
Biological Connection of Psychological Stress and Polytrauma under Intensive Care: The Role of Oxytocin and Hydrogen Sulfide

 , , ,
, , ,
Abstract
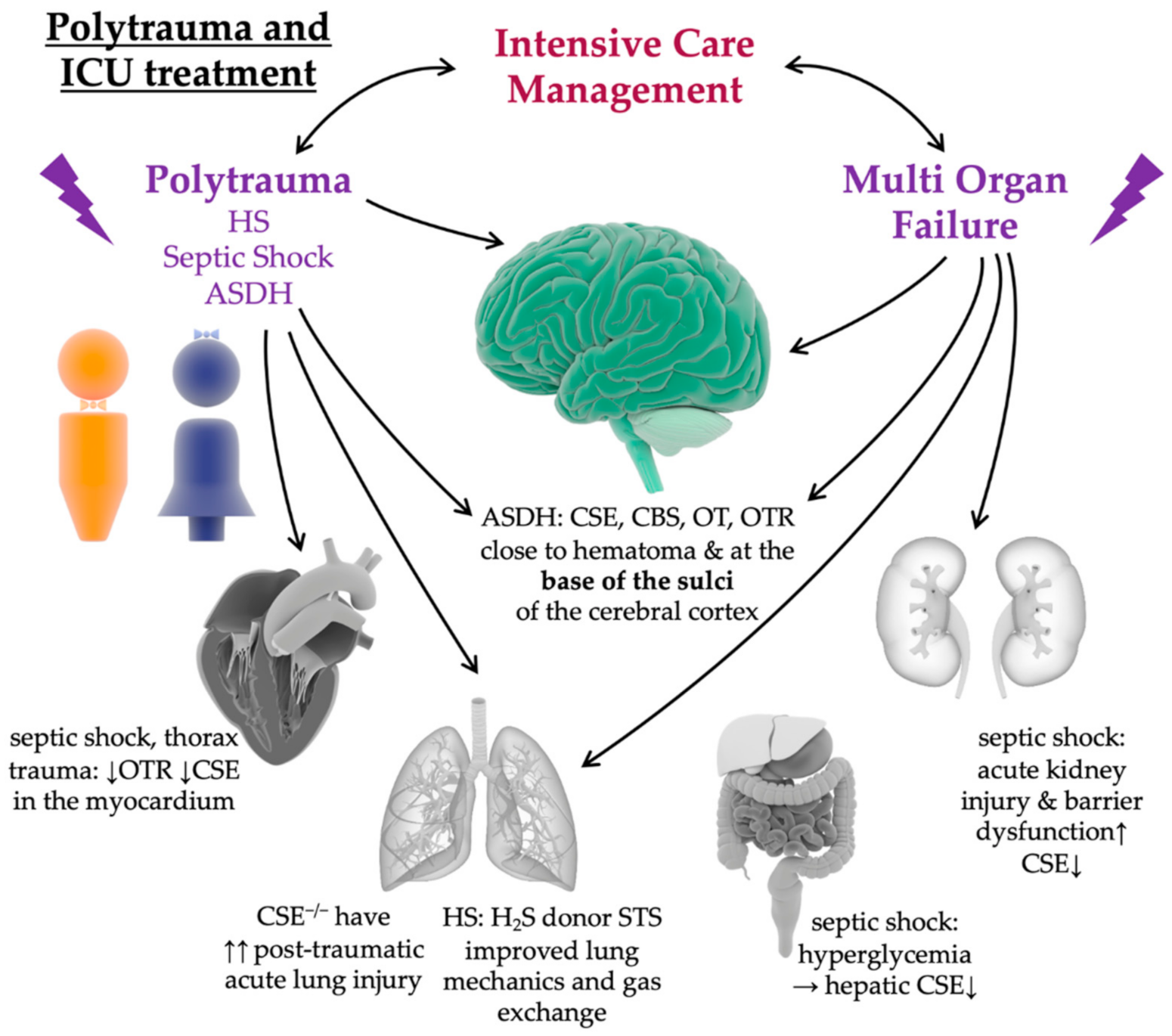
:1. Introduction Polytrauma–Hemorrhage and Brain Injury
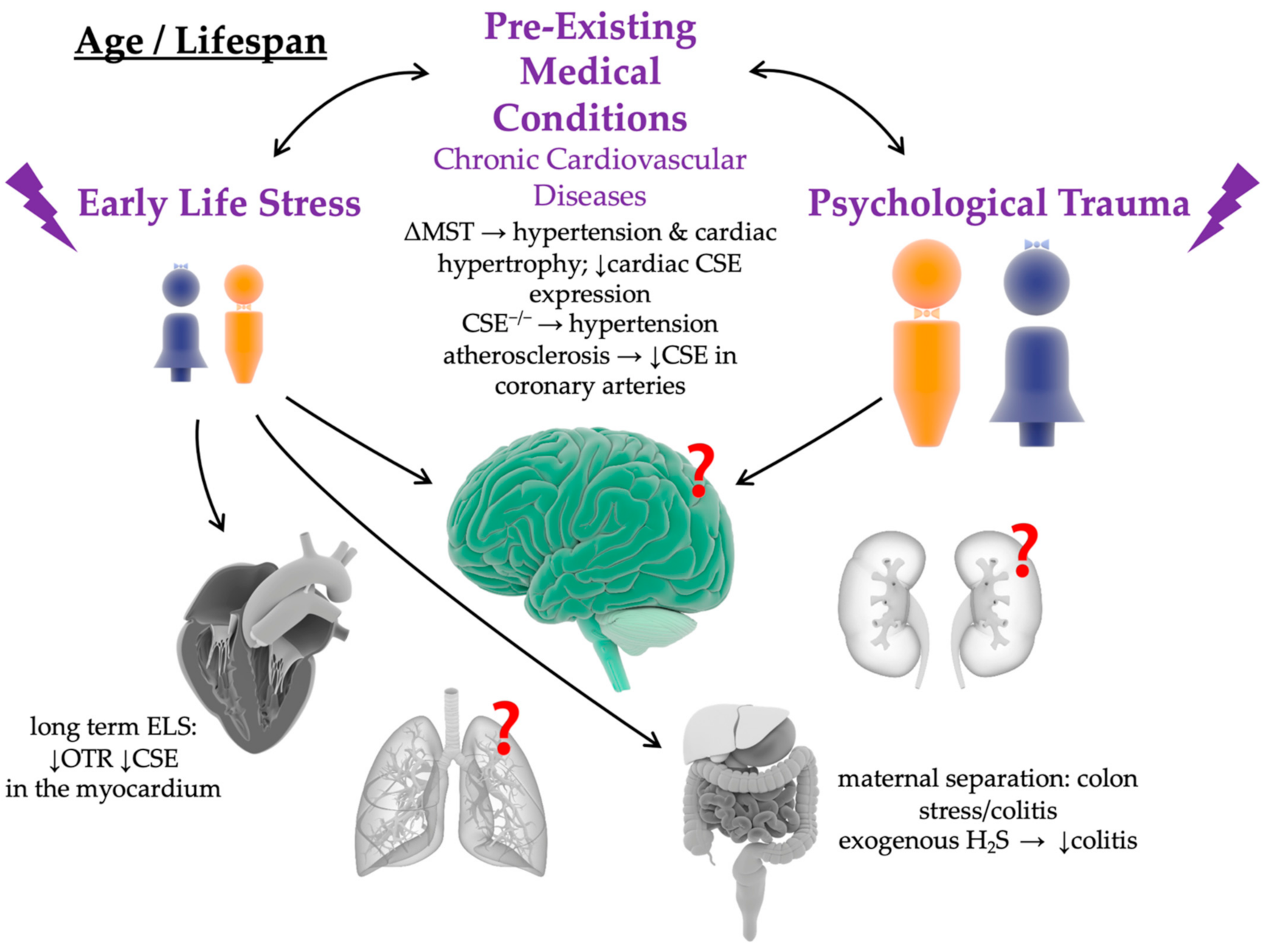
2. Impact of Chronic Cardiovascular and Psychological Pre-Existing Medical Conditions on the Long-Term Patient Outcome
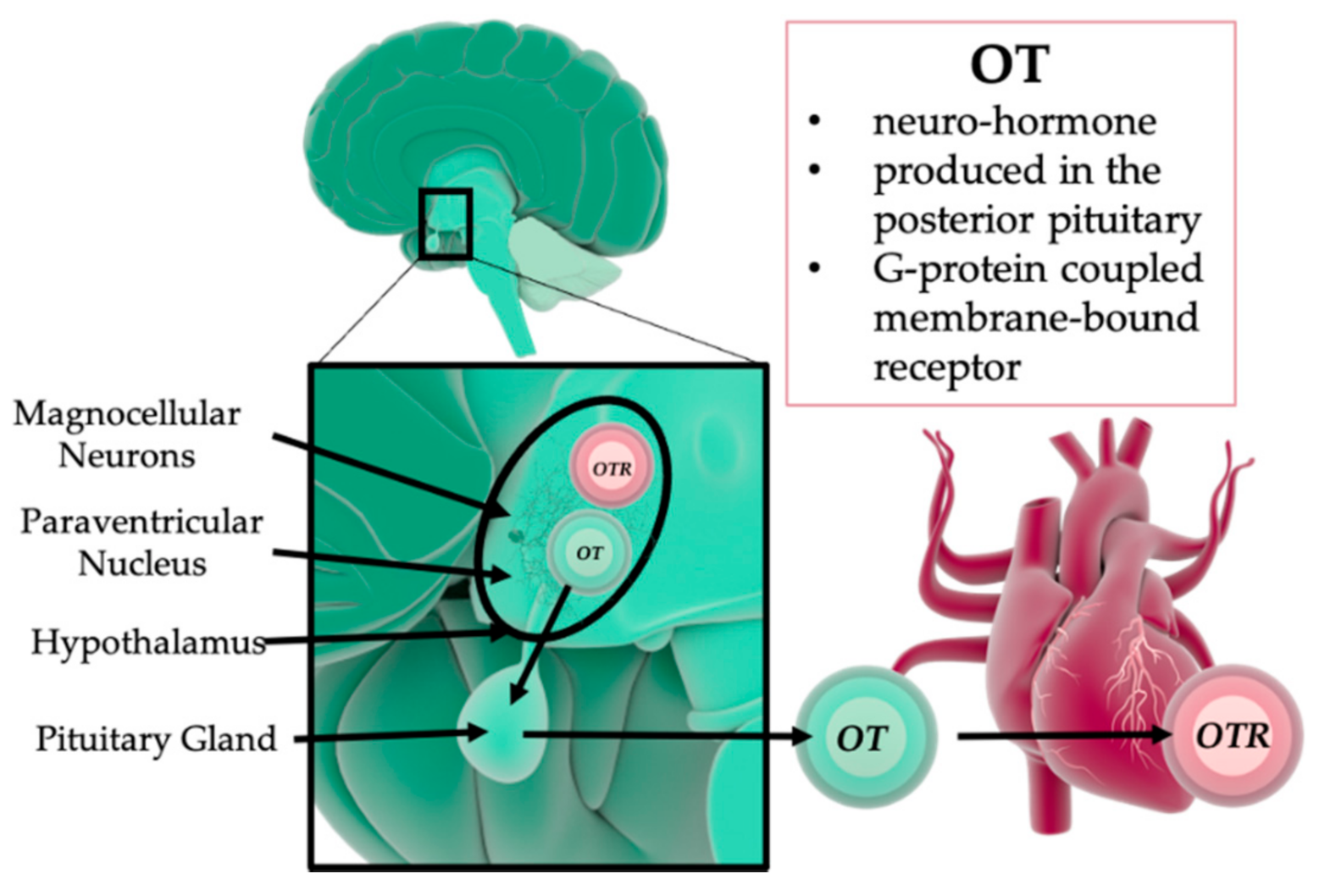
3. The Role of Oxytocin in Psychological and Physical Trauma
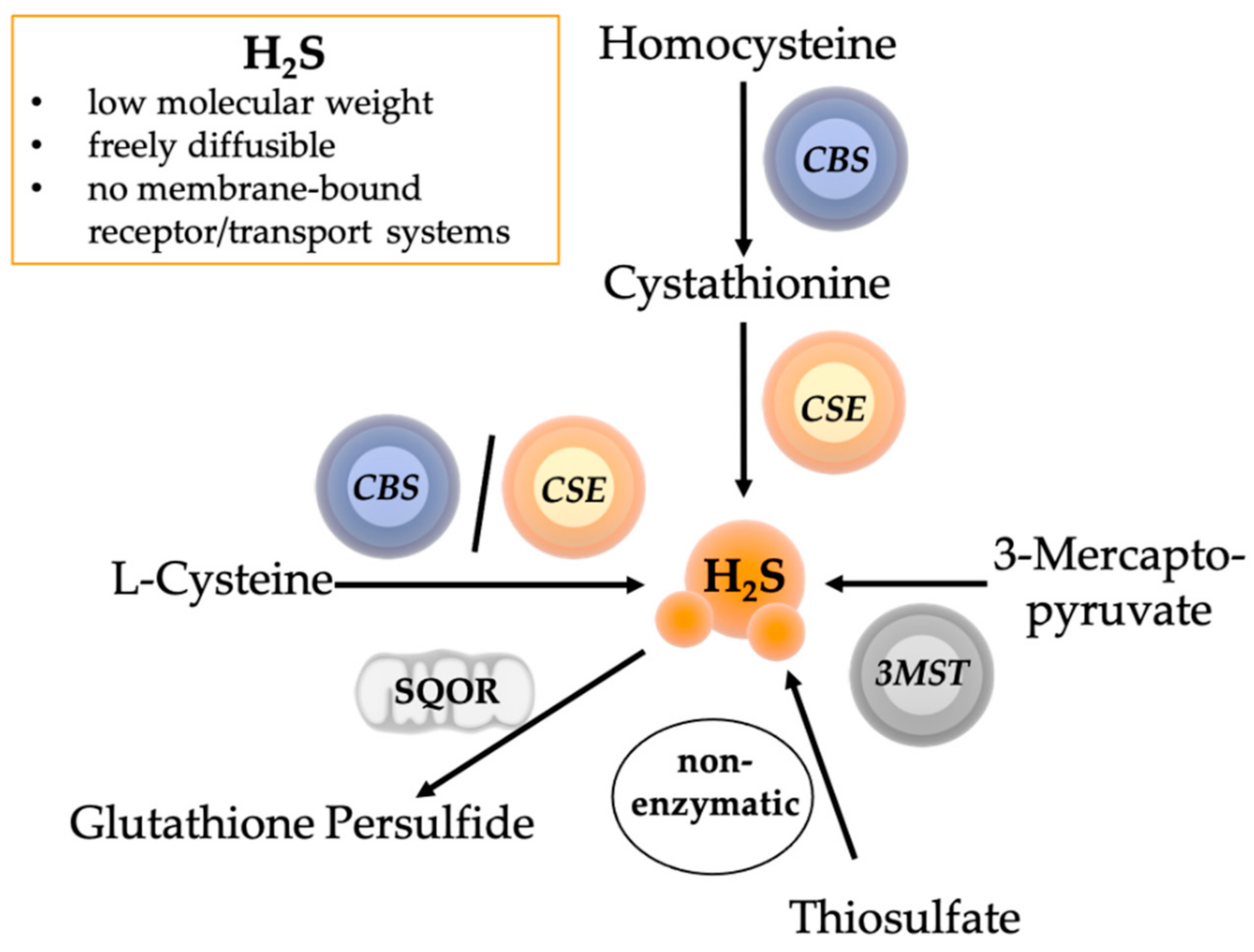
4. The Role of Hydrogen Sulfide in Psychological and Physical Trauma
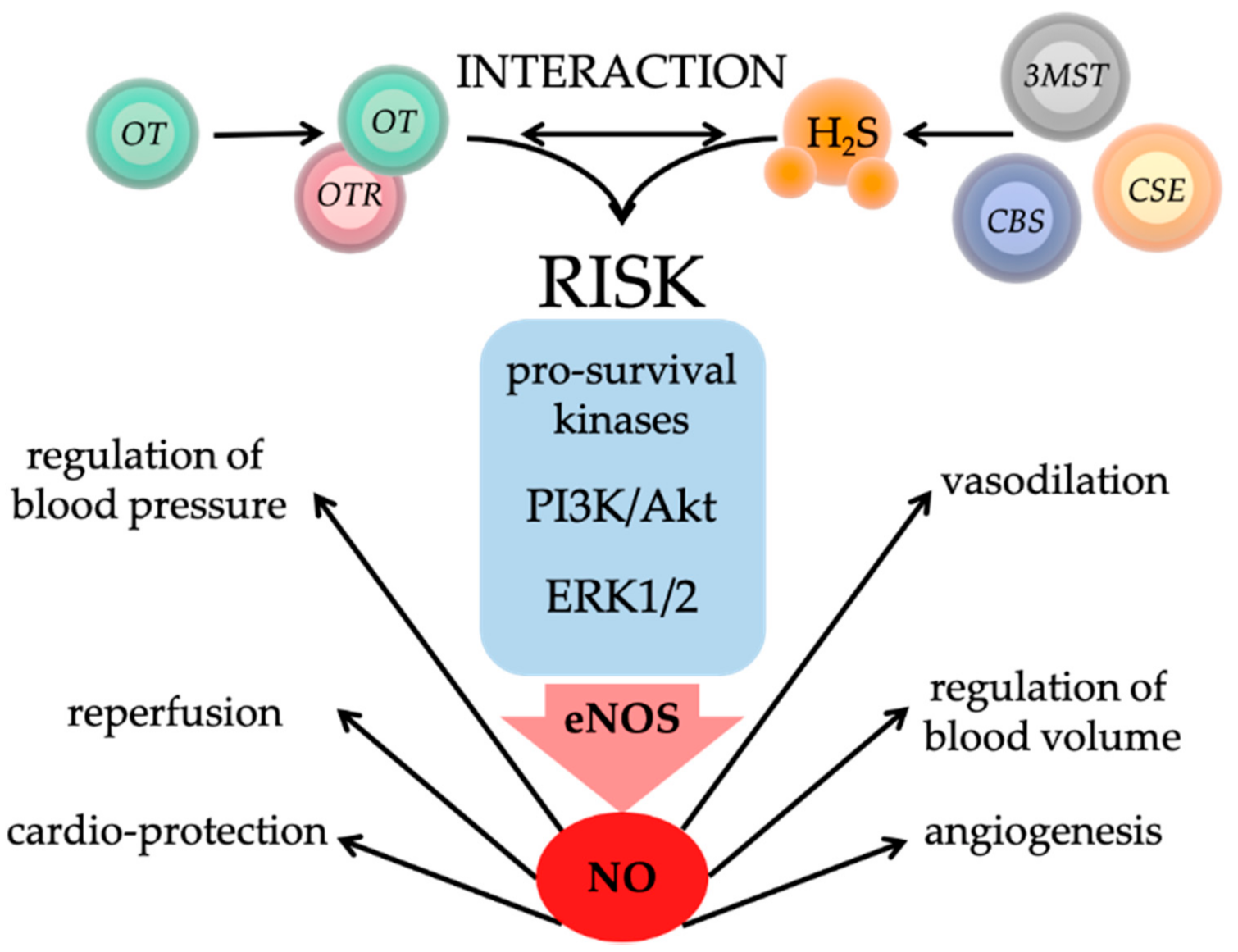
5. Interaction of Oxytocin and Hydrogen Sulfide in Physical and Psychological Trauma
6. Therapeutic Potential of Oxytocin and Hydrogen Sulfide in Trauma
7. Sex
8. Impact of Intensive Care Treatment in Pre-Clinical Animal Models
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cannon, J.W. Hemorrhagic Shock. N. Engl. J. Med. 2018, 378, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Kauvar, D.S.; Lefering, R.; Wade, C.E. Impact of Hemorrhage on Trauma Outcome: An Overview of Epidemiology, Clinical Presentations, and Therapeutic Considerations. J. Trauma Inj. Infect. Crit. Care 2006, 60, S3–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angele, M.K.; Schneider, C.P.; Chaudry, I.H. Bench-to-Bedside Review: Latest Results in Hemorrhagic Shock. Crit. Care 2008, 12, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbgebauer, R.; Braun, C.K.; Denk, S.; Mayer, B.; Cinelli, P.; Radermacher, P.; Wanner, G.A.; Simmen, H.-P.; Gebhard, F.; Rittirsch, D.; et al. Hemorrhagic Shock Drives Glycocalyx, Barrier and Organ Dysfunction Early after Polytrauma. J. Crit. Care 2018, 44, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Messerer, D.A.C.; Halbgebauer, R.; Nilsson, B.; Pavenstädt, H.; Radermacher, P.; Huber-Lang, M. Immunopathophysiology of Trauma-Related Acute Kidney Injury. Nat. Rev. Nephrol. 2021, 17, 91–111. [Google Scholar] [CrossRef]
- Minei, J.P.; Cuschieri, J.; Sperry, J.; Moore, E.E.; West, M.A.; Harbrecht, B.G.; O’Keefe, G.E.; Cohen, M.J.; Moldawer, L.L.; Tompkins, R.G.; et al. The Changing Pattern and Implications of Multiple Organ Failure after Blunt Injury with Hemorrhagic Shock*. Crit. Care Med. 2012, 40, 1129–1135. [Google Scholar] [CrossRef] [Green Version]
- Lahner, D.; Fritsch, G. Pathophysiologie intrakranieller Verletzungen. Unfallchirurg 2017, 120, 728–733. [Google Scholar] [CrossRef]
- Ma, X.; Cheng, Y.; Garcia, R.; Haorah, J. Hemorrhage Associated Mechanisms of Neuroinflammation in Experimental Traumatic Brain Injury. J. Neuroimmune Pharmacol. 2020, 15, 181–195. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 10, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Di Saverio, S.; Gambale, G.; Coccolini, F.; Catena, F.; Giorgini, E.; Ansaloni, L.; Amadori, N.; Coniglio, C.; Giugni, A.; Biscardi, A.; et al. Changes in the Outcomes of Severe Trauma Patients from 15-Year Experience in a Western European Trauma ICU of Emilia Romagna Region (1996–2010). A Population Cross-Sectional Survey Study. Langenbecks Arch. Surg. 2014, 399, 109–126. [Google Scholar] [CrossRef]
- Gross, T.; Schüepp, M.; Attenberger, C.; Pargger, H.; Amsler, F. Outcome in Polytraumatized Patients with and without Brain Injury: Quality of Life Following Polytrauma. Acta Anaesthesiol. Scand. 2012, 56, 1163–1174. [Google Scholar] [CrossRef]
- Andruszkow, H.; Probst, C.; Grün, O.; Krettek, C.; Hildebrand, F. Does Additional Head Trauma Affect the Long-Term Outcome After Upper Extremity Trauma in Multiple Traumatized Patients: Is There an Additional Effect of Traumatic Brain Injury? Clin. Orthop. 2013, 471, 2899–2905. [Google Scholar] [CrossRef] [Green Version]
- Licastro, F.; Hrelia, S.; Porcellini, E.; Malaguti, M.; Di Stefano, C.; Angeloni, C.; Carbone, I.; Simoncini, L.; Piperno, R. Peripheral Inflammatory Markers and Antioxidant Response during the Post-Acute and Chronic Phase after Severe Traumatic Brain Injury. Front. Neurol. 2016, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Shan, H.; Wang, T.; Liu, W.; Wang, Y.; Wang, L.; Zhang, L.; Chang, P.; Dong, W.; Chen, X.; et al. Dynamic Change of Hydrogen Sulfide After Traumatic Brain Injury and Its Effect in Mice. Neurochem. Res. 2013, 38, 714–725. [Google Scholar] [CrossRef]
- Rixen, D.; Siegel, J.H. Bench-to-Bedside Review: Oxygen Debt and Its Metabolic Correlates as Quantifiers of the Severity of Hemorrhagic and Posttraumatic Shock. Crit. Care 2005, 9, 441. [Google Scholar] [CrossRef] [Green Version]
- Barbee, R.W.; Reynolds, P.S.; Ward, K.R. Assesing schock resuscitation strategies by oxygen debt repayment. Shock 2010, 33, 113–122. [Google Scholar] [CrossRef]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.J.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic Brain Injury: Integrated Approaches to Improve Prevention, Clinical Care, and Research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- Ganster, F.; Burban, M.; de la Bourdonnaye, M.; Fizanne, L.; Douay, O.; Loufrani, L.; Mercat, A.; Calès, P.; Radermacher, P.; Henrion, D.; et al. Effects of Hydrogen Sulfide on Hemodynamics, Inflammatory Response and Oxidative Stress during Resuscitated Hemorrhagic Shock in Rats. Crit. Care 2010, 14, R165. [Google Scholar] [CrossRef] [Green Version]
- Brealey, D.; Karyampudi, S.; Jacques, T.S.; Novelli, M.; Stidwill, R.; Taylor, V.; Smolenski, R.T.; Singer, M. Mitochondrial Dysfunction in a Long-Term Rodent Model of Sepsis and Organ Failure. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2004, 286, R491–R497. [Google Scholar] [CrossRef] [Green Version]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between Mitochondrial Dysfunction and Severity and Outcome of Septic Shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Harrois, A.; Huet, O.; Duranteau, J. Alterations of Mitochondrial Function in Sepsis and Critical Illness. Curr. Opin. Anaesthesiol. 2009, 22, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Stolk, R.F.; van der Pasch, E.; Naumann, F.; Schouwstra, J.; Bressers, S.; van Herwaarden, A.E.; Gerretsen, J.; Schambergen, R.; Ruth, M.M.; van der Hoeven, J.G.; et al. Norepinephrine Dysregulates the Immune Response and Compromises Host Defense during Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Stolk, R.F.; van der Poll, T.; Angus, D.C.; van der Hoeven, J.G.; Pickkers, P.; Kox, M. Potentially Inadvertent Immunomodulation: Norepinephrine Use in Sepsis. Am. J. Respir. Crit. Care Med. 2016, 194, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Radermacher, P.; Wepler, M.; Nußbaum, B. Non-Hemodynamic Effects of Catecholamines. Shock 2017, 48, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Wardle, T.D. Co-Morbid Factors in Trauma Patients. Br. Med. Bull. 1999, 55, 744–756. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.C.; Bercz, A.; Niziolek, G.M.; Kassam, F.; Veile, R.; Friend, L.A.; Pritts, T.A.; Makley, A.T.; Goodman, M.D. UCH-L1 Is a Poor Serum Biomarker of Murine Traumatic Brain Injury After Polytrauma. J. Surg. Res. 2019, 244, 63–68. [Google Scholar] [CrossRef]
- Ferraris, V.A.; Ferraris, S.P.; Saha, S.P. The Relationship Between Mortality and Preexisting Cardiac Disease in 5,971 Trauma Patients. J. Trauma Inj. Infect. Crit. Care 2010, 69, 645–652. [Google Scholar] [CrossRef]
- Neal, M.D.; Cushieri, J.; Rosengart, M.R.; Alarcon, L.H.; Moore, E.E.; Maier, R.V.; Minei, J.P.; Billiar, T.R.; Peitzman, A.B.; Sperry, J.L. Preinjury Statin Use Is Associated With a Higher Risk of Multiple Organ Failure After Injury: A Propensity Score Adjusted Analysis. J. Trauma Inj. Infect. Crit. Care 2009, 67, 476–484. [Google Scholar] [CrossRef] [Green Version]
- Sellmann, T.; Miersch, D.; Kienbaum, P.; Flohé, S.; Schneppendahl, J.; Lefering, R.; der DGU, T.R. The Impact of Arterial Hypertension on Polytrauma and Traumatic Brain Injury. Dtsch. Aerzteblatt Online 2012, 109, 849–856. [Google Scholar] [CrossRef]
- Chang, J.-C. Regulatory Role of Mitochondria in Oxidative Stress and Atherosclerosis. World J. Cardiol. 2010, 2, 150. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.P.K.; Bennett, M.R. Mitochondrial DNA Damage and Atherosclerosis. Trends Endocrinol. Metab. 2014, 25, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocchetti, N.; Paternò, R.; Citerio, G.; Beretta, L.; Colombo, A. Traumatic Brain Injury in an Aging Population. J. Neurotrauma 2012, 29, 1119–1125. [Google Scholar] [CrossRef]
- Kumar, A.; Stoica, B.A.; Sabirzhanov, B.; Burns, M.P.; Faden, A.I.; Loane, D.J. Traumatic Brain Injury in Aged Animals Increases Lesion Size and Chronically Alters Microglial/Macrophage Classical and Alternative Activation States. Neurobiol. Aging 2013, 34, 1397–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Veldhoven, L.M.; Sander, A.M.; Struchen, M.A.; Sherer, M.; Clark, A.N.; Hudnall, G.E.; Hannay, H.J. Predictive Ability of Preinjury Stressful Life Events and Post-Traumatic Stress Symptoms for Outcomes Following Mild Traumatic Brain Injury: Analysis in a Prospective Emergency Room Sample. J. Neurol. Neurosurg. Psychiatry 2011, 82, 782–787. [Google Scholar] [CrossRef]
