
Exercise, but Not Metformin Prevents Loss of Muscle Function Due to Doxorubicin in Mice Using an In Situ Method

 ,
,
Abstract
:1. Introduction
2. Results
2.1. DOX Treatment Causes Severe Body Weight Loss at 3 Days
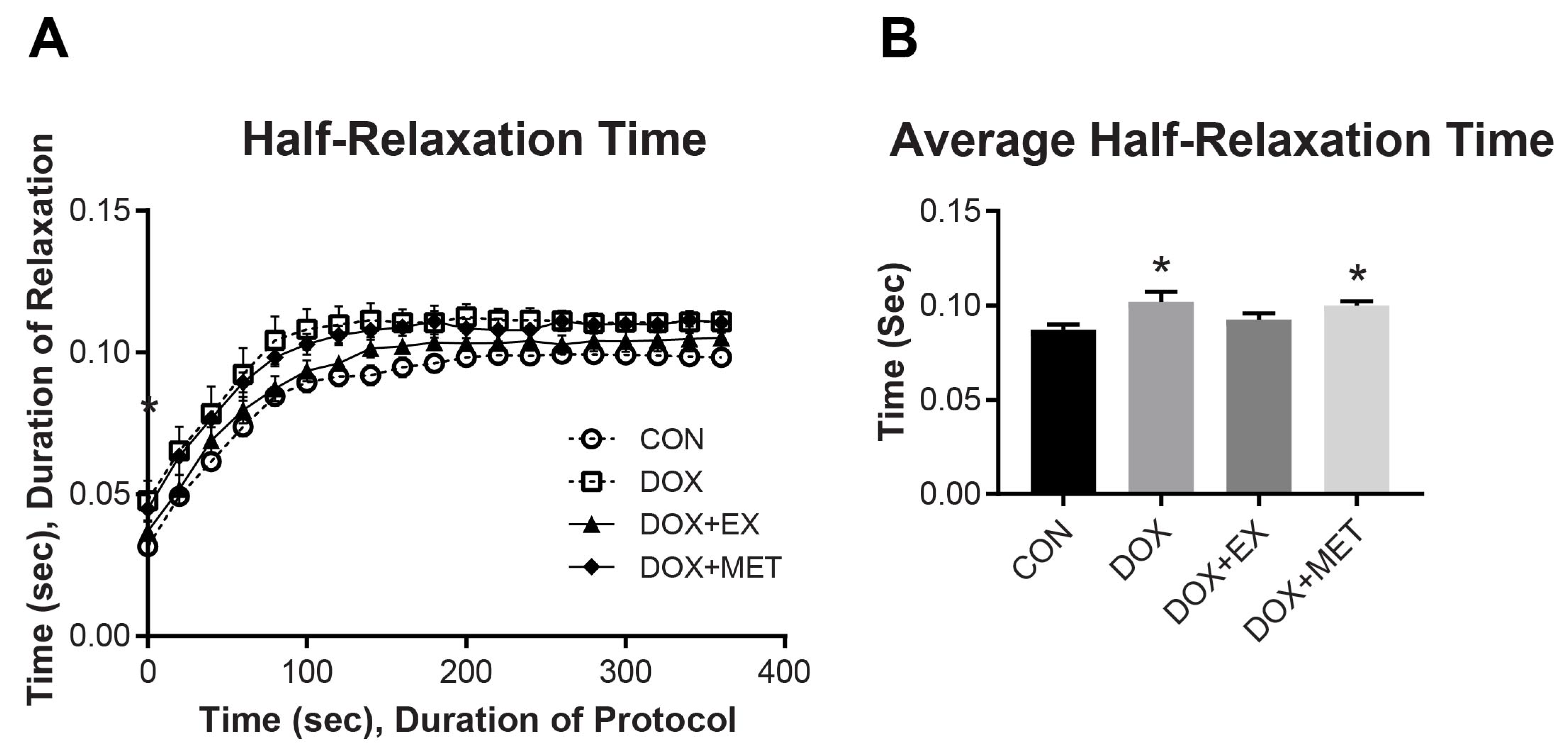
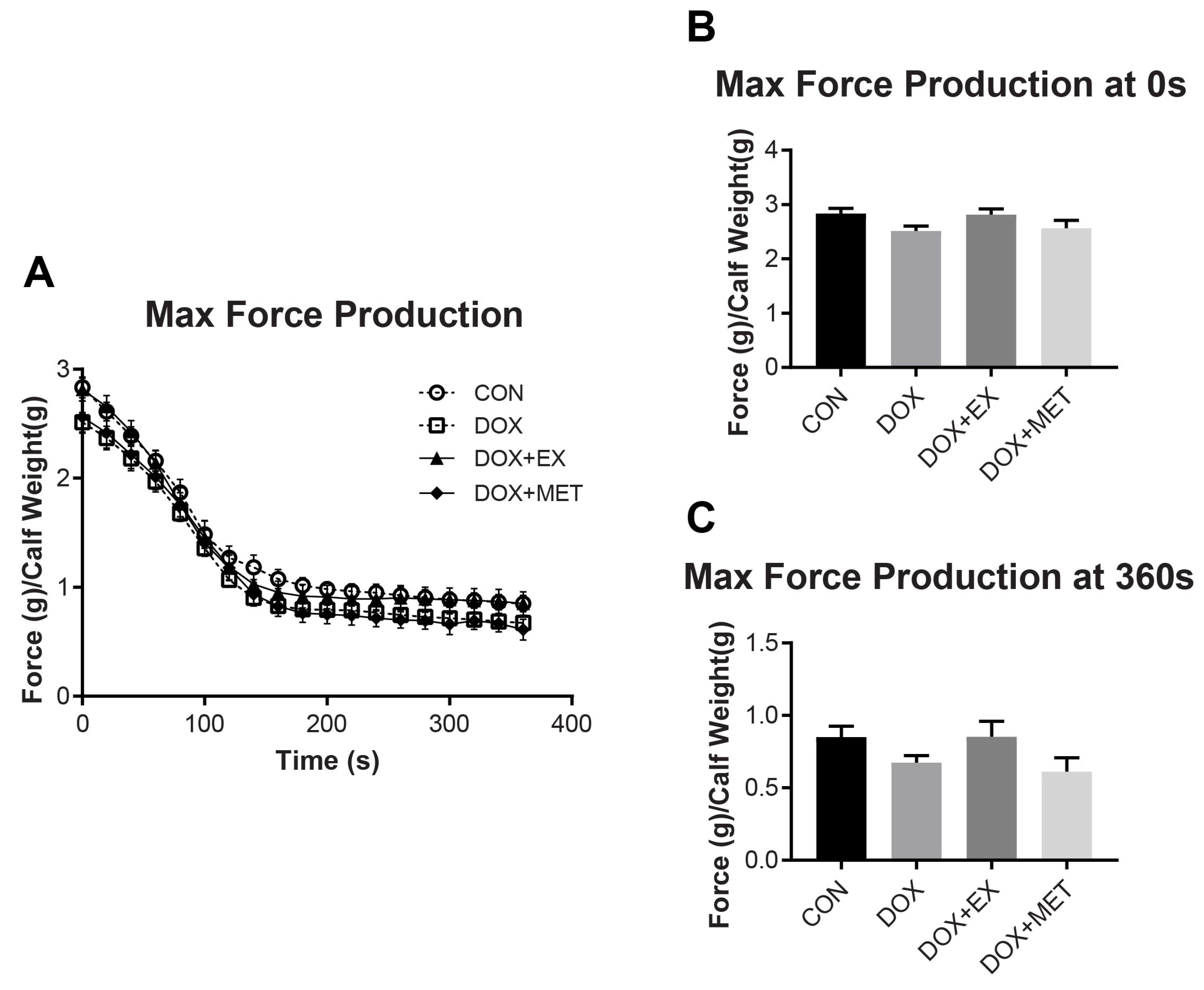
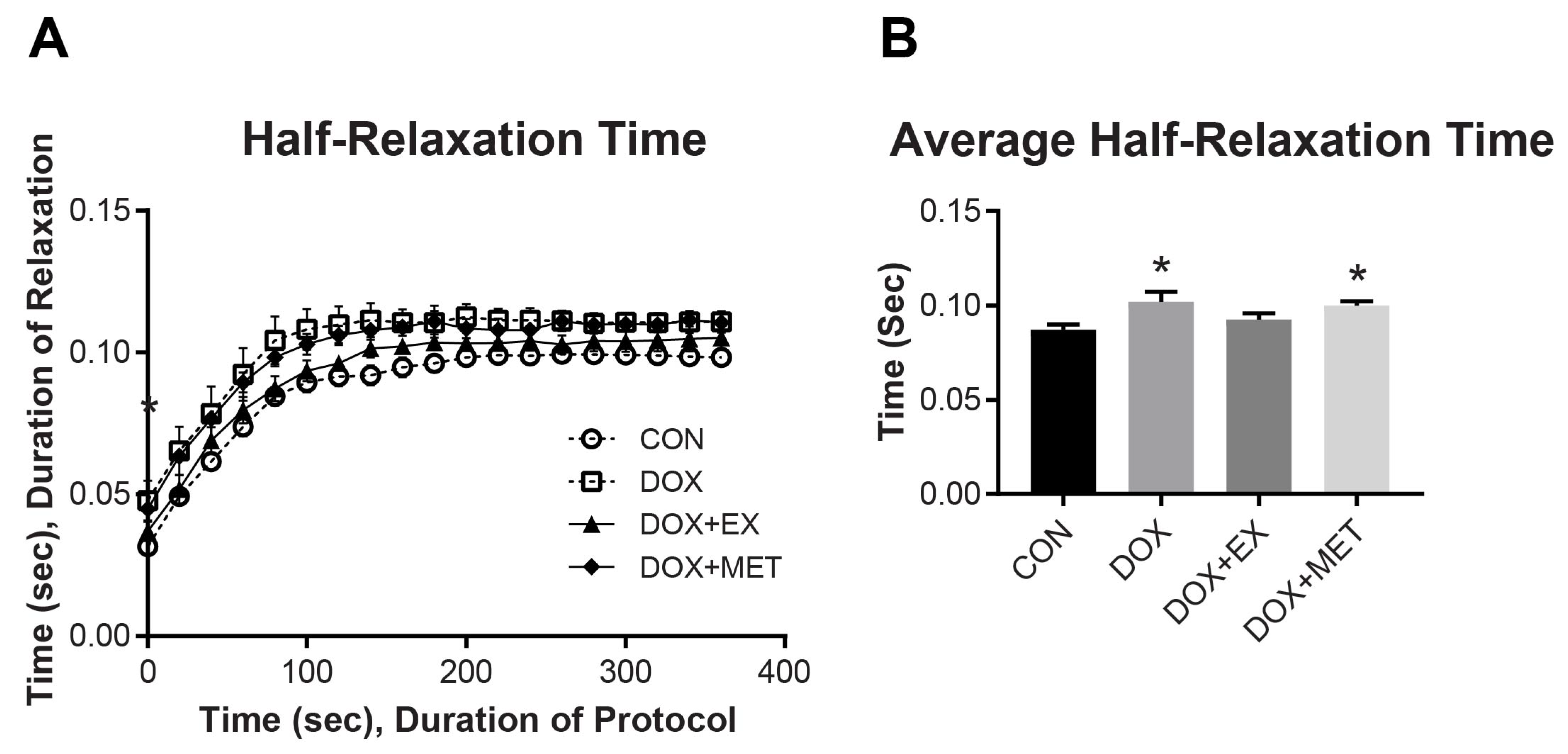
2.2. DOX Treatment and Muscle Function of the GPS Complex
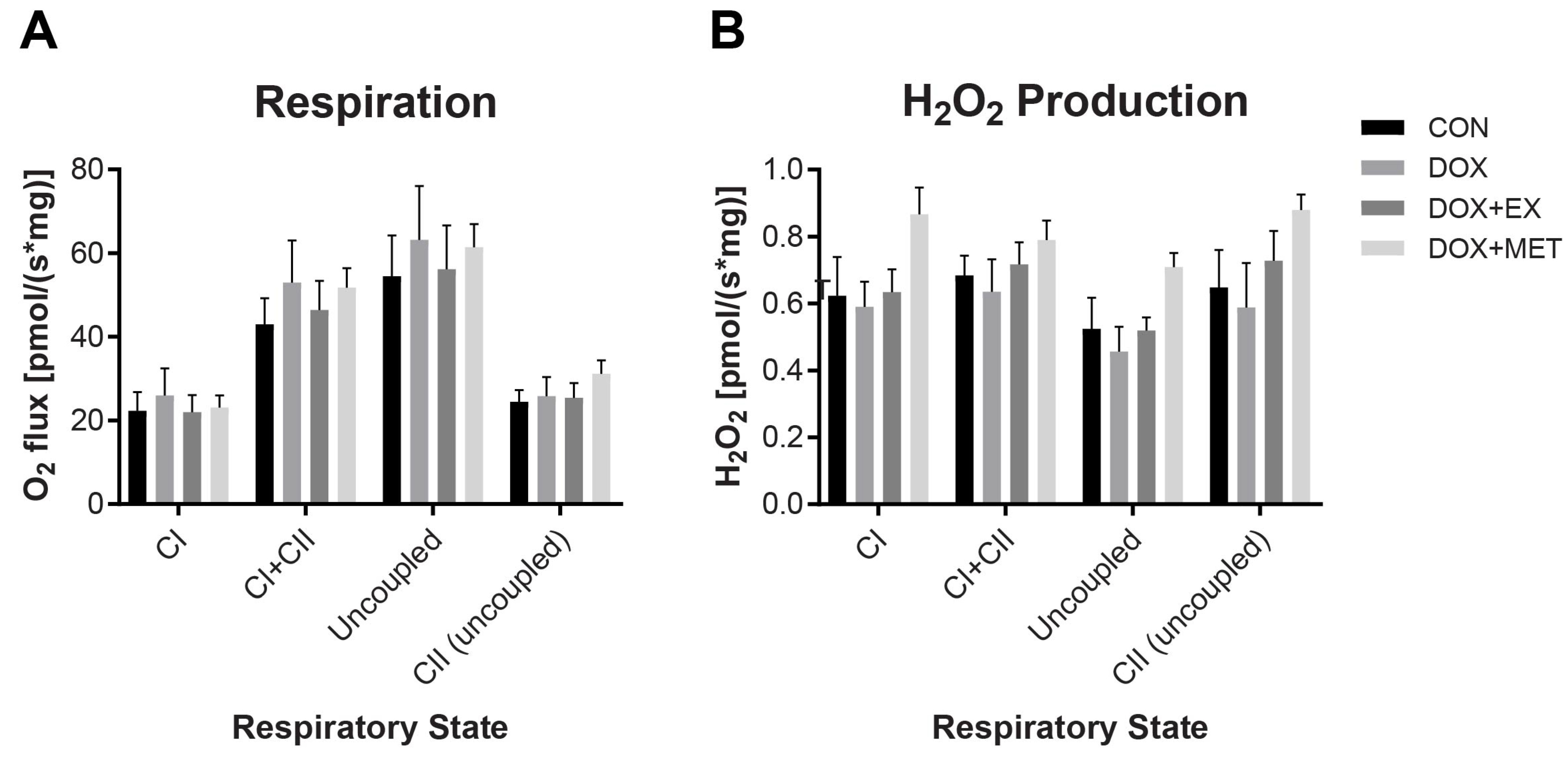
2.3. DOX Treatment Does Not Impair Mitochondrial Respiration
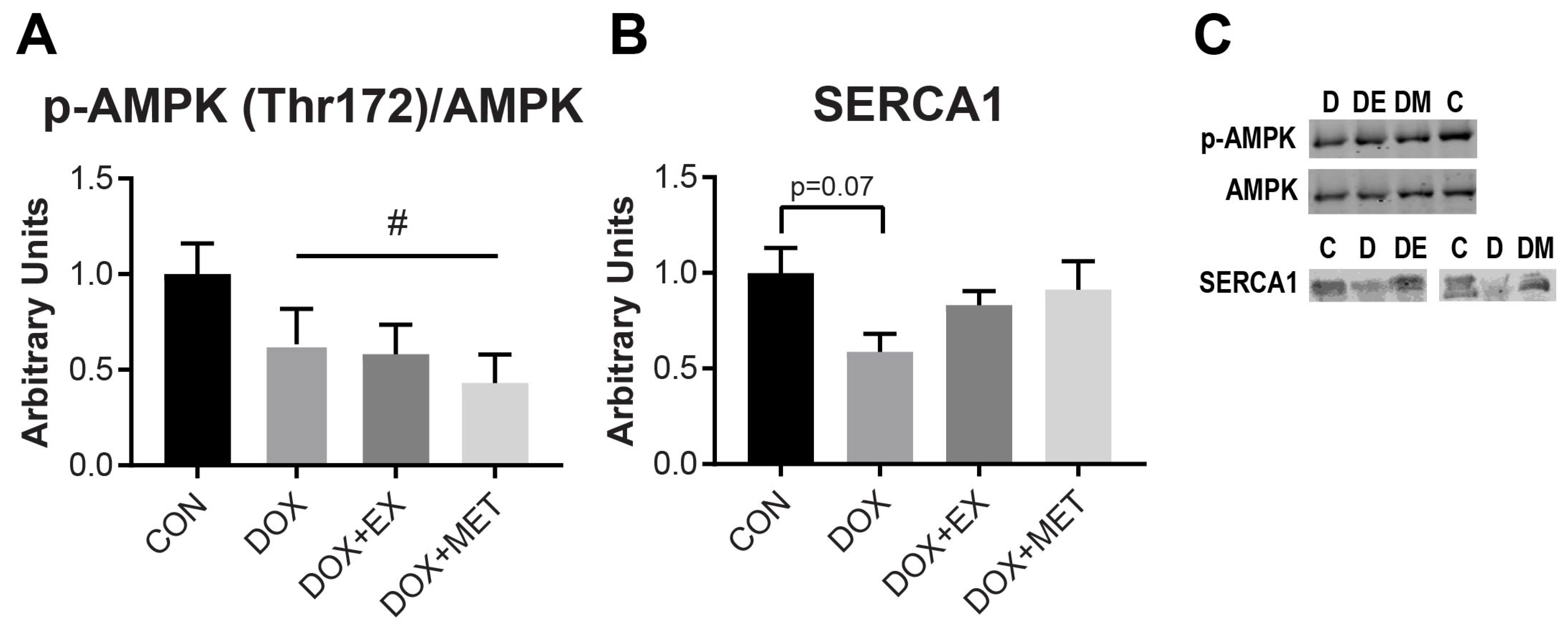
2.4. DOX Treatment Causes a Reduction in AMPK Phosphorylation
2.5. DOX Treatment Causes a Reduction in SERCA1 Protein Expression
3. Discussion
4. Methods
4.1. Animal Care
4.2. In Situ Muscle Function
4.3. Mitochondrial Respiration
4.4. Hydrogen Peroxide Production
4.5. Western Blotting
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Gilliam, L.A.; St Clair, D.K. Chemotherapy-induced weakness and fatigue in skeletal muscle: The role of oxidative stress. Antioxid. Redox Signal. 2011, 15, 2543–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbl, L.; Vasova, I.; Tomaskova, I.; Jedlicka, F.; Kral, Z.; Navratil, M.; Smardova, L.; Wagnerova, B.; Vorlicek, J. Cardiopulmonary exercise testing in the evaluation of functional capacity after treatment of lymphomas in adults. Leuk. Lymphoma 2006, 47, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Villani, F.; Busia, A.; Villani, M.; Laffranchi, A.; Viviani, S.; Bonfante, V. Cardiopulmonary response to exercise in patients with different degrees of lung toxicity after radio-chemotherapy for Hodgkin’s disease. Anticancer Res. 2009, 29, 777–783. [Google Scholar]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Doxorubicin-induced markers of myocardial autophagic signaling in sedentary and exercise trained animals. J. Appl. Physiol. 2013, 115, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavazis, A.N.; Smuder, A.J.; Min, K.; Tümer, N.; Powers, S.K. Short-term exercise training protects against doxorubicin-induced cardiac mitochondrial damage independent of HSP72. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1515–H1524. [Google Scholar] [CrossRef] [Green Version]
