
A Multiplex CRISPR-Screen Identifies PLA2G4A as Prognostic Marker and Druggable Target for HOXA9 and MEIS1 Dependent AML

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
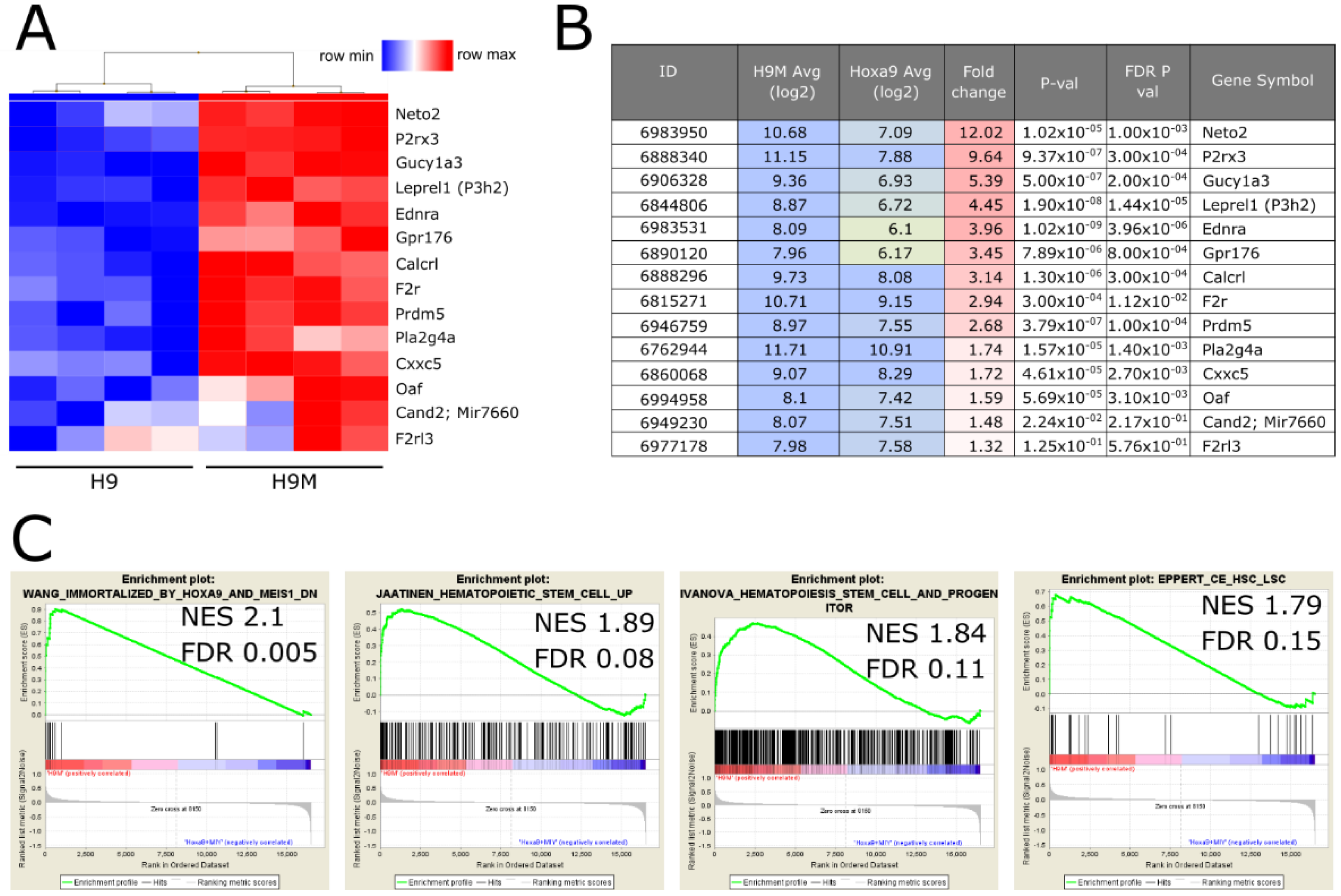