- Chaby, L.E.; Zhang, L.; Liberzon, I. The Effects of Stress in Early Life and Adolescence on Posttraumatic Stress Disorder, Depression, and Anxiety Symptomatology in Adulthood. Curr. Opin. Behav. Sci. 2017, 14, 86–93. [Google Scholar] [CrossRef]
- Alway, Y.; McKay, A.; Gould, K.R.; Johnston, L.; Ponsford, J. Factors associated with posttraumatic stress disorder following moderate to severe traumatic brain injury: A prospective study: Research Article: Predictors of PTSD Following TBI. Depress. Anxiety 2016, 33, 19–26. [Google Scholar] [CrossRef]
- Ponsford, J.; Alway, Y.; Gould, K.R. Epidemiology and Natural History of Psychiatric Disorders After TBI. J. Neuropsychiatry Clin. Neurosci. 2018, 30, 262–270. [Google Scholar] [CrossRef] [Green Version]
- González-Pardo, H.; Arias, J.L.; Gómez-Lázaro, E.; López Taboada, I.; Conejo, N.M. Sex-Specific Effects of Early Life Stress on Brain Mitochondrial Function, Monoamine Levels and Neuroinflammation. Brain Sci. 2020, 10, 447. [Google Scholar] [CrossRef]
- Roque, A.; Ochoa-Zarzosa, A.; Torner, L. Maternal Separation Activates Microglial Cells and Induces an Inflammatory Response in the Hippocampus of Male Rat Pups, Independently of Hypothalamic and Peripheral Cytokine Levels. Brain. Behav. Immun. 2016, 55, 39–48. [Google Scholar] [CrossRef]
- Réus, G.Z.; Fernandes, G.C.; de Moura, A.B.; Silva, R.H.; Darabas, A.C.; de Souza, T.G.; Abelaira, H.M.; Carneiro, C.; Wendhausen, D.; Michels, M.; et al. Early Life Experience Contributes to the Developmental Programming of Depressive-like Behaviour, Neuroinflammation and Oxidative Stress. J. Psychiatr. Res. 2017, 95, 196–207. [Google Scholar] [CrossRef]
- Diaz-Chávez, A.; Lajud, N.; Roque, A.; Cheng, J.P.; Meléndez-Herrera, E.; Valdéz-Alarcón, J.J.; Bondi, C.O.; Kline, A.E. Early Life Stress Increases Vulnerability to the Sequelae of Pediatric Mild Traumatic Brain Injury. Exp. Neurol. 2020, 329, 113318. [Google Scholar] [CrossRef]
- Lajud, N.; Roque, A.; Cheng, J.P.; Bondi, C.O.; Kline, A.E. Early Life Stress Preceding Mild Pediatric Traumatic Brain Injury Increases Neuroinflammation but Does Not Exacerbate Impairment of Cognitive Flexibility during Adolescence. J. Neurotrauma 2021, 38, 411–421. [Google Scholar] [CrossRef]
- Sanchez, C.M.; Titus, D.J.; Wilson, N.M.; Freund, J.E.; Atkins, C.M. Early Life Stress Exacerbates Outcome after Traumatic Brain Injury. J. Neurotrauma 2021, 38, 555–565. [Google Scholar] [CrossRef]
- Corbo, V.; Salat, D.H.; Amick, M.M.; Leritz, E.C.; Milberg, W.P.; McGlinchey, R.E. Reduced Cortical Thickness in Veterans Exposed to Early Life Trauma. Psychiatry Res. Neuroimaging 2014, 223, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Lange, R.T.; Lippa, S.M.; Brickell, T.A.; Yeh, P.-H.; Ollinger, J.; Wright, M.; Driscoll, A.; Sullivan, J.; Braatz, S.; Gartner, R.; et al. Post-Traumatic Stress Disorder Is Associated with Neuropsychological Outcome but Not White Matter Integrity after Mild Traumatic Brain Injury. J. Neurotrauma 2021, 38, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; McLaughlin, K.A.; Misra, S.; Koenen, K.C. Childhood Maltreatment and Health Impact: The Examples of Cardiovascular Disease and Type 2 Diabetes Mellitus in Adults. Clin. Psychol. Sci. Pract. 2017, 24, 125–139. [Google Scholar] [CrossRef]
- Cirulli, F. Interactions between Early Life Stress and Metabolic Stress in Programming of Mental and Metabolic Health. Curr. Opin. Behav. Sci. 2017, 14, 65–71. [Google Scholar] [CrossRef]
- Cruceanu, C.; Matosin, N.; Binder, E.B. Interactions of Early-Life Stress with the Genome and Epigenome: From Prenatal Stress to Psychiatric Disorders. Curr. Opin. Behav. Sci. 2017, 14, 167–171. [Google Scholar] [CrossRef]
- Felitti, V.J.; Anda, R.F.; Nordenberg, D.; Williamson, D.F.; Spitz, A.M.; Edwards, V.; Koss, M.P.; Marks, J.S. Relationship of Childhood Abuse and Household Dysfunction to Many of the Leading Causes of Death in Adults. Am. J. Prev. Med. 1998, 14, 245–258. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Early Life Events and Their Consequences for Later Disease: A Life History and Evolutionary Perspective. Am. J. Hum. Biol. 2007, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.E.; Chen, E.; Parker, K.J. Psychological Stress in Childhood and Susceptibility to the Chronic Diseases of Aging: Moving toward a Model of Behavioral and Biological Mechanisms. Psychol. Bull. 2011, 137, 959–997. [Google Scholar] [CrossRef] [PubMed]
- Shonkoff, J.P.; Garner, A.S.; The Committee on Pyschosocials Aspects of Child and Family Health, Committee on Early Childhood, Adoption, and Dependent Care, and Section on Developmental and Behavioral Pediatrics; Siegel, B.S.; Dobbins, M.I.; Earls, M.F.; Garner, A.S.; McGuinn, L.; Pascoe, J.; Wood, D.L. The Lifelong Effects of Early Childhood Adversity and Toxic Stress. Pediatrics 2012, 129, e232–e246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCook, O.; Denoix, N.; Radermacher, P.; Waller, C.; Merz, T. H2S and Oxytocin Systems in Early Life Stress and Cardiovascular Disease. J. Clin. Med. 2021, 10, 3484. [Google Scholar] [CrossRef]
- Balint, E.M.; Boseva, P.; Schury, K.; Guendel, H.; Rottbauer, W.; Waller, C. High Prevalence of Posttraumatic Stress in Patients with Primary Hypertension. Gen. Hosp. Psychiatry 2016, 38, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Loria, A.S.; Ho, D.H.; Pollock, J.S. A Mechanistic Look at the Effects of Adversity Early in Life on Cardiovascular Disease Risk during Adulthood. Acta Physiol. 2014, 210, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.O.; Cohn, D.M.; Loria, A.S. Developmental Origins of Cardiovascular Disease: Impact of Early Life Stress in Humans and Rodents. Neurosci. Biobehav. Rev. 2017, 74, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Anda, R.F.; Brown, D.W.; Dube, S.R.; Bremner, J.D.; Felitti, V.J.; Giles, W.H. Adverse Childhood Experiences and Chronic Obstructive Pulmonary Disease in Adults. Am. J. Prev. Med. 2008, 34, 396–403. [Google Scholar] [CrossRef]
- Shields, M.; Hovdestad, W.; Gilbert, C.; Tonmyr, L. Childhood Maltreatment as a Risk Factor for COPD: Findings from a Population-Based Survey of Canadian Adults. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Franz, H.M.; Corbo, V.; Fonda, J.R.; Levin, L.K.; Milberg, W.P.; McGlinchey, R.E. The Impact of Interpersonal Early Life Trauma on Cardio-Metabolic Health in Post-9/11 Veterans. Health Psychol. 2019, 38, 113–121. [Google Scholar] [CrossRef]
- Gu, H.; Tang, C.; Yang, Y. Psychological Stress, Immune Response, and Atherosclerosis. Atherosclerosis 2012, 223, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Slopen, N.; Koenen, K.C.; Kubzansky, L.D. Childhood Adversity and Immune and Inflammatory Biomarkers Associated with Cardiovascular Risk in Youth: A Systematic Review. Brain. Behav. Immun. 2012, 26, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Fleshner, M. Stress-Evoked Sterile Inflammation, Danger Associated Molecular Patterns (DAMPs), Microbial Associated Molecular Patterns (MAMPs) and the Inflammasome. Brain. Behav. Immun. 2013, 27, 1–7. [Google Scholar] [CrossRef]
- Elwenspoek, M.M.C.; Hengesch, X.; Leenen, F.A.D.; Schritz, A.; Sias, K.; Schaan, V.K.; Mériaux, S.B.; Schmitz, S.; Bonnemberger, F.; Schächinger, H.; et al. Proinflammatory T Cell Status Associated with Early Life Adversity. J. Immunol. 2017, 199, 4046–4055. [Google Scholar] [CrossRef] [Green Version]
- Keresztes, M.; Rudisch, T.; Tajti, J.; Ocsovszki, I.; Gardi, J. Granulocyte Activation in Humans Is Modulated by Psychological Stress and Relaxation: Research Report. Stress 2007, 10, 271–281. [Google Scholar] [CrossRef]
- Schwaiger, M.; Grinberg, M.; Moser, D.; Zang, J.C.S.; Heinrichs, M.; Hengstler, J.G.; Rahnenführer, J.; Cole, S.; Kumsta, R. Altered Stress-Induced Regulation of Genes in Monocytes in Adults with a History of Childhood Adversity. Neuropsychopharmacology 2016, 41, 2530–2540. [Google Scholar] [CrossRef] [Green Version]
- Prasad, K.N.; Bondy, S.C. Common Biochemical Defects Linkage between Post-Traumatic Stress Disorders, Mild Traumatic Brain Injury (TBI) and Penetrating TBI. Brain Res. 2015, 1599, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Tezcan, E.; Atmaca, M.; Kuloglu, M.; Ustundag, B. Free Radicals in Patients with Post-Traumatic Stress Disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2003, 253, 89–91. [Google Scholar] [CrossRef]
- Waller, C.; Rhee, D.-S.; Gröger, M.; Rappel, M.; Maier, T.; Müller, M.; Rottler, E.; Nerz, K.; Nerz, C.; Brill, S.; et al. Social Stress-Induced Oxidative DNA Damage Is Related to Prospective Cardiovascular Risk. J. Clin. Med. 2020, 9, 3783. [Google Scholar] [CrossRef]