- Kouzi, S.A.; Uddin, M.N. Aerobic Exercise Training as a Potential Cardioprotective Strategy to Attenuate Doxorubicin-Induced Cardiotoxicity. J. Pharm. Pharm. Sci. 2016, 19, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Hayward, R.; Hydock, D.; Gibson, N.; Greufe, S.; Bredahl, E.; Parry, T. Tissue retention of doxorubicin and its effects on cardiac, smooth, and skeletal muscle function. J. Physiol. Biochem. 2013, 69, 177–187. [Google Scholar] [CrossRef]
- Ertunc, M.; Sara, Y.; Korkusuz, P.; Onur, R. Differential contractile impairment of fast- and slow-twitch skeletal muscles in a rat model of doxorubicin-induced congestive heart failure. Pharmacology 2009, 84, 240–248. [Google Scholar] [CrossRef]
- Hayward, R.; Lien, C.Y.; Jensen, B.T.; Hydock, D.S.; Schneider, C.M. Exercise training mitigates anthracycline-induced chronic cardiotoxicity in a juvenile rat model. Pediatr. Blood Cancer 2012, 59, 149–154. [Google Scholar] [CrossRef]
- Jensen, B.T.; Lien, C.Y.; Hydock, D.S.; Schneider, C.M.; Hayward, R. Exercise mitigates cardiac doxorubicin accumulation and preserves function in the rat. J. Cardiovasc. Pharm. 2013, 62, 263–269. [Google Scholar] [CrossRef]
- Chicco, A.J.; Hydock, D.S.; Schneider, C.M.; Hayward, R. Low-intensity exercise training during doxorubicin treatment protects against cardiotoxicity. J. Appl. Physiol. 2006, 100, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthy, C.; Cedraschi, C.; Pugliesi, A.; Di Silvestro, K.; Mugnier-Konrad, B.; Rapiti, E.; Allaz, A.F. Patients’ views about causes and preferences for the management of cancer-related fatigue-a case for non-congruence with the physicians? Support. Care Cancer 2011, 19, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced markers of autophagy signaling in skeletal muscle. J. Appl. Physiol. 