2.1. Identification of H9M-Specific Gene Signatures and Targeting sgRNAs
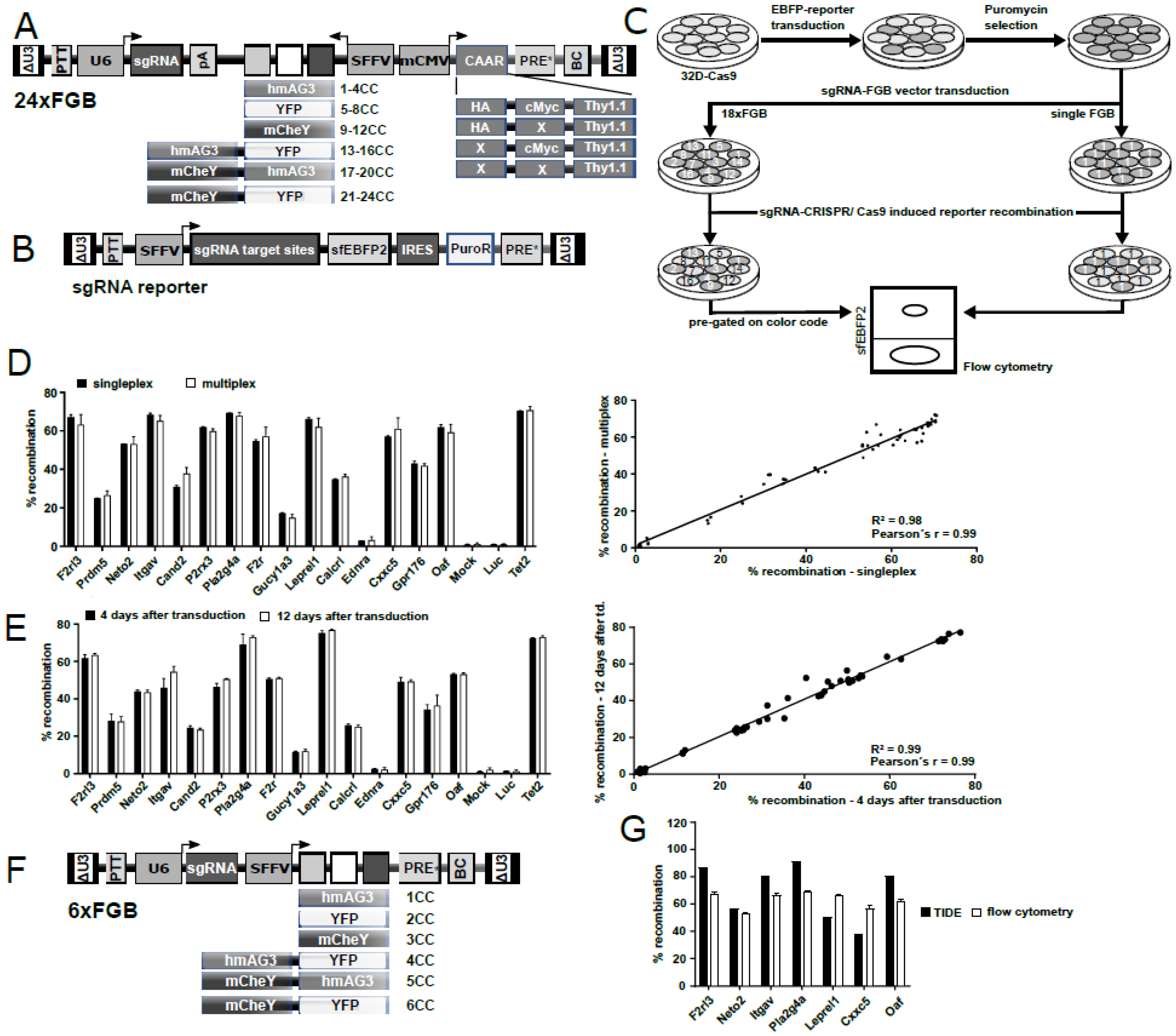
2.2. Generation of a Complex Multiplexing Vector Library for Efficient sgRNA Characterization
