
Multiple Leptin Signalling Pathways in the Control of Metabolism and Fertility: A Means to Different Ends?

Abstract
:1. Introduction
2. Neuronal Targets of Leptin Receptor Signalling in the Brain
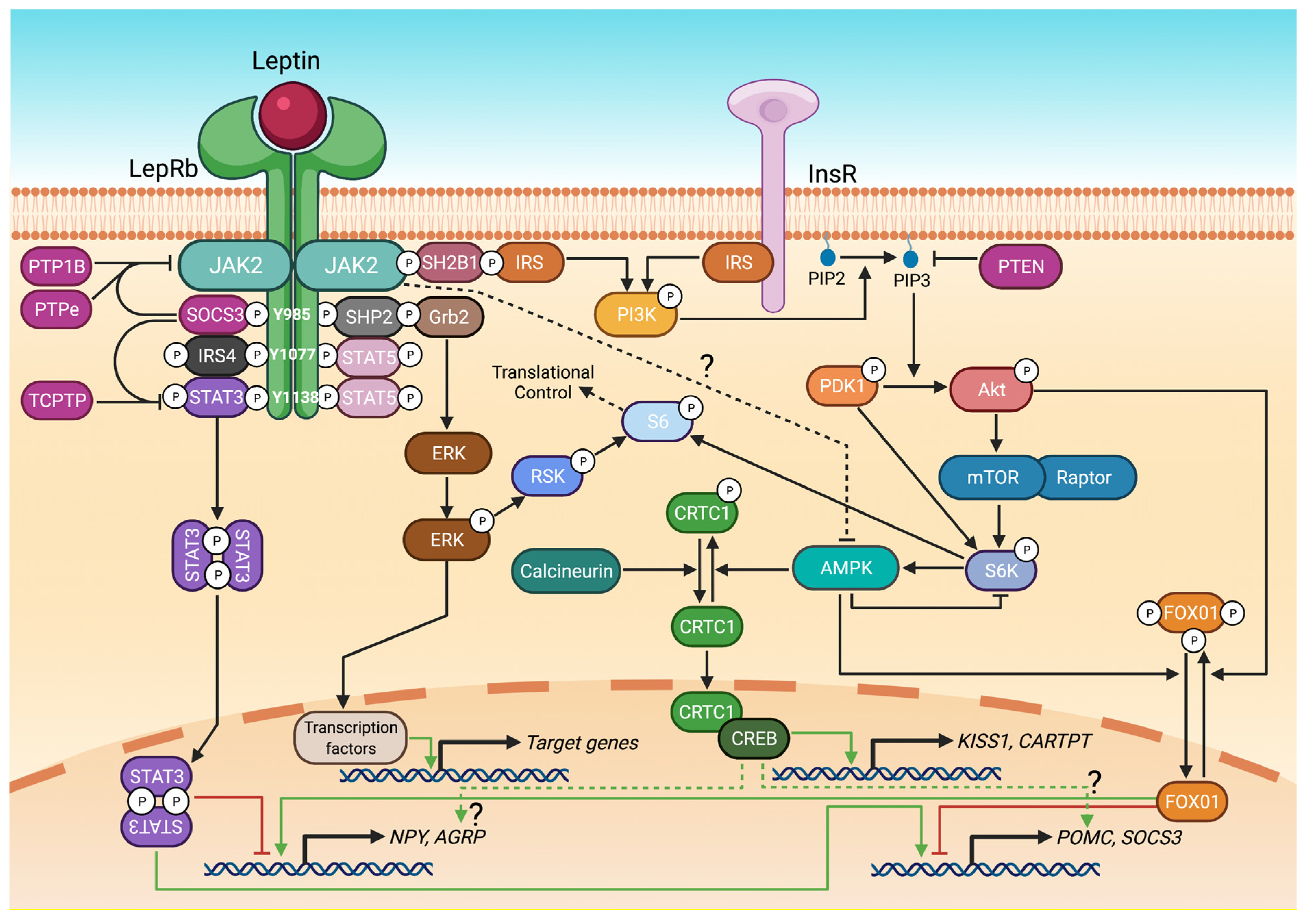
3. Leptin Receptor Signalling Pathways
3.1. STAT3 and STAT5
3.2. PI3K/Akt, FOX01 and mTOR/S6
3.3. ERK1/2
3.4. CRTC
3.5. Interactions between Leptin and Oestrogen Receptor (ER) Signalling
4. The Pathogenesis of Leptin Resistance
4.1. SOCS3
4.2. PTP1B
4.3. PTPe
4.4. TCPTP
4.5. PTEN
5. Summary and Areas for Future Focus

{kind=link}
| Signalling Molecule Deleted | Cell Type Deleted from | Description of Findings | Reference |
|---|---|---|---|
| STAT3 | LepRb cells | Obesity but not delayed puberty or infertility. No effect on oestrogen-induced weight loss. | [32,33,40,83] |
| Brain cells | Obesity and infertility. | [38] | |
| POMC neurons | Mild obesity, defects in compensatory refeeding and decreased Pomc mRNA expression. | [108] | |
| AgRP neurons | Modest weight gain, increased Npy, decreased Socs3 mRNA. | [109] | |
| STAT5 | Global | Increased food intake and altered energy expenditure. | [45] |
| LepRb cells | No effect on bodyweight, puberty timing or fertility. | [33] | |
| PI3K | POMC neurons | Reduced sensitivity to leptin’s anorectic effects. | [59] |
| PI3K catalytic subunits, p110α and p110β | POMC or AgRP neurons | POMC p110β (but not p110α KO) KO: leptin resistance, obesity, loss of insulin- and leptin-stimulated neuronal firing. AgRP p110β (but not p110α KO) KO: increased leptin sensitivity, resistance to diet-induced obesity. POMC PI3K KO mice: disrupted leptin and insulin-induced POMC neuronal firing. | [110] [53] |
| Kisspeptin neurons | Reduced kisspeptin cell number in AVPV (females) and arcuate nucleus (males); reduced female fertility. | [111] | |
| SH2B1 | Global | Hyperphagia, obesity. Resolved by neuronal SB2B1 rescue. | [112,113] |
| LepRb cells | Obesity, insulin resistance. | [114] | |
| PTEN | LepRb cells | Increased PI3K activity, reduced adiposity. | [115] |
| POMC neurons | Leptin resistance, obesity, reduced POMC firing. | [106] | |
| FOX01 | POMC neurons | Increased sensitivity to leptin’s anorectic effects. | [60] |
| AgRP neurons | Reduced food intake, increased glucose and leptin sensitivity. | [116] | |
| S6 kinase | Global | Leptin insensitivity. Protection against obesity, increases insulin sensitivity. | [61] [62] |
| POMC or AgRP neurons | Impaired glucose homeostasis and altered POMC and AgRP neuronal excitability, no effect on food intake or bodyweight. | [117] | |
| SHP2 | Brain neurons | Obesity, reduced leptin-induced pERK1/2 (pSTAT3 was preserved). Reproductive impairment. | [71] |
| AMPKα | POMC or AgRP neurons | POMC KO: obesity but remained sensitive to leptin. AgRP KO: age-dependent lean phenotype. | [118] |
| Kisspeptin neurons | AMPKα2-KO: fasting-induced disruption of oestrous cycles prevented. AMPKα1 KO: subnutrition-induced puberty delay prevented. | [119,120] | |
| CRTC1 | Global | Obesity and infertility (Altarejos) Obesity but normal fertility (Breuillaud). | [37] [79] |
| SOCS3 | Global | +/− mice have reduced diet-induced obesity and leptin resistance. | [93] |
| Brain cells | Protection from diet-induced obesity. | [94] | |
| Brain neurons | Protection from diet-induced obesity (most evident in males) and leptin resistance; delayed the onset of diet-induced infertility in females. | [23] | |
| LepRb cells | Reduced food intake after fasting; reduced Agrp and Npy mRNA. | [121] | |
| POMC neurons | Improved leptin sensitivity and glucose homeostasis. | [95] | |
| PTP1b | Global | Resistance to diabetes and diet-induced obesity. | [98] |
| Brain cells | Resistance to diet-induced obesity, hypersensitive to leptin. | [96,99] | |
| Brain neurons | Resistance to diet-induced obesity, not diet-induced female infertility. | [122] | |
| LepRb cells | Resistance to diet-induced obesity, hypersensitive to leptin. | [100] | |
| POMC neurons | Resistance to diet-induced obesity, hypersensitive to leptin. | [123] | |
| PTPe | Global | Reduced female obesity and improved blood glucose control. | [101] |
| TCPTP | Brain cells | Enhanced leptin sensitivity, reduced leptin-induced food intake. | [104] |
| AgRP neurons | Resistant to diet-induced obesity, increased energy expenditure. | [103] | |