2011, 111, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced oxidative stress and proteolysis in skeletal muscle. J. Appl. Physiol. 2011, 110, 935–942. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, J.M.; D’Lugos, A.C.; Mahmood, T.N.; Ormsby, J.C.; Salvo, L.; Dedmon, W.L.; Patel, S.H.; Katsma, M.S.; Mookadam, F.; Gonzales, R.J.; et al. Exercise Protects Skeletal Muscle during Chronic Doxorubicin Administration. Med. Sci. Sports Exerc. 2017, 49, 2394. [Google Scholar] [CrossRef]
- Bredahl, E.C.; Hydock, D.S. Creatine Supplementation and Doxorubicin-Induced Skeletal Muscle Dysfunction: An Ex Vivo Investigation. Nutr. Cancer 2017, 69, 607–615. [Google Scholar] [CrossRef]
- Bredahl, E.C.; Pfannenstiel, K.B.; Quinn, C.J.; Hayward, R.; Hydock, D.S. Effects of Exercise on Doxorubicin-Induced Skeletal Muscle Dysfunction. Med. Sci. Sports Exerc. 2016, 48, 1468–1473. [Google Scholar] [CrossRef]
- Hydock, D.S.; Lien, C.Y.; Jensen, B.T.; Schneider, C.M.; Hayward, R. Characterization of the effect of in vivo doxorubicin treatment on skeletal muscle function in the rat. Anticancer Res. 2011, 31, 2023–2028. [Google Scholar]
- van Norren, K.; van Helvoort, A.; Argilés, J.M.; van Tuijl, S.; Arts, K.; Gorselink, M.; Laviano, A.; Kegler, D.; Haagsman, H.P.; van der Beek, E.M. Direct effects of doxorubicin on skeletal muscle contribute to fatigue. Br. J. Cancer 2009, 100, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Hancock, C.R.; Janssen, E.; Terjung, R.L. Skeletal muscle contractile performance and ADP accumulation in adenylate kinase-deficient mice. Am. J. Physiol. Cell Physiol. 2005, 288, C1287–C1297. [Google Scholar] [CrossRef]
- Schwartz, A.L.; Winters-Stone, K.; Gallucci, B. Exercise effects on bone mineral density in women with breast cancer receiving adjuvant chemotherapy. Oncol. Nurs. Forum 2007, 34, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.L. Daily fatigue patterns and effect of exercise in women with breast cancer. Cancer Pract. 2000, 8, 16–24. [Google Scholar] [CrossRef]
