elneel, K.; Ramos, E.M.; Mysore, J.S.; Gillis, T.; et al. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadri-Vakili, G.; Cha, J.-H.J. Mechanisms of Disease: Histone modifications in Huntington’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 330–338. [Google Scholar] [CrossRef]
- Giacomello, M.; Oliveros, J.C.; Naranjo, J.; Carafoli, E. Neuronal Ca2+ dyshomeostasis in Huntington disease. Prion 2013, 7, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.Z.; Cattaneo, E.; et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 2004, 304, 100. [Google Scholar] [CrossRef] [Green Version]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pa-thology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar]
- Atwal, R.S.; Desmond, C.R.; Caron, N.; Maiuri, T.; Xia, J.; Sipione, S.; Truant, R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 2011, 7, 453–460. [Google Scholar] [CrossRef]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vicente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef]
- Veliera, J.; Kima, M.; Schwarza, C.; Kim, T.W.; Sappa, E.; Chaseb, K.; Aroninb, N.; Di Figliaa, M. Wild-Type and Mutant Huntingtins Function in Vesicle Trafficking in the Secretory and Endocytic Pathways. Exp. Neurol. 1998, 152, 34–40. [Google Scholar] [CrossRef]
- Glaser, T.; Shimojo, H.; Ribeiro, D.E.; Martins, P.P.L.; Beco, R.P.; Kosinski, M.; Sampaio, V.F.A.; Corrêa-Velloso, J.; Oliveira-Giacomelli, A.; Lameu, C.; et al. ATP and spontaneous calcium oscillations control neural stem cell fate determination in Huntington’s disease: A novel approach for cell clock research. Mol. Psychiatry 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk, M.; Glaser, T.; Ulrich, H.; Slominska, E.M.; Smolenski, R.T. Huntingtin protein maintains balanced energetics in mouse cardiomyocytes. Nucleosides Nucleotides Nucleic Acids 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.B. Calcium Signaling and Neurodegeneration. Acta Nat. 2010, 2, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Toescu, E.C.; Verkhratsky, A. The importance of being subtle: Small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 2007, 6, 267–273. [Google Scholar] [CrossRef]
- Cheung, W.Y. Role of Calmodulin in Brain Function. Prog. Brain Res. 1982, 56, 237–253. [Google Scholar] [CrossRef]
- Dudek, N.L.; Dai, Y.; Muma, N.A. Protective Effects of Interrupting the Binding of Calmodulin to Mutant Huntingtin. J. Neuropathol. Exp. Neurol. 2008, 67, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Kekenes-Huskey, P.M. Assessing the Role of Calmodulin’s Linker Flexibility in Target Binding. Int. J. Mol. Sci. 2021, 22, 4990. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.G.; Agopyan, N.; Gutekunst, C.-A.; Leavitt, B.; LePiane, F.; Singaraja, R.; Smith, D.J.; Bissada, N.; McCutcheon, K.; Nasir, J.; et al. A YAC Mouse Model for Huntington’s Disease with Full-Length Mutant Huntingtin, Cytoplasmic Toxicity, and Selective Striatal Neurodegeneration. Neuron 1999, 23, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Kolobkova, Y.A.; Vigont, V.A.; Shalygin, A.V.; Kaznacheyeva, E.V. Huntington’s Disease: Calcium Dyshomeostasis and Pathology Models. Acta Nat. 2017, 9, 34–46. [Google Scholar] [CrossRef]
- Bao, J.; Sharp, A.H.; Wagster, M.V.; Becher, M.; Schilling, G.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Expansion of polyglutamine repeat in huntingtin leads to abnormal protein interactions involving calmodulin. Proc. Natl. Acad. Sci. USA 1996, 93, 5037–5042. [Google Scholar] [CrossRef] [Green Version]