| PTEN | LepRb cells | Lean phenotype due to increased energy expenditure. | [106] |
| Signalling Pathway | Target Gene | Description of Findings | Reference |
|---|---|---|---|
| STAT3 | Npy | Leptin actions via the 221-bp region of the Npy promotor, which possesses two putative STAT3 binding sites. | [124] |
| Agrp | STAT binding sites upstream of Agrp (but not STAT3 itself) are required for fasting-induced Agrp transcription. | [124,125] | |
| Pomc | Leptin and STAT3 increased Pomc promoter activity. A 30-bp promoter element is required for leptin regulation. | [58,126] | |
| TRH | STAT3-response elements identified in Trh promoter. | [127] | |
| Socs3 | STAT3-bindis to the Socs3 promoter. | [127] | |
| ERα | ERα promoter contains a STAT3 response element. | [85] | |
| FOX01 and STAT3 | Agrp and Pomc | Leptin-induced phospho-STAT3 activates the POMC promoter via an SP1-binding site which overlaps with a FOX01-binding element. FOXO1 binds to STAT3 and prevents it from interacting with the promoter. FOXO1 and STAT3 exert opposing actions on Agrp and Pomc expression. FOXO1 activates Agrp and inhibitis Pomc. | [56,57,58] |
| FOX01 | NPY, AGRP and Pomc | FOXO1 binds upstream of the NPY coding region and increases NPY promoter activity. Foxo1-mediated NPY transcription is negatively regulated by leptin, insulin and PI3K/Akt signalling. Foxo1 increases AGRP promoter activity but not Pomc promoter activity. | [58] |
| AMPK-CRTC1 | Cartpt and Kiss1 | Cartpt and Kiss1 promoters contain CREB binding sites. Leptin recruits CRTC1 to Cart and Kiss1 promoters. | [37] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Mounzih, K.; Lu, R.; Chehab, F.F. Leptin treatment rescues the sterility of genetically obese ob/ob males. Endocrinology 1997, 138, 1190–1193. [Google Scholar] [CrossRef] [PubMed]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.; Lawrence, E.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 1999, 341, 879–884. [Google Scholar] [CrossRef]
- Cohen, P.; Zhao, C.; Cai, X.; Montez, J.M.; Rohani, S.C.; Feinstein, P.; Mombaerts, P.; Friedman, J.M. Selective deletion of leptin receptor in neurons leads to obesity. J. Clin. Investig. 2001, 108, 1113–1121. [Google Scholar] [CrossRef]
- Quennell, J.H.; Mulligan, A.C.; Tups, A.; Liu, X.; Phipps, S.J.; Kemp, C.J.; Herbison, A.E.; Grattan, D.R.; Anderson, G.M. Leptin indirectly regulates gonadotropin-releasing hormone neuronal function. Endocrinology 2009, 150, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Boden, G.; Chen, X.; Mozzoli, M.; Ryan, I. Effect of fasting on serum leptin in normal human subjects. J. Clin. Endocrinol. Metab. 1996, 81, 3419–3423. [Google Scholar]
- Schwartz, M.W.; Baskin, D.G.; Bukowski, T.R.; Kuijper, J.L.; Foster, D.; Lasser, G.; Prunkard, D.E.; Porte, D., Jr.; Woods, S.C.; Seeley, R.J.; et al. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes 1996, 45, 531–535. [Google Scholar] [CrossRef] [Green Version]
- Welt, C.K.; Chan, J.L.; Bullen, J.; Murphy, R.; Smith, P.; DePaoli, A.M.; Karalis, A.; Mantzoros, C.S. Recombinant human leptin in women with hypothalamic amenorrhea. N. Engl. J. Med. 2004, 351, 987–997. [Google Scholar] [CrossRef]
- Saladin, R.; De Vos, P.; Guerre-Millo, M.; Leturque, A.; Girard, J.; Staels, B.; Auwerx, J. Transient increase in obese gene expression after food intake or insulin administration. Nature 1995, 377, 527–529. [Google Scholar] [CrossRef]
- Ahima, R.S.; Prabakaran, D.; Mantzoros, C.; Qu, D.; Lowell, B.; Maratos-Flier, E.; Flier, J.S. Role of leptin in the neuroendocrine response to fasting. Nature 1996, 382, 250–252. [Google Scholar] [CrossRef]
- Munzberg, H.; Morrison, C.D. Structure, production and signalling of leptin. Metabolism 2015, 64, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Licinio, J.; Caglayan, S.; Ozata, M.; Yildiz, B.O.; de Miranda, P.B.; O’Kirwan, F.; Whitby, R.; Liang, L.; Cohen, P.; Bhasin, S.; et al. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behaviour in leptin-deficient adults. Proc. Natl. Acad. Sci. USA 2004, 101, 4531–4536. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, I.S.; Bullmore, E.; Keogh, J.; Gillard, J.; O’Rahilly, S.; Fletcher, P.C. Leptin regulates striatal regions and human eating behaviour. Science 2007, 317, 1355. [Google Scholar] [CrossRef] [Green Version]