- Kavazis, A.N.; Smuder, A.J.; Powers, S.K. Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl. Physiol. 2014, 117, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Gilliam, L.A.; Fisher-Wellman, K.H.; Lin, C.T.; Maples, J.M.; Cathey, B.L.; Neufer, P.D. The anticancer agent doxorubicin disrupts mitochondrial energy metabolism and redox balance in skeletal muscle. Free Radic. Biol. Med. 2013, 65, 988–996. [Google Scholar] [CrossRef] [Green Version]
- Marques-Aleixo, I.; Santos-Alves, E.; Mariani, D.; Rizo-Roca, D.; Padrão, A.I.; Rocha-Rodrigues, S.; Viscor, G.; Torrella, J.R.; Ferreira, R.; Oliveira, P.J.; et al. Physical exercise prior and during treatment reduces sub-chronic doxorubicin-induced mitochondrial toxicity and oxidative stress. Mitochondrion 2015, 20, 22–33. [Google Scholar] [CrossRef] [PubMed]
- El-Ashmawy, N.E.; Khedr, N.F.; El-Bahrawy, H.A.; Abo Mansour, H.E. Metformin augments doxorubicin cytotoxicity in mammary carcinoma through activation of adenosine monophosphate protein kinase pathway. Tumour Biol. 2017, 39, 1010428317692235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asensio-Lopez, M.C.; Sanchez-Mas, J.; Pascual-Figal, D.A.; de Torre, C.; Valdes, M.; Lax, A. Ferritin heavy chain as main mediator of preventive effect of metformin against mitochondrial damage induced by doxorubicin in cardiomyocytes. Free Radic. Biol. Med. 2014, 67, 19–29. [Google Scholar] [CrossRef]
- Asensio-López, M.C.; Sánchez-Más, J.; Pascual-Figal, D.A.; Abenza, S.; Pérez-Martínez, M.T.; Valdés, M.; Lax, A. Involvement of ferritin heavy chain in the preventive effect of metformin against doxorubicin-induced cardiotoxicity. Free Radic. Biol. Med. 2013, 57, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Asensio-López, M.C.; Lax, A.; Pascual-Figal, D.A.; Valdés, M.; Sánchez-Más, J. Metformin protects against doxorubicin-induced cardiotoxicity: Involvement of the adiponectin cardiac system. Free Radic. Biol. Med. 2011, 51, 1861–1871. [Google Scholar] [CrossRef]