- Boeck, C.; Koenig, A.M.; Schury, K.; Geiger, M.L.; Karabatsiakis, A.; Wilker, S.; Waller, C.; Gündel, H.; Fegert, J.M.; Calzia, E.; et al. Inflammation in Adult Women with a History of Child Maltreatment: The Involvement of Mitochondrial Alterations and Oxidative Stress. Mitochondrion 2016, 30, 197–207. [Google Scholar] [CrossRef]
- Boeck, C.; Gumpp, A.M.; Koenig, A.M.; Radermacher, P.; Karabatsiakis, A.; Kolassa, I.-T. The Association of Childhood Maltreatment With Lipid Peroxidation and DNA Damage in Postpartum Women. Front. Psychiatry 2019, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.R.; Leve, L.D.; Levitt, P.; Fisher, P.A. Childhood Adversity, Mental Health, and Oxidative Stress: A Pilot Study. PLoS ONE 2019, 14, e0215085. [Google Scholar] [CrossRef]
- Aguirre, E.; Rodríguez-Juárez, F.; Bellelli, A.; Gnaiger, E.; Cadenas, S. Kinetic Model of the Inhibition of Respiration by Endogenous Nitric Oxide in Intact Cells. Biochim. Biophys. Acta BBA—Bioenerg. 2010, 1797, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Manoli, I.; Alesci, S.; Blackman, M.R.; Su, Y.A.; Rennert, O.M.; Chrousos, G.P. Mitochondria as Key Components of the Stress Response. Trends Endocrinol. Metab. 2007, 18, 190–198. [Google Scholar] [CrossRef]
- Morava, É.; Kozicz, T. Mitochondria and the Economy of Stress (Mal)Adaptation. Neurosci. Biobehav. Rev. 2013, 37, 668–680. [Google Scholar] [CrossRef]
- Hoffmann, A.; Spengler, D. The Mitochondrion as Potential Interface in Early-Life Stress Brain Programming. Front. Behav. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef] [Green Version]
- Boeck, C.; Gumpp, A.M.; Calzia, E.; Radermacher, P.; Waller, C.; Karabatsiakis, A.; Kolassa, I.-T. The Association between Cortisol, Oxytocin, and Immune Cell Mitochondrial Oxygen Consumption in Postpartum Women with Childhood Maltreatment. Psychoneuroendocrinology 2018, 96, 69–77. [Google Scholar] [CrossRef]
- Ho, D.H.; Burch, M.L.; Musall, B.; Musall, J.B.; Hyndman, K.A.; Pollock, J.S. Early Life Stress in Male Mice Induces Superoxide Production and Endothelial Dysfunction in Adulthood. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1267–H1274. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; McManus, M.J.; Gray, J.D.; Nasca, C.; Moffat, C.; Kopinski, P.K.; Seifert, E.L.; McEwen, B.S.; Wallace, D.C. Mitochondrial Functions Modulate Neuroendocrine, Metabolic, Inflammatory, and Transcriptional Responses to Acute Psychological Stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6614–E6623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chan, Y.L.; Nguyen, L.T.; Mao, Y.; de Rosa, A.; Beh, I.T.; Chee, C.; Oliver, B.; Herok, G.; Saad, S.; et al. Moderate Traumatic Brain Injury Is Linked to Acute Behaviour Deficits and Long Term Mitochondrial Alterations. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1107–1114. [Google Scholar] [CrossRef]
- Xing, G.; Barry, E.S.; Benford, B.; Grunberg, N.E.; Li, H.; Watson, W.D.; Sharma, P. Impact of Repeated Stress on Traumatic Brain Injury-Induced Mitochondrial Electron Transport Chain Expression and Behavioral Responses in Rats. Front. Neurol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sannino, S.; Chini, B.; Grinevich, V. Lifespan Oxytocin Signaling: Maturation, Flexibility, and Stability in Newborn, Adolescent, and Aged Brain: Lifespan Oxytocin Signaling. Dev. Neurobiol. 2017, 77, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Alves, E.; Fielder, A.; Ghabriel, N.; Sawyer, M.; Buisman-Pijlman, F.T.A. Early Social Environment Affects the Endogenous Oxytocin System: A Review and Future Directions. Front. Endocrinol. 2015, 6, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veenema, A.H. Toward Understanding How Early-Life Social Experiences Alter Oxytocin- and Vasopressin-Regulated Social Behaviors. Horm. Behav. 2012, 61, 304–312. [Google Scholar] [CrossRef]
- Wigger, D.C.; Gröger, N.; Lesse, A.; Krause, S.; Merz, T.; Gündel, H.; Braun, K.; McCook, O.; Radermacher, P.; Bock, J.; et al. Maternal Separation Induces Long-Term Alterations in the Cardiac Oxytocin Receptor and Cystathionine γ -Lyase Expression in Mice. Oxid. Med. Cell. Longev. 2020, 2020, 4309605. [Google Scholar] [CrossRef] [Green Version]
- Boeck, C.; Krause, S.; Karabatsiakis, A.; Schury, K.; Gündel, H.; Waller, C.; Kolassa, I.-T. History of Child Maltreatment and Telomere Length in Immune Cell Subsets: Associations with Stress- and Attachment-Related Hormones. Dev. Psychopathol. 2018, 30, 539–551. [Google Scholar] [CrossRef]
- Krause, S.; Boeck, C.; Gumpp, A.M.; Rottler, E.; Schury, K.; Karabatsiakis, A.; Buchheim, A.; Gündel, H.; Kolassa, I.-T.; Waller, C. Child Maltreatment Is Associated with a Reduction of the Oxytocin Receptor in Peripheral Blood Mononuclear Cells. Front. Psychol. 2018, 9, 173. [Google Scholar] [CrossRef] [Green Version]
- Ellis, B.J.; Horn, A.J.; Carter, C.S.; van IJzendoorn, M.H.; Bakermans-Kranenburg, M.J. Developmental Programming of Oxytocin through Variation in Early-Life Stress: Four Meta-Analyses and a Theoretical Reinterpretation. Clin. Psychol. Rev. 2021, 86, 101985. [Google Scholar] [CrossRef]
- Szeto, A.; Nation, D.A.; Mendez, A.J.; Dominguez-Bendala, J.; Brooks, L.G.; Schneiderman, N.; McCabe, P.M. Oxytocin Attenuates NADPH-Dependent Superoxide Activity and IL-6 Secretion in Macrophages and Vascular Cells. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1495–E1501. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yang, H.-P.; Tian, S.; Wang, L.; Wang, S.C.; Zhang, F.; Wang, Y.-F. Oxytocin-Secreting System: A Major Part of the Neuroendocrine Center Regulating Immunologic Activity. J. Neuroimmunol. 2015, 289, 152–161. [Google Scholar] [CrossRef]
- Kingsbury, M.A.; Bilbo, S.D. The Inflammatory Event of Birth: How Oxytocin Signaling May Guide the Development of the Brain and Gastrointestinal System. Front. Neuroendocrinol. 2019, 55, 100794. [Google Scholar] [CrossRef]
- Reiss, A.B.; Glass, D.S.; Lam, E.; Glass, A.D.; De Leon, J.; Kasselman, L.J. Oxytocin: Potential to Mitigate Cardiovascular Risk. Peptides 2019, 117, 170089. [Google Scholar] [CrossRef]
- Monstein, H.-J.; Grahn, N.; Truedsson, M.; Ohlsson, B. Oxytocin and Oxytocin-Receptor MRNA Expression in the Human Gastrointestinal Tract: A Polymerase Chain Reaction Study. Regul. Pept. 2004, 119, 39–44. [Google Scholar] [CrossRef]
- Gimpl, G.; Fahrenholz, F. The Oxytocin Receptor System: Structure, Function, and Regulation. Physiol. Rev. 2001, 81, 629–683. [Google Scholar] [CrossRef] [Green Version]
- Szczepanska-Sadowska, E.; Cudnoch-Jedrzejewska, A.; Wsol, A. The Role of Oxytocin and Vasopressin in the Pathophysiology of Heart Failure in Pregnancy and in Fetal and Neonatal Life. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H639–H651. [Google Scholar] [CrossRef]
- Wang, P.; Wang, S.C.; Yang, H.; Lv, C.; Jia, S.; Liu, X.; Wang, X.; Meng, D.; Qin, D.; Zhu, H.; et al. Therapeutic Potential of Oxytocin in Atherosclerotic Cardiovascular Disease: Mechanisms and Signaling Pathways. Front. Neurosci. 2019, 13, 454. [Google Scholar] [CrossRef] [Green Version]
- Moghimian, M.; Faghihi, M.; Karimian, S.M.; Imani, A.; Houshmand, F.; Azizi, Y. The Role of Central Oxytocin in Stress-Induced Cardioprotection in Ischemic-Reperfused Heart Model. J. Cardiol. 2013, 61, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Wsol, A.; Cudnoch-Jedrzejewska, A.; Szczepanska-Sadowska, E.; Kowalewski, S.; Puchalska, L. Oxytocin in the cardiovascular responses to stress. J. Physiol. Pharmacol. 2008, 59, 123–127. [Google Scholar]
- Wsol, A.; Cudnoch-Je˛drzejewska, A.; Szczepanska-Sadowska, E.; Kowalewski, S.; Dobruch, J. Central Oxytocin Modulation of Acute Stress-Induced Cardiovascular Responses after Myocardial Infarction in the Rat. Stress 2009, 12, 517–525. [Google Scholar] [CrossRef]
- Chaves, V.E.; Tilelli, C.Q.; Brito, N.A.; Brito, M.N. Role of Oxytocin in Energy Metabolism. Peptides 2013, 45, 9–14. [Google Scholar] [CrossRef]
- Florian, M.; Jankowski, M.; Gutkowska, J. Oxytocin Increases Glucose Uptake in Neonatal Rat Cardiomyocytes. Endocrinology 2010, 151, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutkowska, J.; Jankowski, M.; Antunes-Rodrigues, J. The Role of Oxytocin in Cardiovascular Regulation. Braz. J. Med. Biol. Res. 2014, 47, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutkowska, J.; Jankowski, M. Oxytocin Revisited: Its Role in Cardiovascular Regulation: Role of OT in Cardiovascular Regulation. J. Neuroendocrinol. 2012, 24, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Amini-Khoei, H.; Mohammadi-Asl, A.; Amiri, S.; Hosseini, M.-J.; Momeny, M.; Hassanipour, M.; Rastegar, M.; Haj-Mirzaian, A.; Mirzaian, A.H.-; Sanjarimoghaddam, H.; et al. Oxytocin Mitigated the Depressive-like Behaviors of Maternal Separation Stress through Modulating Mitochondrial Function and Neuroinflammation. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 76, 169–178. [Google Scholar] [CrossRef]
- Carter, C.S. The Oxytocin–Vasopressin Pathway in the Context of Love and Fear. Front. Endocrinol. 2017, 8, 356. [Google Scholar] [CrossRef] [Green Version]