- Knight, Z.A.; Hannan, K.S.; Greenberg, M.L.; Friedman, J.M. Hyperleptinemia is required for the development of leptin resistance. PLoS ONE 2010, 5, e11376. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signalling. Trends Pharm. Sci. 2015, 36, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Pedroso, J.A.B.; Ramos-Lobo, A.M.; Donato, J., Jr. SOCS3 as a future target to treat metabolic disorders. Hormones (Athens) 2019, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Panzhinskiy, E.; Hua, Y.; Culver, B.; Ren, J.; Nair, S. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia 2013, 56, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Tortoriello, D.V.; McMinn, J.; Chua, S.C. Dietary-induced obesity and hypothalamic infertility in female DBA/2J mice. Endocrinology 2004, 145, 1238–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talmor, A.; Dunphy, B. Female obesity and infertility. Best Pract. Res. Clin. Obs. Gynaecol. 2015, 29, 498–506. [Google Scholar] [CrossRef] [PubMed]
- McMinn, J.E.; Liu, S.M.; Liu, H.; Dragatsis, I.; Dietrich, P.; Ludwig, T.; Boozer, C.N.; Chua, S.C., Jr. Neuronal deletion of Lepr elicits diabesity in mice without affecting cold tolerance or fertility. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E403–E411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwen, H.J.; Inglis, M.A.; Quennell, J.H.; Grattan, D.R.; Anderson, G.M. Deletion of Suppressor of Cytokine Signalling 3 from Forebrain Neurons Delays Infertility and Onset of Hypothalamic Leptin Resistance in Response to a High Caloric Diet. J. Neurosci. 2016, 36, 7142–7153. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.C.; Anderson, G.M. Neuroendocrine integration of nutritional signals on reproduction. J. Mol. Endocrinol. 2017, 58, R107–R128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingham, N.C.; Anderson, K.K.; Reuter, A.L.; Stallings, N.R.; Parker, K.L. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology 2008, 149, 2138–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Wall, E.; Leshan, R.; Xu, A.W.; Balthasar, N.; Coppari, R.; Liu, S.M.; Jo, Y.H.; MacKenzie, R.G.; Allison, D.B.; Dun, N.J.; et al. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 2008, 149, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Zuure, W.A.; Roberts, A.L.; Quennell, J.H.; Anderson, G.M. Leptin signalling in GABA neurons, but not glutamate neurons, is required for reproductive function. J. Neurosci. 2013, 33, 17874–17883. [Google Scholar] [CrossRef] [Green Version]
- Egan, O.K.; Inglis, M.A.; Anderson, G.M. Leptin Signalling in AgRP Neurons Modulates Puberty Onset and Adult Fertility in Mice. J. Neurosci. 2017, 37, 3875–3886. [Google Scholar] [CrossRef]
- Oakley, A.E.; Clifton, D.K.; Steiner, R.A. Kisspeptin signalling in the brain. Endocr. Rev. 2009, 30, 713–743. [Google Scholar] [CrossRef]
- Donato, J., Jr.; Cravo, R.M.; Frazao, R.; Gautron, L.; Scott, M.M.; Lachey, J.; Castro, I.A.; Margatho, L.O.; Lee, S.; Lee, C.; et al. Leptin’s effect on puberty in mice is relayed by the ventral premammillary nucleus and does not require signalling in Kiss1 neurons. J. Clin. Investig. 2011, 121, 355–368. [Google Scholar] [CrossRef]
- Allison, M.B.; Myers, M.G., Jr. 20 years of leptin: Connecting leptin signalling to biological function. J. Endocrinol. 2014, 223, T25–T35. [Google Scholar] [CrossRef]
- Bates, S.H.; Stearns, W.H.; Dundon, T.A.; Schubert, M.; Tso, A.W.; Wang, Y.; Banks, A.S.; Lavery, H.J.; Haq, A.K.; Maratos-Flier, E.; et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 2003, 421, 856–859. [Google Scholar] [CrossRef]
- Singireddy, A.V.; Inglis, M.A.; Zuure, W.A.; Kim, J.S.; Anderson, G.M. Neither signal transducer and activator of transcription 3 (STAT3) or STAT5 signalling pathways are required for leptin’s effects on fertility in mice. Endocrinology 2013, 154, 2434–2445. [Google Scholar] [CrossRef] [Green Version]
- Ladyman, S.R.; Fieldwick, D.M.; Grattan, D.R. Suppression of leptin-induced hypothalamic JAK/STAT signalling and feeding response during pregnancy in the mouse. Reproduction 2012, 144, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Rui, L. Leptin signalling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Wauman, J.; Zabeau, L.; Tavernier, J. The Leptin Receptor Complex: Heavier Than Expected? Front. Endocrinol. (Lausanne) 2017, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Altarejos, J.Y.; Goebel, N.; Conkright, M.D.; Inoue, H.; Xie, J.; Arias, C.M.; Sawchenko, P.E.; Montminy, M. The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat. Med. 2008, 14, 1112–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Wolfgang, M.J.; Neschen, S.; Morino, K.; Horvath, T.L.; Shulman, G.I.; Fu, X.Y. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc. Natl. Acad. Sci. USA 2004, 101, 4661–4666. [Google Scholar] [CrossRef] [Green Version]