- Kobashigawa, L.C.; Xu, Y.C.; Padbury, J.F.; Tseng, Y.T.; Yano, N. Metformin protects cardiomyocyte from doxorubicin induced cytotoxicity through an AMP-activated protein kinase dependent signaling pathway: An in vitro study. PLoS ONE 2014, 9, e104888. [Google Scholar] [CrossRef] [Green Version]
- Sheta, A.; Elsakkar, M.; Hamza, M.; Solaiman, A. Effect of metformin and sitagliptin on doxorubicin-induced cardiotoxicity in adult male albino rats. Hum. Exp. Toxicol 2016, 35, 0960327115627685. [Google Scholar] [CrossRef] [PubMed]
- Kelleni, M.T.; Amin, E.F.; Abdelrahman, A.M. Effect of Metformin and Sitagliptin on Doxorubicin-Induced Cardiotoxicity in Rats: Impact of Oxidative Stress, Inflammation, and Apoptosis. J. Toxicol. 2015, 2015, 424813. [Google Scholar] [CrossRef] [Green Version]
- Argun, M.; Üzüm, K.; Sönmez, M.F.; Özyurt, A.; Derya, K.; Çilenk, K.T.; Unalmış, S.; Pamukcu, Ö.; Baykan, A.; Narin, F.; et al. Cardioprotective effect of metformin against doxorubicin cardiotoxicity in rats. Anatol. J. Cardiol. 2016, 16, 234–241. [Google Scholar] [CrossRef]
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hamm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Fasano, M.; Della Corte, C.M.; Sasso, F.C.; Papaccio, F.; Viscardi, G.; Esposito, G.; Di Liello, R.; Normanno, N.; Capuano, A.; et al. Results of the safety run-in part of the METAL (METformin in Advanced Lung cancer) study: A multicentre, open-label phase I–II study of metformin with erlotinib in second-line therapy of patients with stage IV non-small-cell lung cancer. ESMO Open 2017, 2, e000132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winder, W.W.; Hardie, D.G. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am. J. Physiol. 1996, 270, E299–E304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkholder, T.J.; Fingado, B.; Baron, S.; Lieber, R.L. Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. J. Morphol. 1994, 221, 177–190. [Google Scholar] [CrossRef]