- Zingg, H.H. Vasopressin and Oxytocin Receptors. Bailliere’s Clin. Endocrinol. Metab. 1996, 10, 75–96. [Google Scholar] [CrossRef]
- Vincent, J.-L.; Su, F. Physiology and Pathophysiology of the Vasopressinergic System. Best Pract. Res. Clin. Anaesthesiol. 2008, 22, 243–252. [Google Scholar] [CrossRef]
- Levy, B.; Collin, S.; Sennoun, N.; Ducrocq, N.; Kimmoun, A.; Asfar, P.; Perez, P.; Meziani, F. Vascular Hyporesponsiveness to Vasopressors in Septic Shock: From Bench to Bedside. Intensive Care Med. 2010, 36, 2019–2029. [Google Scholar] [CrossRef]
- Asfar, P.; Hauser, B.; Iványi, Z.; Ehrmann, U.; Kick, J.; Albicini, M.; Vogt, J.; Wachter, U.; Brückner, U.B.; Radermacher, P.; et al. Low-Dose Terlipressin during Long-Term Hyperdynamic Porcine Endotoxemia: Effects on Hepatosplanchnic Perfusion, Oxygen Exchange, and Metabolism*. Crit. Care Med. 2005, 33, 373–380. [Google Scholar] [CrossRef]
- Asfar, P.; Russell, J.A.; Tuckermann, J.; Radermacher, P. Selepressin in Septic Shock: A Step Toward Decatecholaminization?*. Crit. Care Med. 2016, 44, 234–236. [Google Scholar] [CrossRef]
- Beloncle, F.; Meziani, F.; Lerolle, N.; Radermacher, P.; Asfar, P. Does Vasopressor Therapy Have an Indication in Hemorrhagic Shock? Ann. Intensive Care 2013, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Demiselle, J.; Fage, N.; Radermacher, P.; Asfar, P. Vasopressin and Its Analogues in Shock States: A Review. Ann. Intensive Care 2020, 10, 9. [Google Scholar] [CrossRef]
- Simon, F.; Giudici, R.; Scheuerle, A.; Gröger, M.; Asfar, P.; Vogt, J.A.; Wachter, U.; Ploner, F.; Georgieff, M.; Möller, P.; et al. Comparison of Cardiac, Hepatic, and Renal Effects of Arginine Vasopressin and Noradrenaline during Porcine Fecal Peritonitis: A Randomized Controlled Trial. Crit. Care 2009, 13, R113. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Radeos, M. Severe Delayed Postpartum Hemorrhage after Cesarean Section. J. Emerg. Med. 2018, 55, 408–410. [Google Scholar] [CrossRef]
- Düşünceli, F.; İşeri, S.Ö.; Ercan, F.; Gedik, N.; Yeğen, C.; Yeğen, B.Ç. Oxytocin Alleviates Hepatic Ischemia–Reperfusion Injury in Rats. Peptides 2008, 29, 1216–1222. [Google Scholar] [CrossRef]
- Erbaş, O.; Ergenoglu, A.M.; Akdemir, A.; Yeniel, A.Ö.; Taskiran, D. Comparison of Melatonin and Oxytocin in the Prevention of Critical Illness Polyneuropathy in Rats with Experimentally Induced Sepsis. J. Surg. Res. 2013, 183, 313–320. [Google Scholar] [CrossRef]
- İşeri, S.Ö.; Şener, G.; Saǧlam, B.; Gedik, N.; Ercan, F.; Yeǧen, B.Ç. Oxytocin Protects Against Sepsis-Induced Multiple Organ Damage: Role of Neutrophils. J. Surg. Res. 2005, 126, 73–81. [Google Scholar] [CrossRef]
- Carter, C.S.; Kenkel, W.M.; MacLean, E.L.; Wilson, S.R.; Perkeybile, A.M.; Yee, J.R.; Ferris, C.F.; Nazarloo, H.P.; Porges, S.W.; Davis, J.M.; et al. Is Oxytocin “Nature’s Medicine”? Pharmacol. Rev. 2020, 72, 829–861. [Google Scholar] [CrossRef]
- Flanagan, J.C.; Sippel, L.M.; Wahlquist, A.; Moran-Santa Maria, M.M.; Back, S.E. Augmenting Prolonged Exposure Therapy for PTSD with Intranasal Oxytocin: A Randomized, Placebo-Controlled Pilot Trial. J. Psychiatr. Res. 2018, 98, 64–69. [Google Scholar] [CrossRef]
- Flanagan, J.C.; Mitchell, J.M.; Baker, N.L.; Woolley, J.; Wangelin, B.; Back, S.E.; McQuaid, J.R.; Neylan, T.C.; Wolfe, W.R.; Brady, K.T. Enhancing Prolonged Exposure Therapy for PTSD among Veterans with Oxytocin: Design of a Multisite Randomized Controlled Trial. Contemp. Clin. Trials 2020, 95, 106074. [Google Scholar] [CrossRef]
- Rault, J.-L.; Carter, C.S.; Garner, J.P.; Marchant-Forde, J.N.; Richert, B.T.; Lay, D.C. Repeated Intranasal Oxytocin Administration in Early Life Dysregulates the HPA Axis and Alters Social Behavior. Physiol. Behav. 2013, 112–113, 40–48. [Google Scholar] [CrossRef]
- Navarra, P.; Dello Russo, C.; Mancuso, C.; Preziosi, P.; Grossman, A. Gaseous Neuromodulators in the Control of Neuroendocrine Stress Axis. Ann. N. Y. Acad. Sci. 2000, 917, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.J.; Calvert, J.W.; Butler, J.; Lefer, D.J. The Cardioprotective Actions of Hydrogen Sulfide in Acute Myocardial Infarction and Heart Failure. Scientifica 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Gaseotransmitters: New Frontiers for Translational Science. Sci. Transl. Med. 2010, 2, 59ps54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Kimura, H. The Possible Role of Hydrogen Sulfide as an Endogenous Neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine-Lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, C.; Hafner, S.; Scheuerle, A.; Möller, P.; Huber-Lang, M.; Jung, B.; Nubaum, B.; McCook, O.; Gröger, M.; Wagner, F.; et al. The Role of Cystathionine-γ-Lyase In Blunt Chest Trauma in Cigarette Smoke Exposed Mice. Shock 2017, 47, 491–499. [Google Scholar] [CrossRef]
- McCook, O.; Radermacher, P.; Volani, C.; Asfar, P.; Ignatius, A.; Kemmler, J.; Möller, P.; Szabó, C.; Whiteman, M.; Wood, M.E.; et al. H2S during Circulatory Shock: Some Unresolved Questions. Nitric Oxide 2014, 41, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Wagner, F.; Scheuerle, A.; Weber, S.; Stahl, B.; McCook, O.; Knöferl, M.W.; Huber-Lang, M.; Seitz, D.H.; Thomas, J.; Asfar, P.; et al. Cardiopulmonary, Histologic, and Inflammatory Effects of Intravenous Na2S After Blunt Chest Trauma-Induced Lung Contusion in Mice. J. Trauma Inj. Infect. Crit. Care 2011, 71, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Stenzel, T.; Weidgang, C.; Wagner, K.; Wagner, F.; Gröger, M.; Weber, S.; Stahl, B.; Wachter, U.; Vogt, J.; Calzia, E.; et al. Association of Kidney Tissue Barrier Disrupture and Renal Dysfunction in Resuscitated Murine Septic Shock. Shock 2016, 46, 398–404. [Google Scholar] [CrossRef]
- Merz, T.; Vogt, J.A.; Wachter, U.; Calzia, E.; Szabo, C.; Wang, R.; Radermacher, P.; McCook, O. Impact of Hyperglycemia on Cystathionine-γ-Lyase Expression during Resuscitated Murine Septic Shock. Intensive Care Med. Exp. 2017, 5, 30. [Google Scholar] [CrossRef]
- Merz, T.; Wepler, M.; Nußbaum, B.; Vogt, J.; Calzia, E.; Wang, R.; Szabo, C.; Radermacher, P.; McCook, O. Cystathionine-γ-Lyase Expression Is Associated with Mitochondrial Respiration during Sepsis-Induced Acute Kidney Injury in Swine with Atherosclerosis. Intensive Care Med. Exp. 2018, 6, 43. [Google Scholar] [CrossRef]
- Peleli, M.; Bibli, S.-I.; Li, Z.; Chatzianastasiou, A.; Varela, A.; Katsouda, A.; Zukunft, S.; Bucci, M.; Vellecco, V.; Davos, C.H.; et al. Cardiovascular Phenotype of Mice Lacking 3-Mercaptopyruvate Sulfurtransferase. Biochem. Pharmacol. 2020, 176, 113833. [Google Scholar] [CrossRef]
- Trautwein, B.; Merz, T.; Denoix, N.; Szabo, C.; Calzia, E.; Radermacher, P.; McCook, O. ΔMST and the Regulation of Cardiac CSE and OTR Expression in Trauma and Hemorrhage. Antioxidants 2021, 10, 233. [Google Scholar] [CrossRef]
- Latorre, E.; Torregrossa, R.; Wood, M.E.; Whiteman, M.; Harries, L.W. Mitochondria-Targeted Hydrogen Sulfide Attenuates Endothelial Senescence by Selective Induction of Splicing Factors HNRNPD and SRSF2. Aging 2018, 10, 1666–1681. [Google Scholar] [CrossRef]
- Xu, K.; Wu, F.; Xu, K.; Li, Z.; Wei, X.; Lu, Q.; Jiang, T.; Wu, F.; Xu, X.; Xiao, J.; et al. NaHS Restores Mitochondrial Function and Inhibits Autophagy by Activating the PI3K/Akt/MTOR Signalling Pathway to Improve Functional Recovery after Traumatic Brain Injury. Chem. Biol. Interact. 2018, 286, 96–105. [Google Scholar] [CrossRef]
- Guan, R.; Cai, Z.; Wang, J.; Ding, M.; Li, Z.; Xu, J.; Li, Y.; Li, J.; Yao, H.; Liu, W.; et al. Hydrogen Sulfide Attenuates Mitochondrial Dysfunction-Induced Cellular Senescence and Apoptosis in Alveolar Epithelial Cells by Upregulating Sirtuin 1. Aging 2019, 11, 11844–11864. [Google Scholar] [CrossRef]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of Mitochondrial Bioenergetic Function by Hydrogen Sulfide. Part I. Biochemical and Physiological Mechanisms: Biochemistry of H 2 S and Mitochondrial Function. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [Green Version]
- Gröger, M.; Scheuerle, A.; Wagner, F.; Simon, F.; Matallo, J.; McCook, O.; Seifritz, A.; Stahl, B.; Wachter, U.; Vogt, J.A.; et al. Effects of Pretreatment Hypothermia During Resuscitated Porcine Hemorrhagic Shock. Crit. Care Med. 2013, 41, e105–e117. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, K.; Wagner, F.; Gröger, M.; Weber, S.; Barth, E.; Vogt, J.A.; Wachter, U.; Huber-Lang, M.; Knöferl, M.W.; Albuszies, G.; et al. Cardiac and Metabolic Effects of Hypothermia and Inhaled Hydrogen Sulfide in Anesthetized and Ventilated Mice*. Crit. Care Med. 2010, 38, 588–595. [Google Scholar] [CrossRef] [Green Version]