- Buettner, C.; Pocai, A.; Muse, E.D.; Etgen, A.M.; Myers, M.G., Jr.; Rossetti, L. Critical role of STAT3 in leptin’s metabolic actions. Cell Metab. 2006, 4, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Piper, M.L.; Unger, E.K.; Myers, M.G., Jr.; Xu, A.W. Specific physiological roles for signal transducer and activator of transcription 3 in leptin receptor-expressing neurons. Mol. Endocrinol. 2008, 22, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.B.; Uotani, S.; Pierroz, D.D.; Flier, J.S.; Kahn, B.B. In vivo administration of leptin activates signal transduction directly in insulin-sensitive tissues: Overlapping but distinct pathways from insulin. Endocrinology 2000, 141, 2328–2339. [Google Scholar] [CrossRef]
- Carvalheira, J.B.; Siloto, R.M.; Ignacchitti, I.; Brenelli, S.L.; Carvalho, C.R.; Leite, A.; Velloso, L.A.; Gontijo, J.A.; Saad, M.J. Insulin modulates leptin-induced STAT3 activation in rat hypothalamus. FEBS Lett. 2001, 500, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.W.; Elias, C.F.; Fukuda, M.; Williams, K.W.; Berglund, E.D.; Holland, W.L.; Cho, Y.R.; Chuang, J.C.; Xu, Y.; Choi, M.; et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010, 11, 286–297. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Dao, H.; Wang, M.; Heston, A.; Garcia, K.M.; Sangal, A.; Dowling, A.R.; Faulkner, L.D.; Molitor, S.C.; Elias, C.F.; et al. Insulin and Leptin Signalling Interact in the Mouse Kiss1 Neuron during the Peripubertal Period. PLoS ONE 2015, 10, e0121974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Muenzberg, H.; Gavrilova, O.; Reed, J.A.; Berryman, D.; Villanueva, E.C.; Louis, G.W.; Leinninger, G.M.; Bertuzzi, S.; Seeley, R.J.; et al. Loss of cytokine-STAT5 signalling in the CNS and pituitary gland alters energy balance and leads to obesity. PLoS ONE 2008, 3, e1639. [Google Scholar] [CrossRef] [Green Version]
- Grattan, D.R.; Xu, J.J.; McLachlan, M.J.; Kokay, I.C.; Bunn, S.J.; Hovey, R.C.; Davey, H.W. Feedback regulation of PRL secretion is mediated by the transcription factor, signal transducer, and activator of transcription 5b. Endocrinology 2001, 142, 3935–3940. [Google Scholar] [CrossRef]
- Cave, B.J.; Norman, M.; Flynn, A.; Townsend, J.; Wakerley, J.B.; Tortonese, D.J. Prolactin-induced activation of STAT5 within the hypothalamic arcuate nucleus. Neuroreport 2005, 16, 1423–1426. [Google Scholar] [CrossRef]
- Bennett, E.; McGuinness, L.; Gevers, E.F.; Thomas, G.B.; Robinson, I.C.; Davey, H.W.; Luckman, S.M. Hypothalamic STAT proteins: Regulation of somatostatin neurones by growth hormone via STAT5b. J. Neuroendocrinol. 2005, 17, 186–194. [Google Scholar] [CrossRef]
- Patterson, C.M.; Villanueva, E.C.; Greenwald-Yarnell, M.; Rajala, M.; Gonzalez, I.E.; Saini, N.; Jones, J.; Myers, M.G., Jr. Leptin action via LepR-b Tyr1077 contributes to the control of energy balance and female reproduction. Mol. Metab. 2012, 1, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Galiano, D.; Borges, B.C.; Allen, S.J.; Elias, C.F. PI3K signalling in leptin receptor cells: Role in growth and reproduction. J. Neuroendocrinol. 2019, 31, e12685. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, Y.; Carter-Su, C.; Myers, M.G., Jr.; Rui, L. SH2B1 enhances leptin signalling by both Janus kinase 2 Tyr813 phosphorylation-dependent and -independent mechanisms. Mol. Endocrinol. 2007, 21, 2270–2281. [Google Scholar] [CrossRef]
- Niswender, K.D.; Morton, G.J.; Stearns, W.H.; Rhodes, C.J.; Myers, M.G., Jr.; Schwartz, M.W. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature 2001, 413, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Williams, K.W.; Ye, C.; Luo, J.; Balthasar, N.; Coppari, R.; Cowley, M.A.; Cantley, L.C.; Lowell, B.B.; Elmquist, J.K. Acute effects of leptin require PI3K signalling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Investig. 2008, 118, 1796–1805. [Google Scholar] [CrossRef] [Green Version]