- Wu, K.D.; Lytton, J. Molecular cloning and quantification of sarcoplasmic reticulum Ca(2+)-ATPase isoforms in rat muscles. Am. J. Physiol. 1993, 264, C333–C341. [Google Scholar] [CrossRef]
- Punkt, K.; Naupert, A.; Asmussen, G. Differentiation of rat skeletal muscle fibres during development and ageing. Acta Histochem. 2004, 106, 145–154. [Google Scholar] [CrossRef]
- Kristensen, A.M.; MacDougall, K.B.; MacIntosh, B.R.; Overgaard, K. Is curvature of the force-velocity relationship affected by oxygen availability? Evidence from studies in ex vivo and in situ rat muscles. Pflügers Arch.-Eur. J. Physiol. 2020, 472, 597–608. [Google Scholar] [CrossRef]
- Hancock, C.R.; Brault, J.J.; Wiseman, R.W.; Terjung, R.L.; Meyer, R.A. 31P-NMR observation of free ADP during fatiguing, repetitive contractions of murine skeletal muscle lacking AK1. Am. J. Physiol. Cell Physiol. 2005, 288, C1298–C1304. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.J.; Gadian, D.G.; Wilkie, D.R. Mechanical relaxation rate and metabolism studied in fatiguing muscle by phosphorus nuclear magnetic resonance. J. Physiol. 1980, 299, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, M.D.; Amorese, A.J.; Balestrieri, N.P.; Fisher-Wellman, K.H.; Spangenburg, E.E. Doxorubicin causes lesions in the electron transport system of skeletal muscle mitochondria that are associated with a loss of contractile function. J. Biol. Chem. 2019, 294, 19709–19722. [Google Scholar] [CrossRef]