- Asfar, P.; Calzia, E.; Radermacher, P. Is Pharmacological, H2S-Induced ‘Suspended Animation’ Feasible in the ICU? Crit. Care 2014, 18, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Módis, K.; Bos, E.M.; Calzia, E.; van Goor, H.; Coletta, C.; Papapetropoulos, A.; Hellmich, M.R.; Radermacher, P.; Bouillaud, F.; Szabo, C. Regulation of Mitochondrial Bioenergetic Function by Hydrogen Sulfide. Part II. Pathophysiological and Therapeutic Aspects: Pathophysiology of H 2 S and Mitochondrial Function. Br. J. Pharmacol. 2014, 171, 2123–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marutani, E.; Ichinose, F. Emerging pharmacological tools to control hydrogen sulfide signaling in critical illness. Intensive Care Med. Exp. 2020, 8, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, K.R. Hydrogen sulfide as an oxygen sensor. Clin. Chem. Lab. Med. 2013, 51, 623–632. [Google Scholar] [CrossRef]
- Bauer, M.; Radermacher, P.; Wepler, M. Sodium Thiosulfate: A New Player for Circulatory Shock and Ischemia/Reperfusion Injury? In Annual Update in Intensive Care and Emergency Medicine; Vincent, J.L., Ed.; Springer: Cham, Switzerland, 2019; pp. 167–178. [Google Scholar]
- Jiang, X.; Huang, Y.; Lin, W.; Gao, D.; Fei, Z. Protective Effects of Hydrogen Sulfide in a Rat Model of Traumatic Brain Injury via Activation of Mitochondrial Adenosine Triphosphate–Sensitive Potassium Channels and Reduction of Oxidative Stress. J. Surg. Res. 2013, 184, e27–e35. [Google Scholar] [CrossRef]
- Zhang, M.; Shan, H.; Chang, P.; Wang, T.; Dong, W.; Chen, X.; Tao, L. Hydrogen Sulfide Offers Neuroprotection on Traumatic Brain Injury in Parallel with Reduced Apoptosis and Autophagy in Mice. PLoS ONE 2014, 9, e87241. [Google Scholar] [CrossRef]
- Sun, J.; Li, X.; Gu, X.; Du, H.; Zhang, G.; Wu, J.; Wang, F. Neuroprotective Effect of Hydrogen Sulfide against Glutamate-Induced Oxidative Stress Is Mediated via the P53/Glutaminase 2 Pathway after Traumatic Brain Injury. Aging 2021, 13, 7180–7189. [Google Scholar] [CrossRef]
- Satterly, S.A.; Salgar, S.; Hoffer, Z.; Hempel, J.; DeHart, M.J.; Wingerd, M.; Raywin, H.; Stallings, J.D.; Martin, M. Hydrogen Sulfide Improves Resuscitation via Non-Hibernatory Mechanisms in a Porcine Shock Model. J. Surg. Res. 2015, 199, 197–210. [Google Scholar] [CrossRef]
- Datzmann, T.; Hoffmann, A.; McCook, O.; Merz, T.; Wachter, U.; Preuss, J.; Vettorazzi, S.; Calzia, E.; Gröger, M.; Kohn, F.; et al. Effects of Sodium Thiosulfate (Na2S2O3) during Resuscitation from Hemorrhagic Shock in Swine with Preexisting Atherosclerosis. Pharmacol. Res. 2020, 151, 104536. [Google Scholar] [CrossRef]
- Morrison, M.L.; Blackwood, J.E.; Lockett, S.L.; Iwata, A.; Winn, R.K.; Roth, M.B. Surviving Blood Loss Using Hydrogen Sulfide. J. Trauma Inj. Infect. Crit. Care 2008, 65, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Chai, W.; Wang, Y.; Lin, J.-Y.; Sun, X.-D.; Yao, L.-N.; Yang, Y.-H.; Zhao, H.; Jiang, W.; Gao, C.-J.; Ding, Q. Exogenous Hydrogen Sulfide Protects Against Traumatic Hemorrhagic Shock Via Attenuation of Oxidative Stress. J. Surg. Res. 2012, 176, 210–219. [Google Scholar] [CrossRef]
- Gao, C.; Xu, D.-Q.; Gao, C.-J.; Ding, Q.; Yao, L.-N.; Li, Z.-C.; Chai, W. An Exogenous Hydrogen Sulphide Donor, NaHS, Inhibits the Nuclear Factor ΚB Inhibitor Kinase/Nuclear Factor ΚB Inhibitor/Nuclear Factor-ΚB Signaling Pathway and Exerts Cardioprotective Effects in a Rat Hemorrhagic Shock Model. Biol. Pharm. Bull. 2012, 35, 1029–1034. [Google Scholar] [CrossRef] [Green Version]
- Issa, K.; Kimmoun, A.; Collin, S.; Ganster, F.; Fremont-Orlowski, S.; Asfar, P.; Mertes, P.-M.; Levy, B. Compared Effects of Inhibition and Exogenous Administration of Hydrogen Sulphide in Ischaemia-Reperfusion Injury. Crit. Care 2013, 17, R129. [Google Scholar] [CrossRef] [Green Version]
- Dyson, A.; Dal-Pizzol, F.; Sabbatini, G.; Lach, A.B.; Galfo, F.; dos Santos Cardoso, J.; Pescador Mendonça, B.; Hargreaves, I.; Bollen Pinto, B.; Bromage, D.I.; et al. Ammonium Tetrathiomolybdate Following Ischemia/Reperfusion Injury: Chemistry, Pharmacology, and Impact of a New Class of Sulfide Donor in Preclinical Injury Models. PLoS Med. 2017, 14, e1002310. [Google Scholar] [CrossRef] [Green Version]
- Mok, Y.-Y.P.; Mohammed Atan, M.S.B.; Ping, C.Y.; Jing, W.Z.; Bhatia, M.; Moochhala, S.; Moore, P.K. Role of Hydrogen Sulphide in Haemorrhagic Shock in the Rat: Protective Effect of Inhibitors of Hydrogen Sulphide Biosynthesis: Hydrogen Sulphide and Shock. Br. J. Pharmacol. 2004, 143, 881–889. [Google Scholar] [CrossRef]
- Mok, Y.-Y.P.; Moore, P.K. Hydrogen Sulphide Is Pro-Inflammatory in Haemorrhagic Shock. Inflamm. Res. 2008, 57, 512–518. [Google Scholar] [CrossRef]
- Drabek, T.; Kochanek, P.M.; Stezoski, J.; Wu, X.; Bayr, H.; Morhard, R.C.; Stezoski, S.W.; Tisherman, S.A. Intravenous Hydrogen Sulfide Does Not Induce Hypothermia or Improve Survival from Hemorrhagic Shock in Pigs. Shock 2011, 35, 67–73. [Google Scholar] [CrossRef]
- Bracht, H.; Scheuerle, A.; Gröger, M.; Hauser, B.; Matallo, J.; McCook, O.; Seifritz, A.; Wachter, U.; Vogt, J.A.; Asfar, P.; et al. Effects of Intravenous Sulfide during Resuscitated Porcine Hemorrhagic Shock*. Crit. Care Med. 2012, 40, 2157–2167. [Google Scholar] [CrossRef]
- Whiteman, M.; Li, L.; Rose, P.; Tan, C.-H.; Parkinson, D.B.; Moore, P.K. The Effect of Hydrogen Sulfide Donors on Lipopolysaccharide-Induced Formation of Inflammatory Mediators in Macrophages. Antioxid. Redox Signal. 2010, 12, 1147–1154. [Google Scholar] [CrossRef]
- Wepler, M.; Merz, T.; Wachter, U.; Vogt, J.; Calzia, E.; Scheuerle, A.; Möller, P.; Gröger, M.; Kress, S.; Fink, M.; et al. The Mitochondria-Targeted H2S-Donor AP39 in a Murine Model of Combined Hemorrhagic Shock and Blunt Chest Trauma. Shock 2019, 52, 230–239. [Google Scholar] [CrossRef]
- Mendonça, B.P.; Cardoso, J.D.S.; Michels, M.; Vieira, A.C.; Wendhausen, D.; Manfredini, A.; Singer, M.; Dal-Pizzol, F.; Dyson, A. Neuroprotective Effects of Ammonium Tetrathiomolybdate, a Slow-Release Sulfide Donor, in a Rodent Model of Regional Stroke. Intensive Care Med. Exp. 2020, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeer, P. Medical Uses of Sodium Thiosulfate. J. Neurol. Neuromedicine 2016, 1, 28–30. [Google Scholar] [CrossRef] [Green Version]
- Paris, E.I.; Pelisov, M.G. Use of Sodium Thiosulfate in Shock Due to Burns. Voen. Med. Zhurnal 1966, 5, 38–40. [Google Scholar]
- Oksman, T.M.; Levandovskii, I.V.; Epishin, Y.N.; Vrana, M.; Blažek, Z. Sodium Thiosulfate in the Treatment of Early Postischemic Disorders. Bull. Exp. Biol. Med. 1981, 92, 1160–1163. [Google Scholar] [CrossRef]
- Broner, C.W.; Shenep, J.L.; Stidham, G.L.; Stokes, D.C.; Fairclough, D.; Schonbaum, G.R.; Rehg, J.E.; Hildner, W.K. Effect of Antioxidants in Experimental Escherichia Coli Septicemia. Circ. Shock 1989, 29, 77–92. [Google Scholar]
- Sakaguchi, M.; Marutani, E.; Shin, H.; Chen, W.; Hanaoka, K.; Xian, M.; Ichinose, F. Sodium Thiosulfate Attenuates Acute Lung Injury in Mice. Anesthesiology 2014, 121, 1248–1257. [Google Scholar] [CrossRef] [Green Version]
- Shirozu, K.; Tokuda, K.; Marutani, E.; Lefer, D.J.; Wang, R.; Ichinose, F. OP05 Cystathionine γ-Lyase Deficiency Protects Mice from Galactosamine/Lipopolysaccharide-Induced Acute Liver Failure. Nitric Oxide 2013, 31, S21. [Google Scholar] [CrossRef]
- Renieris, G.; Droggiti, D.-E.; Katrini, K.; Koufargyris, P.; Gkavogianni, T.; Karakike, E.; Antonakos, N.; Damoraki, G.; Karageorgos, A.; Sabracos, L.; et al. Host Cystathionine-γ Lyase Derived Hydrogen Sulfide Protects against Pseudomonas Aeruginosa Sepsis. PLoS Pathog. 2021, 17, e1009473. [Google Scholar] [CrossRef]
- Acero, G.; Nava Catorce, M.; González-Mendoza, R.; Meraz-Rodríguez, M.A.; Hernández-Zimbron, L.F.; González-Salinas, R.; Gevorkian, G. Sodium Thiosulphate Attenuates Brain Inflammation Induced by Systemic Lipopolysaccharide Administration in C57BL/6J Mice. Inflammopharmacology 2017, 25, 585–593. [Google Scholar] [CrossRef]
- Marutani, E.; Yamada, M.; Ida, T.; Tokuda, K.; Ikeda, K.; Kai, S.; Shirozu, K.; Hayashida, K.; Kosugi, S.; Hanaoka, K.; et al. Thiosulfate Mediates Cytoprotective Effects of Hydrogen Sulfide Against Neuronal Ischemia. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Snijder, P.M.; Frenay, A.R.; de Boer, R.A.; Pasch, A.; Hillebrands, J.L.; Leuvenink, H.G.D.; van Goor, H. Exogenous Administration of Thiosulfate, a Donor of Hydrogen Sulfide, Attenuates Angiotensin II-Induced Hypertensive Heart Disease in Rats: Sulfide and Hypertensive Heart Disease. Br. J. Pharmacol. 2015, 172, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, I.T.N.; Wiggenhauser, L.M.; Bulthuis, M.; Hillebrands, J.-L.; Feelisch, M.; Verhaar, M.C.; van Goor, H.; Joles, J.A. Cardiac Protection by Oral Sodium Thiosulfate in a Rat Model of L-NNA-Induced Heart Disease. Front. Pharmacol. 2021, 12, 650968. [Google Scholar] [CrossRef]
- Snijder, P.M.; Frenay, A.-R.S.; Koning, A.M.; Bachtler, M.; Pasch, A.; Kwakernaak, A.J.; van den Berg, E.; Bos, E.M.; Hillebrands, J.-L.; Navis, G.; et al. Sodium Thiosulfate Attenuates Angiotensin II-Induced Hypertension, Proteinuria and Renal Damage11These Authors Contributed Equally to This Manuscript. Nitric Oxide 2014, 42, 87–98. [Google Scholar] [CrossRef]
- Nguyen, I.T.N.; Klooster, A.; Minnion, M.; Feelisch, M.; Verhaar, M.C.; van Goor, H.; Joles, J.A. Sodium Thiosulfate Improves Renal Function and Oxygenation in L-NNA–Induced Hypertension in Rats. Kidney Int. 2020, 98, 366–377. [Google Scholar] [CrossRef]