- Tups, A.; Anderson, G.M.; Rizwan, M.; Augustine, R.A.; Chaussade, C.; Shepherd, P.R.; Grattan, D.R. Both p110alpha and p110beta isoforms of phosphatidylinositol 3-OH-kinase are required for insulin signalling in the hypothalamus. J. Neuroendocrinol. 2010, 22, 534–542. [Google Scholar] [CrossRef]
- Garcia-Galiano, D.; Borges, B.C.; Donato, J., Jr.; Allen, S.J.; Bellefontaine, N.; Wang, M.; Zhao, J.J.; Kozloff, K.M.; Hill, J.W.; Elias, C.F. PI3Kalpha inactivation in leptin receptor cells increases leptin sensitivity but disrupts growth and reproduction. JCI Insight 2017, 2, e96728. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Feng, Y.; Kitamura, Y.I.; Chua, S.C., Jr.; Xu, A.W.; Barsh, G.S.; Rossetti, L.; Accili, D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat. Med. 2006, 12, 534–540. [Google Scholar] [CrossRef]
- Yang, G.; Lim, C.Y.; Li, C.; Xiao, X.; Radda, G.K.; Li, C.; Cao, X.; Han, W. FoxO1 inhibits leptin regulation of pro-opiomelanocortin promoter activity by blocking STAT3 interaction with specificity protein 1. J. Biol. Chem. 2009, 284, 3719–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Pak, Y.K.; Jang, P.G.; Namkoong, C.; Choi, Y.S.; Won, J.C.; Kim, K.S.; Kim, S.W.; Kim, H.S.; Park, J.Y.; et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat. Neurosci. 2006, 9, 901–906. [Google Scholar] [CrossRef]
- Belgardt, B.F.; Husch, A.; Rother, E.; Ernst, M.B.; Wunderlich, F.T.; Hampel, B.; Klockener, T.; Alessi, D.; Kloppenburg, P.; Bruning, J.C. PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell Metab. 2008, 7, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Plum, L.; Lin, H.V.; Dutia, R.; Tanaka, J.; Aizawa, K.S.; Matsumoto, M.; Kim, A.J.; Cawley, N.X.; Paik, J.H.; Loh, Y.P.; et al. The obesity susceptibility gene Cpe links FoxO1 signalling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat. Med. 2009, 15, 1195–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cota, D.; Matter, E.K.; Woods, S.C.; Seeley, R.J. The role of hypothalamic mammalian target of rapamycin complex 1 signalling in diet-induced obesity. J. Neurosci. 2008, 28, 7202–7208. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar] [CrossRef]
- Roa, J.; Garcia-Galiano, D.; Varela, L.; Sanchez-Garrido, M.A.; Pineda, R.; Castellano, J.M.; Ruiz-Pino, F.; Romero, M.; Aguilar, E.; Lopez, M.; et al. The mammalian target of rapamycin as novel central regulator of puberty onset via modulation of hypothalamic Kiss1 system. Endocrinology 2009, 150, 5016–5026. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signalling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, M.G., Jr. Leptin receptor signalling and the regulation of mammalian physiology. Recent Prog. Horm. Res. 2004, 59, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G., Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, L.; Catalano, S.; Bossi, G.; Pellegrino, M.; Barone, I.; Morales, S.; Giordano, C.; Bartella, V.; Casaburi, I.; Ando, S. Evidences that leptin up-regulates E-cadherin expression in breast cancer: Effects on tumor growth and progression. Cancer Res. 2007, 67, 3412–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorbaek, C.; Buchholz, R.M.; Davis, S.M.; Bates, S.H.; Pierroz, D.D.; Gu, H.; Neel, B.G.; Myers, M.G., Jr.; Flier, J.S. Divergent roles of SHP-2 in ERK activation by leptin receptors. J. Biol. Chem. 2001, 276, 4747–4755. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Ishida-Takahashi, R.; Villanueva, E.C.; Fingar, D.C.; Munzberg, H.; Myers, M.G., Jr. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J. Biol. Chem. 2007, 282, 31019–31027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmouni, K.; Sigmund, C.D.; Haynes, W.G.; Mark, A.L. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes 2009, 58, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, E.E.; Chapeau, E.; Hagihara, K.; Feng, G.S. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 16064–16069. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. (Berl.) 1996, 74, 589–607. [Google Scholar] [CrossRef]
- Kim, G.H.; Szabo, A.; King, E.M.; Ayala, J.; Ayala, J.E.; Altarejos, J.Y. Leptin recruits Creb-regulated transcriptional coactivator 1 to improve hyperglycemia in insulin-deficient diabetes. Mol. Metab. 2015, 4, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011, 12, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferre, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef]