- De Lima Junior, E.A.; Yamashita, A.S.; Pimentel, G.D.; De Sousa, L.G.; Santos, R.V.; Goncalves, C.L.; Streck, E.L.; de Lira, F.S.; Rosa Neto, J.C. Doxorubicin caused severe hyperglycaemia and insulin resistance, mediated by inhibition in AMPk signalling in skeletal muscle. J. Cachexia Sarcopenia Muscle 2016, 7, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Gratia, S.; Kay, L.; Potenza, L.; Seffouh, A.; Novel-Chate, V.; Schnebelen, C.; Sestili, P.; Schlattner, U.; Tokarska-Schlattner, M. Inhibition of AMPK signalling by doxorubicin: At the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovasc. Res. 2012, 95, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Timm, K.N.; Tyler, D.J. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Drugs 2020, 34, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Arai, M.; Yoguchi, A.; Takizawa, T.; Yokoyama, T.; Kanda, T.; Kurabayashi, M.; Nagai, R. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum Ca(2+)-ATPase gene transcription. Circ. Res. 2000, 86, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, A.D.; Marchant, E.D.; Munk, D.J.; Watt, R.K.; Hansen, J.M.; Thomson, D.M.; Hancock, C.R. Multitissue analysis of exercise and metformin on doxorubicin-induced iron dysregulation. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E922–E930. [Google Scholar] [CrossRef]
- Fu, X.; Kong, L.; Tang, M.; Zhang, J.; Zhou, X.; Li, G.; Wang, H.; Fu, F. Protective effect of ocotillol against doxorubicininduced acute and chronic cardiac injury. Mol. Med. Rep. 2014, 9, 360–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.F.; Stone, J.R.; Schuldt, A.J.; Okoshi, K.; Okoshi, M.P.; Nakayama, M.; Ho, K.K.; Manning, W.J.; Marchionni, M.A.; Lorell, B.H.; et al. Heterozygous knockout of neuregulin-1 gene in mice exacerbates doxorubicin-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H660–H666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Y.; Sun, M.; Silver, M.; Ho, K.K.; Marchionni, M.A.; Caggiano, A.O.; Stone, J.R.; Amende, I.; Hampton, T.G.; Morgan, J.P.; et al. Neuregulin-1 attenuated doxorubicin-induced decrease in cardiac troponins. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1974–H1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardman, S.E.; Hall, D.E.; Cabrera, A.J.; Hancock, C.R.; Thomson, D.M. The effects of age and muscle contraction on AMPK activity and heterotrimer composition. Exp. Gerontol. 2014, 55, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchant, E.D.; Marchant, N.D.; Hyldahl, R.D.; Gifford, J.R.; Smith, M.W.; Hancock, C.R. Skeletal Muscle Mitochondrial Function following a 100-km Ultramarathon: A Case Study in Monozygotic Twins. Med. Sci. Sports Exerc. 2021. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CON (n = 23) | DOX (n = 20) | DOX + EX (n = 25) | DOX + MET (n = 19) | |
|---|---|---|---|---|
| Starting BW (g) | 19.8 ± 0.3 | 20.0 ± 0.3 | 20.3 ± 0.3 | 19.7 ± 0.4 |
| Final BW (g) | 20.2 ± 0.4 a | 16.7 ± 0.3 b* | 17.7 ± 0.4 b* | 17.0 ± 0.3 b* |
| % Change in BW | 2.2 ± 0.8 a | −16.4 ± 0.9 b* | −12.6 ± 1.4 b* | −13.6 ± 1.1 b* |
| Final CW (mg) | 135.6 ± 4.7 a | 123.6 ± 5.7 ab | 119.5 ± 2.6 b | 108.5 ± 3.2 b |
| CW as % of BW | 0.65 ± 0.02 ab | 0.71 ± 0.02 a | 0.71 ± 0.02 a | 0.62 ± 0.02 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mackay, A.D.; Marchant, E.D.; Louw, M.; Thomson, D.M.; Hancock, C.R. Exercise, but Not Metformin Prevents Loss of Muscle Function Due to Doxorubicin in Mice Using an In Situ Method. Int. J. Mol. Sci. 2021, 22, 9163. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179163
Mackay AD, Marchant ED, Louw M, Thomson DM, Hancock CR. Exercise, but Not Metformin Prevents Loss of Muscle Function Due to Doxorubicin in Mice Using an In Situ Method. International Journal of Molecular Sciences. 2021; 22(17):9163. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179163
Chicago/Turabian StyleMackay, Amy D., Erik D. Marchant, Makensie Louw, David M. Thomson, and Chad R. Hancock. 2021. "Exercise, but Not Metformin Prevents Loss of Muscle Function Due to Doxorubicin in Mice Using an In Situ Method" International Journal of Molecular Sciences 22, no. 17: 9163. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179163