- Gröger, M.; Hogg, M.; Abdelsalam, E.; Kress, S.; Hoffmann, A.; Stahl, B.; Saub, V.; Denoix, N.; McCook, O.; Calzia, E.; et al. Effects of Sodium Thiosulfate During Resuscitation from Trauma-and-Hemorrhage in Cystathionine γ-Lyase (CSE) Knockout Mice. Shock 2021. [Google Scholar] [CrossRef]
- Coletti, R.; Almeida-Pereira, G.; Elias, L.L.K.; Antunes-Rodrigues, J. Effects of Hydrogen Sulfide (H2S) on Water Intake and Vasopressin and Oxytocin Secretion Induced by Fluid Deprivation. Horm. Behav. 2015, 67, 12–20. [Google Scholar] [CrossRef]
- Flannigan, K.L.; Agbor, T.A.; Blackler, R.W.; Kim, J.J.; Khan, W.I.; Verdu, E.F.; Ferraz, J.G.P.; Wallace, J.L. Impaired Hydrogen Sulfide Synthesis and IL-10 Signaling Underlie Hyperhomocysteinemia-Associated Exacerbation of Colitis. Proc. Natl. Acad. Sci. USA 2014, 111, 13559–13564. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Lee, C.; Martin, Z.; Li, X.; Koike, Y.; Hock, A.; Zani-Ruttenstock, E.; Zani, A.; Pierro, A. Intestinal Epithelial Injury Induced by Maternal Separation Is Protected by Hydrogen Sulfide. J. Pediatr. Surg. 2017, 52, 40–44. [Google Scholar] [CrossRef]
- Mani, S.; Li, H.; Untereiner, A.; Wu, L.; Yang, G.; Austin, R.C.; Dickhout, J.G.; Lhoták, Š.; Meng, Q.H.; Wang, R. Decreased Endogenous Production of Hydrogen Sulfide Accelerates Atherosclerosis. Circulation 2013, 127, 2523–2534. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.; Untereiner, A.; Wu, L.; Wang, R. Hydrogen Sulfide and the Pathogenesis of Atherosclerosis. Antioxid. Redox Signal. 2014, 20, 805–817. [Google Scholar] [CrossRef]
- Wang, R. Physiological Implications of Hydrogen Sulfide: A Whiff Exploration That Blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merz, T.; Lukaschewski, B.; Wigger, D.; Rupprecht, A.; Wepler, M.; Gröger, M.; Hartmann, C.; Whiteman, M.; Szabo, C.; Wang, R.; et al. Interaction of the Hydrogen Sulfide System with the Oxytocin System in the Injured Mouse Heart. Intensive Care Med. Exp. 2018, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nußbaum, B.L.; McCook, O.; Hartmann, C.; Matallo, J.; Wepler, M.; Antonucci, E.; Kalbitz, M.; Huber-Lang, M.; Georgieff, M.; Calzia, E.; et al. Left Ventricular Function during Porcine-Resuscitated Septic Shock with Pre-Existing Atherosclerosis. Intensive Care Med. Exp. 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merz, T.; Denoix, N.; Wigger, D.; Waller, C.; Wepler, M.; Vettorazzi, S.; Tuckermann, J.; Radermacher, P.; McCook, O. The Role of Glucocorticoid Receptor and Oxytocin Receptor in the Septic Heart in a Clinically Relevant, Resuscitated Porcine Model With Underlying Atherosclerosis. Front. Endocrinol. 2020, 11, 299. [Google Scholar] [CrossRef]
- Datzmann, T.; Kapapa, T.; Scheuerle, A.; McCook, O.; Merz, T.; Unmuth, S.; Hoffmann, A.; Mathieu, R.; Mayer, S.; Mauer, U.M.; et al. In-Depth Characterization of a Long-Term, Resuscitated Model of Acute Subdural Hematoma–Induced Brain Injury. J. Neurosurg. 2021, 134, 223–234. [Google Scholar] [CrossRef]
- McCook, O.; Scheuerle, A.; Denoix, N.; Kapapa, T.; Radermacher, P.; Merz, T. Localization of the Hydrogen Sulfide and Oxytocin Systems at the Depth of the Sulci in a Porcine Model of Acute Subdural Hematoma. Neural Regen. Res. 2021, 16, 2376. [Google Scholar] [CrossRef]
- Denoix, N.; Merz, T.; Unmuth, S.; Hoffmann, A.; Nespoli, E.; Scheuerle, A.; Huber-Lang, M.; Gündel, H.; Waller, C.; Radermacher, P.; et al. Cerebral Immunohistochemical Characterization of the H2S and the Oxytocin Systems in a Porcine Model of Acute Subdural Hematoma. Front. Neurol. 2020, 11, 649. [Google Scholar] [CrossRef]
- Denoix, N.; McCook, O.; Ecker, S.; Wang, R.; Waller, C.; Radermacher, P.; Merz, T. The Interaction of the Endogenous Hydrogen Sulfide and Oxytocin Systems in Fluid Regulation and the Cardiovascular System. Antioxidants 2020, 9, 748. [Google Scholar] [CrossRef]
- Matallo, J.; Vogt, J.; McCook, O.; Wachter, U.; Tillmans, F.; Groeger, M.; Szabo, C.; Georgieff, M.; Radermacher, P.; Calzia, E. Sulfide-Inhibition of Mitochondrial Respiration at Very Low Oxygen Concentrations. Nitric Oxide 2014, 41, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Nußbaum, B.L.; Vogt, J.; Wachter, U.; McCook, O.; Wepler, M.; Matallo, J.; Calzia, E.; Gröger, M.; Georgieff, M.; Wood, M.E.; et al. Metabolic, Cardiac, and Renal Effects of the Slow Hydrogen Sulfide-Releasing Molecule GYY4137 During Resuscitated Septic Shock in Swine with Pre-Existing Coronary Artery Disease. Shock 2017, 48, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Hösli, I.; Büchel, J. Stellenwert von Kontraktionsmitteln bei der postpartalen Hämorrhagie. Gynäkologe 2019, 52, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Heesen, M.; Carvalho, B.; Carvalho, J.C.A.; Duvekot, J.J.; Dyer, R.A.; Lucas, D.N.; McDonnell, N.; Orbach-Zinger, S.; Kinsella, S.M. International Consensus Statement on the Use of Uterotonic Agents during Caesarean Section. Anaesthesia 2019, 74, 1305–1319. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.R.; Shnitko, T.A.; Blue, S.W.; Kaucher, A.V.; Winchell, A.J.; Erikson, D.W.; Grant, K.A.; Leggio, L. Labeled Oxytocin Administered via the Intranasal Route Reaches the Brain in Rhesus Macaques. Nat. Commun. 2020, 11, 2783. [Google Scholar] [CrossRef]
- Martins, D.A.; Mazibuko, N.; Zelaya, F.; Vasilakopoulou, S.; Loveridge, J.; Oates, A.; Maltezos, S.; Mehta, M.; Wastling, S.; Howard, M.; et al. Effects of Route of Administration on Oxytocin-Induced Changes in Regional Cerebral Blood Flow in Humans. Nat. Commun. 2020, 11, 1160. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.R.; Scheidweiler, K.B.; Diao, X.X.; Akhlaghi, F.; Cummins, A.; Huestis, M.A.; Leggio, L.; Averbeck, B.B. Oxytocin by Intranasal and Intravenous Routes Reaches the Cerebrospinal Fluid in Rhesus Macaques: Determination Using a Novel Oxytocin Assay. Mol. Psychiatry 2018, 23, 115–122. [Google Scholar] [CrossRef]
- Ventriglio, A.; Bellomo, A.; Ricci, F.; Magnifico, G.; Rinaldi, A.; Borraccino, L.; Piccininni, C.; Cuoco, F.; Gianfelice, G.; Fornaro, M.; et al. New Pharmacological Targets for the Treatment of Schizophrenia: A Literature Review. Curr. Top. Med. Chem. 2021. [Google Scholar] [CrossRef]
- De Berardis, D.; Marini, S.; Iasevoli, F.; Tomasetti, C.; de Bartolomeis, A.; Mazza, M.; Valchera, A.; Fornaro, M.; Cavuto, M.; Srinivasan, V.; et al. The role of intranasal oxytocin in the treatment of patients with schizophrenia: A systematic review. CNS Neurol. Disord. Drug Targets 2013, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Li, S.; Wang, C.; Liu, Y.; Li, W.; Yan, X.; Chen, Q.; Han, S. Distinct Oxytocin Effects on Belief Updating in Response to Desirable and Undesirable Feedback. Proc. Natl. Acad. Sci. USA 2016, 113, 9256–9261. [Google Scholar] [CrossRef] [Green Version]
- Saphire-Bernstein, S.; Way, B.M.; Kim, H.S.; Sherman, D.K.; Taylor, S.E. Oxytocin Receptor Gene (OXTR) Is Related to Psychological Resources. Proc. Natl. Acad. Sci. USA 2011, 108, 15118–15122. [Google Scholar] [CrossRef] [Green Version]
- Olff, M.; Frijling, J.L.; Kubzansky, L.D.; Bradley, B.; Ellenbogen, M.A.; Cardoso, C.; Bartz, J.A.; Yee, J.R.; van Zuiden, M. The Role of Oxytocin in Social Bonding, Stress Regulation and Mental Health: An Update on the Moderating Effects of Context and Interindividual Differences. Psychoneuroendocrinology 2013, 38, 1883–1894. [Google Scholar] [CrossRef] [Green Version]
- Donadon, M.F.; Martin-Santos, R.; Osório, F. de L. The Associations Between Oxytocin and Trauma in Humans: A Systematic Review. Front. Pharmacol. 2018, 9, 154. [Google Scholar] [CrossRef] [Green Version]
- Domes, G.; Lischke, A.; Berger, C.; Grossmann, A.; Hauenstein, K.; Heinrichs, M.; Herpertz, S.C. Effects of Intranasal Oxytocin on Emotional Face Processing in Women. Psychoneuroendocrinology 2010, 35, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.; Ford, E.; Croft, J.; Merrick, M.; Rolle, I.; Giles, W. Sex-Specific Relationships between Adverse Childhood Experiences and Chronic Obstructive Pulmonary Disease in Five States. Int. J. Chron. Obstruct. Pulmon. Dis. 2014, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batten, S.V.; Aslan, M.; Maciejewski, P.K.; Mazure, C.M. Childhood Maltreatment as a Risk Factor for Adult Cardiovascular Disease and Depression. J. Clin. Psychiatry 2004, 65, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lacey, R.E. Does the Association of Child Maltreatment with Adult Cardiovascular Disease Differ by Gender? Heart 2020, 106, 1289–1290. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.L.G.; Hammerton, G.; Howe, L.D.; Rich-Edwards, J.; Halligan, S.; Fraser, A. Sex Differences in the Association between Childhood Maltreatment and Cardiovascular Disease in the UK Biobank. Heart 2020, 106, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Ladwig, K.-H.; Waller, C. Geschlechtsspezifische Aspekte bei der koronaren Herzkrankheit. Bundesgesundheitsblatt-Gesundh.-Gesundh. 2014, 57, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Worth, H.; Buhl, R.; Criée, C.-P.; Kardos, P.; Mailänder, C.; Vogelmeier, C. The ‘Real-Life’ COPD Patient in Germany: The DACCORD Study. Respir. Med. 2016, 111, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Mikolić, A.; van Klaveren, D.; Oude Groeniger, J.; Wiegers, E.J.A.; Lingsma, H.F.; Zeldovich, M.; von Steinbüchel, N.; Maas, A.I.R.; Roeters van Lennep, J.E.; Polinder, S.; et al. Differences between Men and Women in Treatment and Outcome after Traumatic Brain Injury. J. Neurotrauma 2020, 38, 235–251. [Google Scholar] [CrossRef]