- Koo, S.H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005, 437, 1109–1111. [Google Scholar] [CrossRef]
- Hu, Y.; Lv, J.; Fang, Y.; Luo, Q.; He, Y.; Li, L.; Fan, M.; Wang, Z. Crtc1 Deficiency Causes Obesity Potentially via Regulating PPARgamma Pathway in White Adipose. Front. Cell Dev. Biol. 2021, 9, 602529. [Google Scholar] [CrossRef] [PubMed]
- Breuillaud, L.; Halfon, O.; Magistretti, P.J.; Pralong, F.P.; Cardinaux, J.R. Mouse fertility is not dependent on the CREB coactivator Crtc1. Nat. Med. 2009, 15, 989–990; author reply 991. [Google Scholar] [CrossRef]
- Elias, C.F.; Purohit, D. Leptin signalling and circuits in puberty and fertility. Cell. Mol. Life Sci. 2013, 70, 841–862. [Google Scholar] [CrossRef] [Green Version]
- Bjornstrom, L.; Sjoberg, M. Signal transducers and activators of transcription as downstream targets of nongenomic oestrogen receptor actions. Mol. Endocrinol. 2002, 16, 2202–2214. [Google Scholar] [CrossRef]
- Gao, Q.; Mezei, G.; Nie, Y.; Rao, Y.; Choi, C.S.; Bechmann, I.; Leranth, C.; Toran-Allerand, D.; Priest, C.A.; Roberts, J.L.; et al. Anorectic oestrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signalling in obese animals. Nat. Med. 2007, 13, 89–94. [Google Scholar] [CrossRef]
- Kim, J.S.; Rizwan, M.Z.; Clegg, D.J.; Anderson, G.M. Leptin Signalling Is Not Required for Anorexigenic Oestradiol Effects in Female Mice. Endocrinology 2016, 157, 1991–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, S.; Mauro, L.; Marsico, S.; Giordano, C.; Rizza, P.; Rago, V.; Montanaro, D.; Maggiolini, M.; Panno, M.L.; Ando, S. Leptin induces, via ERK1/ERK2 signal, functional activation of oestrogen receptor alpha in MCF-7 cells. J. Biol. Chem. 2004, 279, 19908–19915. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Lee, K.T.; Leung, P.C. Oestrogen receptor alpha pathway is involved in leptin-induced ovarian cancer cell growth. Carcinogenesis 2011, 32, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasor, J.; Park, K.; Byers, M.; Telleria, C.; Kitamura, T.; Yu-Lee, L.Y.; Djiane, J.; Park-Sarge, O.K.; Gibori, G. Differential roles for signal transducers and activators of transcription 5a and 5b in PRL stimulation of ERalpha and ERbeta transcription. Mol. Endocrinol. 2001, 15, 2172–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Lollmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- El-Haschimi, K.; Pierroz, D.D.; Hileman, S.M.; Bjorbaek, C.; Flier, J.S. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Investig. 2000, 105, 1827–1832. [Google Scholar] [CrossRef]
- Bjorbak, C.; Lavery, H.J.; Bates, S.H.; Olson, R.K.; Davis, S.M.; Flier, J.S.; Myers, M.G., Jr. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J. Biol. Chem. 2000, 275, 40649–40657. [Google Scholar] [CrossRef] [Green Version]
- Dunn, S.L.; Bjornholm, M.; Bates, S.H.; Chen, Z.; Seifert, M.; Myers, M.G., Jr. Feedback inhibition of leptin receptor/Jak2 signalling via Tyr1138 of the leptin receptor and suppressor of cytokine signalling 3. Mol. Endocrinol. 2005, 19, 925–938. [Google Scholar] [CrossRef]
- Ernst, M.B.; Wunderlich, C.M.; Hess, S.; Paehler, M.; Mesaros, A.; Koralov, S.B.; Kleinridders, A.; Husch, A.; Munzberg, H.; Hampel, B.; et al. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signalling in obesity. J. Neurosci. 2009, 29, 11582–11593. [Google Scholar] [CrossRef] [Green Version]
- Bjorbaek, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1998, 1, 619–625. [Google Scholar] [CrossRef]
- Howard, J.K.; Cave, B.J.; Oksanen, L.J.; Tzameli, I.; Bjorbaek, C.; Flier, J.S. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat. Med. 2004, 10, 734–738. [Google Scholar] [CrossRef]