- Kreutz, M.R.; Naranjo, J.R.; Ekoch, K.-W.; Eschwaller, B. The Neuronal Functions of EF-Hand Ca2+-Binding Proteins. Front. Mol. Neurosci. 2012, 5, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, F.M.; Garcia-Arencibia, M.; Imarisio, S.; O’Sullivan, N.C.; Ricketts, T.H.; Kent, B.A.; Rao, M.V.; Lam, W.; Green-Thompson, Z.W.; Nixon, R.A.; et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 2014, 22, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthi-Carter, R.; Strand, A.D.; Hanson, S.A.; Kooperberg, C.; Schilling, G.; La Spada, A.R.; Merry, D.E.; Young, A.B.; Ross, C.A.; Borchelt, D.R.; et al. Polyglutamine and transcription: Gene expression changes shared by DRPLA and Huntington’s disease mouse models reveal context-independent effects. Hum. Mol. Genet. 2002, 11, 1927–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef]
- Kuhn, A.; Goldstein, D.R.; Hodges, A.; Strand, A.D.; Sengstag, T.; Kooperberg, C.; Becanovic, K.; Pouladi, M.A.; Sathasivam, K.; Cha, J.-H.J.; et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 2007, 16, 1845–1861. [Google Scholar] [CrossRef] [Green Version]
- Czeredys, M.; Gruszczynska-Biegala, J.; Schacht, T.; Methner, A.; Kuznicki, J. Expression of genes encoding the calcium signalosome in cellular and transgenic models of Huntington’s disease. Front. Mol. Neurosci. 2013, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Zainelli, G.M.; Ross, C.A.; Troncoso, J.C.; Fitzgerald, J.K.; Muma, N.A. Calmodulin Regulates Transglutaminase 2 Cross-Linking of Huntingtin. J. Neurosci. 2004, 24, 1954–1961. [Google Scholar] [CrossRef]
- Delong, M.R.; Wichmann, T. Circuits and Circuit Disorders of the Basal Ganglia. Arch. Neurol. 2007, 64, 20–24. [Google Scholar] [CrossRef]
- Panchal, K.; Tiwari, A.K. Mitochondrial dynamics, a key executioner in neurodegenerative diseases. Mitochondrion 2018, 47, 151–173. [Google Scholar] [CrossRef]
- Intihar, T.A.; Martinez, E.A.; Gomez-Pastor, R. Mitochondrial Dysfunction in Huntington’s Disease; Interplay Between HSF1, p53 and PGC-1α Transcription Factors. Front. Cell. Neurosci. 2019, 13, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joviano-Santos, J.V.; Santos-Miranda, A.; Botelho, A.F.M.; De Jesus, I.C.G.; Andrade, J.N.; Barreto, T.D.O.; Magalhães-Gomes, M.P.S.; Valadão, P.A.C.; Cruz, J.; Melo, M.M.; et al. Increased oxidative stress and CaMKIIactivity contribute to electro-mechanical defects in cardiomyocytes from a murine model of Huntington’s disease. FEBS J. 2018, 286, 110–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dridi, H.; Liu, X.; Yuan, Q.; Reiken, S.; Yehia, M.; Sittenfeld, L.; Apostolou, P.; Buron, J.; Sicard, P.; Matecki, S.; et al. Role of defective calcium regulation in cardiorespiratory dysfunction in Huntington’s disease. JCI Insight 2020, 5, e140614. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Ikura, M. Calmodulin in Action: Diversity in Target Recognition and Activation Mechanisms. Cell 2002, 108, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Yamniuk, A.P.; Vogel, H.J. Calmodulin’s Flexibility Allows for Promiscuity in Its Interactions with Target Proteins and Peptides. Mol. Biotechnol. 2004, 27, 33–58. [Google Scholar] [CrossRef]
- Shepherd, C.M.; Vogel, H.J. A Molecular Dynamics Study of Ca2+-Calmodulin: Evidence of Interdomain Coupling and Structural Collapse on the Nanosecond Timescale. Biophys. J. 2004, 87, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.A.; Brunger, A. The 1.0 Å crystal structure of Ca2+-bound calmodulin: An analysis of disorder and implications for functionally relevant plasticity. J. Mol. Biol. 2000, 301, 1237–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faga, L.A.; Sorensen, B.R.; VanScyoc, W.S.; Shea, M.A. Basic interdomain boundary residues in calmodulin decrease calcium affinity of sites I and II by stabilizing helix-helix interactions. Proteins Struct. Funct. Bioinform. 2003, 50, 381–391. [Google Scholar] [CrossRef]
- Chou, J.J.; Li, S.; Klee, C.B.; Bax, A. Solution structure of Ca(2+)-calmodulin reveals flexible hand-like properties of its domains. Nat. Genet. 2001, 8, 990–997. [Google Scholar] [CrossRef]
- Yuan, T.; Walsh, M.P.; Sutherland, C.; Fabian, A.H.; Vogel, H.J. Calcium-Dependent and -Independent Interactions of the Calmodulin-Binding Domain of Cyclic Nucleotide Phosphodiesterase with Calmodulin. Biochemistry 1999, 38, 1446–1455. [Google Scholar] [CrossRef]