- Medland, J.E.; Pohl, C.S.; Edwards, L.L.; Frandsen, S.; Bagley, K.; Li, Y.; Moeser, A.J. Early Life Adversity in Piglets Induces Long-Term Upregulation of the Enteric Cholinergic Nervous System and Heightened, Sex-Specific Secretomotor Neuron Responses. Neurogastroenterol. Motil. 2016, 28, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Pohl, C.S.; Medland, J.E.; Mackey, E.; Edwards, L.L.; Bagley, K.D.; DeWilde, M.P.; Williams, K.J.; Moeser, A.J. Early Weaning Stress Induces Chronic Functional Diarrhea, Intestinal Barrier Defects, and Increased Mast Cell Activity in a Porcine Model of Early Life Adversity. Neurogastroenterol. Motil. 2017, 29, e13118. [Google Scholar] [CrossRef]
- Tucker, L.B.; Fu, A.H.; McCabe, J.T. Hippocampal-Dependent Cognitive Dysfunction Following Repeated Diffuse Rotational Brain Injury in Male and Female Mice. J. Neurotrauma 2021, 38, 1585–1606. [Google Scholar] [CrossRef]
- Gwarzo, I.H.; Perez-Patron, M.; Xu, X.; Radcliff, T.; Horney, J. Traumatic Brain Injury Related Hospitalizations: Factors Associated with In-Hospital Mortality among Elderly Patients Hospitalized with a TBI. Brain Inj. 2021, 35, 554–562. [Google Scholar] [CrossRef]
- Miller, G.F.; Daugherty, J.; Waltzman, D.; Sarmiento, K. Predictors of Traumatic Brain Injury Morbidity and Mortality: Examination of Data from the National Trauma Data Bank: Predictors of TBI Morbidity & Mortality. Injury 2021, 52, 1138–1144. [Google Scholar] [CrossRef]
- Kerezoudis, P.; Goyal, A.; Puffer, R.C.; Parney, I.F.; Meyer, F.B.; Bydon, M. Morbidity and Mortality in Elderly Patients Undergoing Evacuation of Acute Traumatic Subdural Hematoma. Neurosurg. Focus 2020, 49, E22. [Google Scholar] [CrossRef]
- Sharma, S.R.; Gonda, X.; Dome, P.; Tarazi, F.I. What’s Love Got to Do with It: Role of Oxytocin in Trauma, Attachment and Resilience. Pharmacol. Ther. 2020, 214, 107602. [Google Scholar] [CrossRef]
- Newell, E.A.; Todd, B.P.; Luo, Z.; Evans, L.P.; Ferguson, P.J.; Bassuk, A.G. A Mouse Model for Juvenile, Lateral Fluid Percussion Brain Injury Reveals Sex-Dependent Differences in Neuroinflammation and Functional Recovery. J. Neurotrauma 2020, 37, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Levin, H.S.; Temkin, N.R.; Barber, J.; Nelson, L.D.; Robertson, C.; Brennan, J.; Stein, M.B.; Yue, J.K.; Giacino, J.T.; McCrea, M.A.; et al. Association of Sex and Age With Mild Traumatic Brain Injury–Related Symptoms: A TRACK-TBI Study. JAMA Netw. Open 2021, 4, e213046. [Google Scholar] [CrossRef]
- Daglas, M.; Galle, A.; Draxler, D.F.; Ho, H.; Liu, Z.; Sashindranath, M.; Medcalf, R.L. Sex-dependent Effects of Tranexamic Acid on Blood-brain Barrier Permeability and the Immune Response Following Traumatic Brain Injury in Mice. J. Thromb. Haemost. 2020, 18, 2658–2671. [Google Scholar] [CrossRef]
- Chesnut, R.; Videtta, W.; Vespa, P.; Le Roux, P. Intracranial Pressure Monitoring: Fundamental Considerations and Rationale for Monitoring. Neurocrit. Care 2014, 21, 64–84. [Google Scholar] [CrossRef]
- Leach, M.R.; Shutter, L.A. How Much Oxygen for the Injured Brain—Can Invasive Parenchymal Catheters Help? Curr. Opin. Crit. Care 2021, 27, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Oddo, M.; Bösel, J. Monitoring of Brain and Systemic Oxygenation in Neurocritical Care Patients. Neurocrit. Care 2014, 21, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Talving, P.; Karamanos, E.; Teixeira, P.G.; Skiada, D.; Lam, L.; Belzberg, H.; Inaba, K.; Demetriades, D. Intracranial Pressure Monitoring in Severe Head Injury: Compliance with Brain Trauma Foundation Guidelines and Effect on Outcomes: A Prospective Study: Clinical Article. J. Neurosurg. 2013, 119, 1248–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettervik, T.S.; Engquist, H.; Howells, T.; Lenell, S.; Rostami, E.; Hillered, L.; Enblad, P.; Lewén, A. Arterial Oxygenation in Traumatic Brain Injury—Relation to Cerebral Energy Metabolism, Autoregulation, and Clinical Outcome. J. Intensive Care Med. 2020, 36, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.-Y.; Robertson, C.S. Monitoring Biomarkers of Cellular Injury and Death in Acute Brain Injury. Neurocrit. Care 2014, 21, 187–214. [Google Scholar] [CrossRef]
- Lazaridis, C.; Andrews, C.M. Brain Tissue Oxygenation, Lactate-Pyruvate Ratio, and Cerebrovascular Pressure Reactivity Monitoring in Severe Traumatic Brain Injury: Systematic Review and Viewpoint. Neurocrit. Care 2014, 21, 345–355. [Google Scholar] [CrossRef]
- Makarenko, S.; Griesdale, D.E.; Gooderham, P.; Sekhon, M.S. Multimodal Neuromonitoring for Traumatic Brain Injury: A Shift towards Individualized Therapy. J. Clin. Neurosci. 2016, 26, 8–13. [Google Scholar] [CrossRef]
- Denoix, N.; McCook, O.; Scheuerle, A.; Kapapa, T.; Hoffmann, A.; Gündel, H.; Waller, C.; Szabo, C.; Radermacher, P.; Merz, T. Brain Histology and Immunohistochemistry after Resuscitation from Hemorrhagic Shock (HS) in Swine with Pre-Existing Atherosclerosis: Effects of Sodium Thiosulfate (Na2S2O3). Intensive Care Med. Exp. 2021, submitted. [Google Scholar]
- Neuwelt, E.A.; Brummett, R.E.; Doolittle, N.D.; Muldoon, L.L.; Kroll, R.A.; Pagel, M.A.; Dojan, R.; Church, V.; Remsen, L.G.; Bubalo, J.S. First Evidence of Otoprotection Against Carboplatin-Induced Hearing Loss with a Two-Compartment System in Patients with Central Nervous System Malignancy Using Sodium Thiosulfate. J. Pharmacol. Exp. Ther. 1998, 286, 8. [Google Scholar]
- Mizuta, Y.; Tokuda, K.; Guo, J.; Zhang, S.; Narahara, S.; Kawano, T.; Murata, M.; Yamaura, K.; Hoka, S.; Hashizume, M.; et al. Sodium Thiosulfate Prevents Doxorubicin-Induced DNA Damage and Apoptosis in Cardiomyocytes in Mice. Life Sci. 2020, 257, 118074. [Google Scholar] [CrossRef]
- Bebarta, V.S.; Pitotti, R.L.; Dixon, P.; Lairet, J.R.; Bush, A.; Tanen, D.A. Hydroxocobalamin Versus Sodium Thiosulfate for the Treatment of Acute Cyanide Toxicity in a Swine (Sus scrofa) Model. Ann. Emerg. Med. 2012, 59, 532–539. [Google Scholar] [CrossRef]
- Bebarta, V.S.; Brittain, M.; Chan, A.; Garrett, N.; Yoon, D.; Burney, T.; Mukai, D.; Babin, M.; Pilz, R.B.; Mahon, S.B.; et al. Sodium Nitrite and Sodium Thiosulfate Are Effective Against Acute Cyanide Poisoning When Administered by Intramuscular Injection. Ann. Emerg. Med. 2017, 69, 718–725.e4. [Google Scholar] [CrossRef]
- Bebarta, V.S.; Tanen, D.A.; Lairet, J.; Dixon, P.S.; Valtier, S.; Bush, A. Hydroxocobalamin and Sodium Thiosulfate Versus Sodium Nitrite and Sodium Thiosulfate in the Treatment of Acute Cyanide Toxicity in a Swine (Sus scrofa) Model. Ann. Emerg. Med. 2010, 55, 345–351. [Google Scholar] [CrossRef]
- Merz, T.; Denoix, N.; Wepler, M.; Gäßler, H.; Messerer, D.A.C.; Hartmann, C.; Datzmann, T.; Radermacher, P.; McCook, O. H2S in Acute Lung Injury: A Therapeutic Dead End(?). Intensive Care Med. Exp. 2020, 8, 33. [Google Scholar] [CrossRef]
- Dominic, P.; Ahmad, J.; Bhandari, R.; Pardue, S.; Solorzano, J.; Jaisingh, K.; Watts, M.; Bailey, S.R.; Orr, A.W.; Kevil, C.G.; et al. Decreased availability of nitric oxide and hydrogen sulfide is a hallmark of COVID-19. Redox Biol. 2021, 43, 101982. [Google Scholar] [CrossRef]
- Radermacher, P.; Calzia, E.; McCook, O.; Wachter, U.; Szabo, C. To the Editor. Shock 2021, 55, 138–139. [Google Scholar] [CrossRef]
- Renieris, G.; Katrini, K.; Damoulari, C.; Akinosoglou, K.; Psarrakis, C.; Kyriakopoulou, M.; Dimopoulos, G.; Lada, M.; Koufargyris, P.; Giamarellos-Bourboulis, E.J. Serum Hydrogen Sulfide and Outcome Association in Pneumonia by the SARS-CoV-2 Coronavirus. Shock 2020. [Google Scholar] [CrossRef]
- Citi, V.; Martelli, A.; Brancaleone, V.; Brogi, S.; Gojon, G.; Montanaro, R.; Morales, G.; Testai, L.; Calderone, V. Anti-inflammatory and antiviral roles of hydrogen sulfide: Rationale for considering H2S donors in COVID-19 therapy. Br. J. Pharmacol. 2020, 177, 4931–4941. [Google Scholar] [CrossRef]
- Evgen’ev, M.B.; Frenkel, A. Possible application of H2S-producing compounds in therapy of coronavirus (COVID-19) infection and pneumonia. Cell Stress Chaperones. 2020, 25, 713–715. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author and Year | Species | Experimental Challenge/Trauma/ Treatment | Interaction of OT and H2S |
|---|---|---|---|