- Mori, H.; Hanada, R.; Hanada, T.; Aki, D.; Mashima, R.; Nishinakamura, H.; Torisu, T.; Chien, K.R.; Yasukawa, H.; Yoshimura, A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med. 2004, 10, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Kievit, P.; Howard, J.K.; Badman, M.K.; Balthasar, N.; Coppari, R.; Mori, H.; Lee, C.E.; Elmquist, J.K.; Yoshimura, A.; Flier, J.S. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signalling-3 in POMC-expressing cells. Cell Metab. 2006, 4, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Bittner-Kowalczyk, A.; White, M.F.; Harbeck, M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein. J. Biol. Chem. 2000, 275, 4283–4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Uetani, N.; Simoncic, P.D.; Chaubey, V.P.; Lee-Loy, A.; McGlade, C.J.; Kennedy, B.P.; Tremblay, M.L. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev. Cell 2002, 2, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Tsou, R.C.; Rak, K.S.; Zimmer, D.J.; Bence, K.K. Improved metabolic phenotype of hypothalamic PTP1B-deficiency is dependent upon the leptin receptor. Mol. Metab. 2014, 3, 301–312. [Google Scholar] [CrossRef]
- Tsou, R.C.; Zimmer, D.J.; De Jonghe, B.C.; Bence, K.K. Deficiency of PTP1B in leptin receptor-expressing neurons leads to decreased body weight and adiposity in mice. Endocrinology 2012, 153, 4227–4237. [Google Scholar] [CrossRef] [Green Version]
- Rousso-Noori, L.; Knobler, H.; Levy-Apter, E.; Kuperman, Y.; Neufeld-Cohen, A.; Keshet, Y.; Akepati, V.R.; Klinghoffer, R.A.; Chen, A.; Elson, A. Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signalling in a phosphorylation-dependent manner. Cell Metab. 2011, 13, 562–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledano-Katchalski, H.; Kraut, J.; Sines, T.; Granot-Attas, S.; Shohat, G.; Gil-Henn, H.; Yung, Y.; Elson, A. Protein tyrosine phosphatase epsilon inhibits signalling by mitogen-activated protein kinases. Mol. Cancer Res. 2003, 1, 541–550. [Google Scholar]
- Dodd, G.T.; Andrews, Z.B.; Simonds, S.E.; Michael, N.J.; DeVeer, M.; Bruning, J.C.; Spanswick, D.; Cowley, M.A.; Tiganis, T. A Hypothalamic Phosphatase Switch Coordinates Energy Expenditure with Feeding. Cell Metab. 2017, 26, 577. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Fukushima, A.; Zhang, X.; Galic, S.; Briggs, D.; Enriori, P.J.; Simonds, S.; Wiede, F.; Reichenbach, A.; Hauser, C.; et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 2011, 14, 684–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, N.R.; Downes, C.P. PTEN: The down side of PI 3-kinase signalling. Cell. Signal. 2002, 14, 285–295. [Google Scholar] [CrossRef]
- Plum, L.; Ma, X.; Hampel, B.; Balthasar, N.; Coppari, R.; Munzberg, H.; Shanabrough, M.; Burdakov, D.; Rother, E.; Janoschek, R.; et al. Enhanced PIP3 signalling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J. Clin. Investig. 2006, 116, 1886–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, B.; Hakansson, M.L. Leptin receptors in hypothalamus and circumventricular organs. Clin. Exp. Pharm. Physiol. 2001, 28, 610–617. [Google Scholar] [CrossRef]
- Xu, A.W.; Ste-Marie, L.; Kaelin, C.B.; Barsh, G.S. Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes decreased pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology 2007, 148, 72–80. [Google Scholar] [CrossRef]
- Gong, L.; Yao, F.; Hockman, K.; Heng, H.H.; Morton, G.J.; Takeda, K.; Akira, S.; Low, M.J.; Rubinstein, M.; MacKenzie, R.G. Signal transducer and activator of transcription-3 is required in hypothalamic agouti-related protein/neuropeptide Y neurons for normal energy homeostasis. Endocrinology 2008, 149, 3346–3354. [Google Scholar] [CrossRef]
- Al-Qassab, H.; Smith, M.A.; Irvine, E.E.; Guillermet-Guibert, J.; Claret, M.; Choudhury, A.I.; Selman, C.; Piipari, K.; Clements, M.; Lingard, S.; et al. Dominant role of the p110beta isoform of PI3K over p110alpha in energy homeostasis regulation by POMC and AgRP neurons. Cell Metab. 2009, 10, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Beymer, M.; Negron, A.L.; Yu, G.; Wu, S.; Mayer, C.; Lin, R.Z.; Boehm, U.; Acosta-Martinez, M. Kisspeptin cell-specific PI3K signalling regulates hypothalamic kisspeptin expression and participates in the regulation of female fertility. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E969–E982. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Li, M.; Duan, C.; Rui, L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab. 2005, 2, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, D.; Zhou, Y.; Morris, D.; Li, M.; Li, Z.; Rui, L. Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J. Clin. Investig. 2007, 117, 397–406. [Google Scholar] [CrossRef]