- Garrido, E.; Jaumot, M.; Agell, N.; Granadino-Roldán, J.M.; Rubio-Martinez, J. Toward understanding calmodulin plasticity by molecular dynamics. Futur. Med. Chem. 2019, 11, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Jurado, L.A.; Chockalingam, P.S.; Jarrett, H.W. Apocalmodulin. Physiol. Rev. 1999, 79, 661–682. [Google Scholar] [CrossRef]
- Evenäs, J.; Malmendal, A.; Akke, M. Dynamics of the Transition between Open and Closed Conformations in a Calmodulin C-Terminal Domain Mutant. Structure 2001, 9, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Celio, M.R.; Pauls, T.; Schwaller, B. Guidebook to the Calcium-Binding Proteins; Oxford University Press: Oxford, UK, 1996. [Google Scholar]
- Vogel, H.J. Calmodulin: A versatile calcium mediator protein. Biochem. Cell Biol. 1994, 72, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Ikura, M.; Clore, G.M.; Gronenborn, A.M.; Zhu, G.; Klee, C.B.; Bax, A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 1992, 256, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Moldovean, S.N.; Chiş, V. Specific Key-Point Mutations along the Helical Conformation of Huntingtin-Exon 1 Protein Might Have an Antagonistic Effect on the Toxic Helical Content’s Formation. ACS Chem. Neurosci. 2020, 11, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Milner-White, E.J.; Ross, B.M.; Ismail, R.; Belhadj-Mostefa, K.; Poet, R. One type of gamma turn, rather than the other, gives rise to chain reversal in proteins. J. Mol. Biol. 1983, 204, 777–782. [Google Scholar] [CrossRef]
- Hsu, W.-L.; Oldfield, C.J.; Xue, B.; Meng, J.; Huang, F.; Romero, P.; Uversky, V.N.; Dunker, A.K. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2012, 22, 258–273. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Billeter, S.R.; Eking, A.A.; Hiinenberger, P.H.; Kriiger, P.; Mark, A.E.; Scott, W.R.P.; Tironi, I.G. Biomolecular Simulation, The GROMOS96 Manual and User Guide; VdfHochschulverlag AG an der ETH Zürich: Zürich, Switzerland, 1996; pp. 1–1042. [Google Scholar]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Chattopadhyaya, R.; Meador, W.E.; Means, A.R.; Quiocho, F.A. Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 1992, 228, 1177–1192. [Google Scholar] [CrossRef]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Zacharias, M. Protein-protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci. 2003, 12, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Chen, F.; Kang, Y.; Sun, H.; Liu, H.; Li, D.; Zhu, F.; Hou, T. HawkRank: A new scoring function for protein–protein docking based on weighted energy terms. J. Cheminform. 2017, 9, 66–66:15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Hermans, J. Intermolecular Forces, Interaction models for water in relation to protein hydration. In The Jerusalem Symposia on Quantum Chemistry and Biochemistry; Pullman, B., Ed.; D. Reidel Publishing Company: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Darden, T.A.; York, D.M.; Pedersen, L.G. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moldovean, S.N.; Chiş, V. Decreased Interactions between Calmodulin and a Mutant Huntingtin Model Might Reduce the Cytotoxic Level of Intracellular Ca2+: A Molecular Dynamics Study. Int. J. Mol. Sci. 2021, 22, 9025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22169025
Moldovean SN, Chiş V. Decreased Interactions between Calmodulin and a Mutant Huntingtin Model Might Reduce the Cytotoxic Level of Intracellular Ca2+: A Molecular Dynamics Study. International Journal of Molecular Sciences. 2021; 22(16):9025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22169025
Chicago/Turabian StyleMoldovean, Sanda Nastasia, and Vasile Chiş. 2021. "Decreased Interactions between Calmodulin and a Mutant Huntingtin Model Might Reduce the Cytotoxic Level of Intracellular Ca2+: A Molecular Dynamics Study" International Journal of Molecular Sciences 22, no. 16: 9025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22169025