| Trautwein et al., 2021 [134] | Mice | Naïve ΔMST animals Hemorrhagic Shock wt Hemorrhagic Shock & Blunt Chest Trauma wt | Constitutive CSE & OTR in cardiomyocytes CSE & OTR↓ CSE & OTR↓ CSE &OTR↓↓ |
| Wigger et al., 2020 [85] | Mice | Maternal Separation (Early Life Stress) LTSS (long) STSS (short) | CSE & OTR↓↓ CSE↓ & OTR↑↑ |
| Flannigan et al., 2014 [178] | Rats (vs. wt) | Diet for 6 weeks: “B-Def” lacked vitamins B6, B9, and B12 Colitis induction: 1. Drinking water supplemented with dextran sodium sulfate 2. Intracolonic administration of the hapten dinitrobenzene sulfonic acid 3. IL-10–deficient mice intra-colonical diallyl disulfide administration | In 1., 2., and 3., diet-induced hyperhomocysteinemia ↑colitis diallyl disulfide administration: ↓severity of colitis IL-10-deficient mice on a normal diet had ↓colonic H2S synthesis, a 40% ↑serum homocysteine IL-10–deficient mice fed the vitamin B-deficient diet exhibited ↑↑colonic inflammation Administration of IL-10 to the IL-10–deficient mice restored colonic H2S synthesis ↓serum homocysteine |
| Li et al., 2017 [179] | Mice | Maternal Seperation (vs. control animals) intraperitoneal NaHS administration (vs. vehicle) | Maternal Separation led to: ↓Crypt lengths, ↓goblet cells per crypt, ↓glutathione peroxidase activity, ↑expression of thiobarbituric acid reactive substances, ↑inducible nitric oxide synthase mRNA, ↑IL-6, ↑TNFα ↑myeloperoxidase Administration of NaHS: counteracted negative effects of maternal separation |
| Mani et al., 2013 [180] | Mice CSE−/− (vs. wt) | Knock out and atherogenic diet intraperitoneal NaHS administration (vs. PBS injection) | Early fatty streak lesions in the aortic root ↑Plasma levels of cholesterol, ↑low-density lipoprotein cholesterol Hyperhomocysteinemia ↑Lesional oxidative stress and adhesion molecule expression ↑aortic intimal proliferation CSE−/− treated with NaHS: inhibited the accelerated atherosclerosis development |
| Merz et al., 2018 [183] | Mice CSE−/− (vs. wt) | Native wt Blunt Chest Trauma (and cigarette smoke exposure (CS)) Blunt Chest Trauma CSE−/− (& CS) Blunt Chest Trauma CSE−/− and GYY4137 administration (and CS) | Constitutive OTR in cardiomyocytes OTR↓ OTR↓↓ OTR↑↑ |
| Nußbaum et al., 2016 [184] | Swine (hypercholesteremic vs. sham animals) | Septic Shock | Systemic Troponin↑ ↓ Cardiac output Cardiac CSE↓ |
| Merz et al., 2020 [185] | Swine (hypercholesteremic vs. sham animals) | Septic Shock | Cardiac OTR↓ |
| Coletti et al., 2015 [177] | Rats | Water deprivation for 12 and 24 h intra cerebroventricular Na2S | 24 h water deprivation: ↑Activity of sulfide-generating enzymes in the medial basal hypothalamus Na2S administration: ↓Water intake, ↑arginine vasopressin, OT and corticosterone in plasma, ↓medial basal hypothalamus nitrate/nitrite content |
| Denoix et al., 2020 [188] | Swine | ASDH | CSE, CBS, OTR, and OT were localized to: (i) Cortical neurons in the gyri and at the base of sulci, where pressure-induced injury leads to maximal stress in the gyrencephalic brain (ii) In the parenchyma at the base of the sulci (iii) microvasculature and pial arteries (iv) Resident and infiltrating immune cells. |
| Author and Year | Species | Experimental Challenge | Therapeutic Potential of OT and H2S in Trauma |
|---|---|---|---|
| Ellis et al., 2021 [88] | Humans | ELS Intranasally administered OT | People who grew up under more adverse conditions tend to have ↓endogenous OT Early adversity is associated with higher levels of methylation of the OTR gene Adults who report ↓levels of childhood adversity tend to show ↑positive responses to intranasal OT |
| Flanagan et al., 2018 [119] | Humans | Posttraumatic Stress Disorder (PTSD) Treatment: Prolonged Exposure Therapy and intranasal OT (vs. placebo) | OT group: ↓PTSD & depression symptoms during Prolonged Exposure Therapy ↑Working alliance scores |
| Bracht et al., 2012 [159] | Swine | Hemorrhagic Shock Intravenous Na2S administration 1. 2 h before hemorrhage 2. Simultaneously with blood removal 3. At the beginning of retransfusion of shed blood | 2. simultaneous treatment group: ↓Progressive kidney, liver, and cardiocirculatory dysfunction ↓Histological damage of lung, liver, and kidney Na2S: ↓mortality irrespective of the timing of its administration |
| Whiteman et al., 2010 [160] | Murine RAW264.7 macrophages | Lipopolysaccharide (LPS) treatment NaHS or GYY4137 administration | GYY4137 led to: Concentration-dependently ↓LPS-induced release of proinflammatory mediators (IL-1β, IL-6, TNF⍺, NO, and PGE(2)), ↑synthesis of the antiinflammatory IL-10 NaHSlet to: Biphasic effect on proinflammatory mediators, at high concentrations, ↑synthesis of IL-1β, IL-6, NO, PGE(2) and TNF⍺ |
| Wepler et al., 2019 [161] | Mice | Wave-induced thorax trauma and hemorrhagic shock (vs. sham) Intravenous bolus injection high and low dose of AP39 (vs. vehicle) | High-dose AP39 in thorax trauma: ↓Systemic inflammation, ↓inducible nitric oxide synthase and IκBα in lung tissue thorax trauma and hemorrhagic shock: High-dose AP39: ↓Mean arterial pressure, ↑norepinephrine requirements, ↑mortality Low-dose AP39: no effects |
| Matallo et al., 2014 [190] | Immortalized cell line (AMJ2-C11) | Na2S solution stimulation | Mitochondria analysis: The onset of inhibition of cell respiration by sulfide occurs earlier under a continuous exposure when approaching the anoxic condition. |
| Nußbaum et al., 2017 [191] | Swine (Pre-existing coronary artery disease) | Septic Shock (vs. sham) intravenous GYY4137 administration | GYY4137 led to: ↑Aerobic glucose oxidation, ↑requirements of exogenous glucose to maintain normoglycemia, ↓arterial pH, ↓base excess ↓Cardiac eNOS expression, ↑troponin levels no effect on cardiac and kidney function or the systemic inflammatory response |
| Lee et al., 2020 [194] | Rhesus Macaques | Labelled OT administration nebulizer/intravenous infusion/intranasal | 2 h after OT administration: Labeled OT is found after intranasal administration in orbitofrontal cortex, striatum, brainstem, and thalamus (these lie in the trajectories of the olfactory and trigeminal nerves, bypassing the blood-brain barrier) |
| Martins et al., 2020 [195] | Humans | healthy volunteers OT administration nebulizer/intravenous infusion/standard nasal spray (vs. placebo or saline) | OT-induced: ↓Amygdala perfusion (a key hub of the OT central circuitry)due to OT ↑in systemic circulation following both intranasal and intravenous OT administration Robust evidence confirming the validity of the intranasal route to target specific brain regions |
| Lee et al., 2018 [196] | Rhesus Macaques | Labelled OT administration: intravenous infusion/intranasal (vs. intranasal saline as control) | Cerebro-spinal fluid penetrance of labelled OT exogenous OT delivered by intranasal and intravenous administration Intravenous administration of labelled OT did not lead to increased endogenous OT or endogenous OT in the cerebro-spinal fluid |
| Ma et al., 2016 [199] | Humans | Intranasally administered OT | ↑Optimistic belief updating by facilitating updates of desirable feedback, but ↓updates of undesirable feedback ↑Learning rate (the strength of association between estimation error and subsequent update) of desirable feedback ↑Participants’ confidence in their estimates after receiving desirable but not undesirable feedback |
| Saphire-Bernstein et al., 2011 [200] | Humans | Genotype of OTR | Link between the OTR SNP rs53576 and psychological resources “A” allele carriers have ↓levels of optimism, mastery, and self-esteem, relative to G/G homozygotes |
| Domes et al., 2010 [203] | Humans | Presented with fearful, angry, happy and neutral facial expressions after a single dose of intranasal OT or placebo administration | Blood-oxygen-level-dependent signal was ↑in the left amygdala, the fusiform gyrus & the superior temporal gyrus in response to fearful faces & in the inferior frontal gyrus in response to angry and happy faces following OT treatment. independent of basal plasma levels of OT, estradiol, and progesterone |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merz, T.; McCook, O.; Denoix, N.; Radermacher, P.; Waller, C.; Kapapa, T. Biological Connection of Psychological Stress and Polytrauma under Intensive Care: The Role of Oxytocin and Hydrogen Sulfide. Int. J. Mol. Sci. 2021, 22, 9192. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179192
Merz T, McCook O, Denoix N, Radermacher P, Waller C, Kapapa T. Biological Connection of Psychological Stress and Polytrauma under Intensive Care: The Role of Oxytocin and Hydrogen Sulfide. International Journal of Molecular Sciences. 2021; 22(17):9192. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179192
Chicago/Turabian StyleMerz, Tamara, Oscar McCook, Nicole Denoix, Peter Radermacher, Christiane Waller, and Thomas Kapapa. 2021. "Biological Connection of Psychological Stress and Polytrauma under Intensive Care: The Role of Oxytocin and Hydrogen Sulfide" International Journal of Molecular Sciences 22, no. 17: 9192. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179192