- Jiang, L.; Su, H.; Wu, X.; Shen, H.; Kim, M.H.; Li, Y.; Myers, M.G., Jr.; Owyang, C.; Rui, L. Leptin receptor-expressing neuron Sh2b1 supports sympathetic nervous system and protects against obesity and metabolic disease. Nat. Commun. 2020, 11, 1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plum, L.; Rother, E.; Munzberg, H.; Wunderlich, F.T.; Morgan, D.A.; Hampel, B.; Shanabrough, M.; Janoschek, R.; Konner, A.C.; Alber, J.; et al. Enhanced leptin-stimulated Pi3k activation in the CNS promotes white adipose tissue transdifferentiation. Cell Metab. 2007, 6, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Orozco, I.J.; Su, Y.; Suyama, S.; Gutierrez-Juarez, R.; Horvath, T.L.; Wardlaw, S.L.; Plum, L.; Arancio, O.; Accili, D. FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 2012, 149, 1314–1326. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Katsouri, L.; Irvine, E.E.; Hankir, M.K.; Pedroni, S.M.; Voshol, P.J.; Gordon, M.W.; Choudhury, A.I.; Woods, A.; Vidal-Puig, A.; et al. Ribosomal S6K1 in POMC and AgRP Neurons Regulates Glucose Homeostasis but Not Feeding Behaviour in Mice. Cell Rep. 2015, 11, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Claret, M.; Smith, M.A.; Batterham, R.L.; Selman, C.; Choudhury, A.I.; Fryer, L.G.; Clements, M.; Al-Qassab, H.; Heffron, H.; Xu, A.W.; et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Investig. 2007, 117, 2325–2336. [Google Scholar] [CrossRef] [Green Version]
- Torsoni, M.A.; Borges, B.C.; Cote, J.L.; Allen, S.J.; Mahany, E.; Garcia-Galiano, D.; Elias, C.F. AMPKalpha2 in Kiss1 Neurons Is Required for Reproductive Adaptations to Acute Metabolic Challenges in Adult Female Mice. Endocrinology 2016, 157, 4803–4816. [Google Scholar] [CrossRef]
- Roa, J.; Barroso, A.; Ruiz-Pino, F.; Vazquez, M.J.; Seoane-Collazo, P.; Martinez-Sanchez, N.; Garcia-Galiano, D.; Ilhan, T.; Pineda, R.; Leon, S.; et al. Metabolic regulation of female puberty via hypothalamic AMPK-kisspeptin signalling. Proc. Natl. Acad. Sci. USA 2018, 115, E10758–E10767. [Google Scholar] [CrossRef] [Green Version]
- Pedroso, J.A.; Silveira, M.A.; Lima, L.B.; Furigo, I.C.; Zampieri, T.T.; Ramos-Lobo, A.M.; Buonfiglio, D.C.; Teixeira, P.D.; Frazao, R.; Donato, J., Jr. Changes in Leptin Signalling by SOCS3 Modulate Fasting-Induced Hyperphagia and Weight Regain in Mice. Endocrinology 2016, 157, 3901–3914. [Google Scholar] [CrossRef]
- Ancel, C.M.; Evans, M.C.; Anderson, G.M. Deletion of protein-tyrosine phosphatase 1B from forebrain neurons protects mice from diet-induced obesity but not infertility. Endocrinology. in revision.
- Banno, R.; Zimmer, D.; De Jonghe, B.C.; Atienza, M.; Rak, K.; Yang, W.; Bence, K.K. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J. Clin. Investig. 2010, 120, 720–734. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, H.; Hasegawa, A.; Yamaguchi, T. Transcriptional regulation of neuronal genes and its effect on neural functions: Transcriptional regulation of neuropeptide Y gene by leptin and its effect on feeding. J. Pharmacol. Sci. 2005, 98, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, C.B.; Cooper, G.M.; Sidow, A.; Barsh, G.S. Mammalian comparative sequence analysis of the Agrp locus. PLoS ONE 2007, 2, e702. [Google Scholar] [CrossRef] [Green Version]
- Munzberg, H.; Huo, L.; Nillni, E.A.; Hollenberg, A.N.; Bjorbaek, C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology 2003, 144, 2121–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, M.; Aschkenasi, C.; Elias, C.F.; Chandrankunnel, A.; Nillni, E.A.; Bjoorbaek, C.; Elmquist, J.K.; Flier, J.S.; Hollenberg, A.N. Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signalling. J. Clin. Investig. 2001, 107, 111–120. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, M.C.; Lord, R.A.; Anderson, G.M. Multiple Leptin Signalling Pathways in the Control of Metabolism and Fertility: A Means to Different Ends? Int. J. Mol. Sci. 2021, 22, 9210. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179210
Evans MC, Lord RA, Anderson GM. Multiple Leptin Signalling Pathways in the Control of Metabolism and Fertility: A Means to Different Ends? International Journal of Molecular Sciences. 2021; 22(17):9210. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179210
Chicago/Turabian StyleEvans, Maggie C., Rebecca A. Lord, and Greg M. Anderson. 2021. "Multiple Leptin Signalling Pathways in the Control of Metabolism and Fertility: A Means to Different Ends?" International Journal of Molecular Sciences 22, no. 17: 9210. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179210