2.3. sgRNAs Identified in 32D-Cas9 Reporter Assays Are also Highly Active in H9M Cells
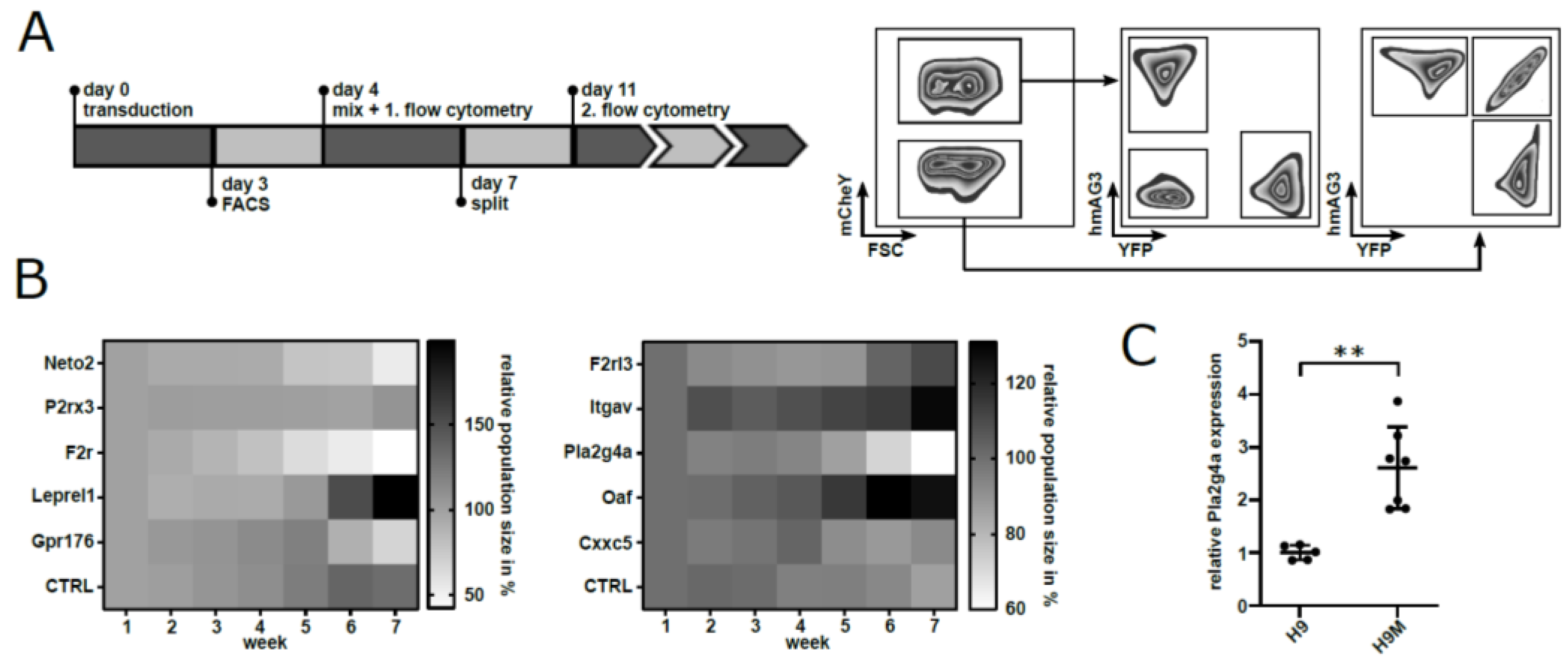
2.4. Competition Assays Identify Pla2g4a as a Putative Target for Cancer Therapy
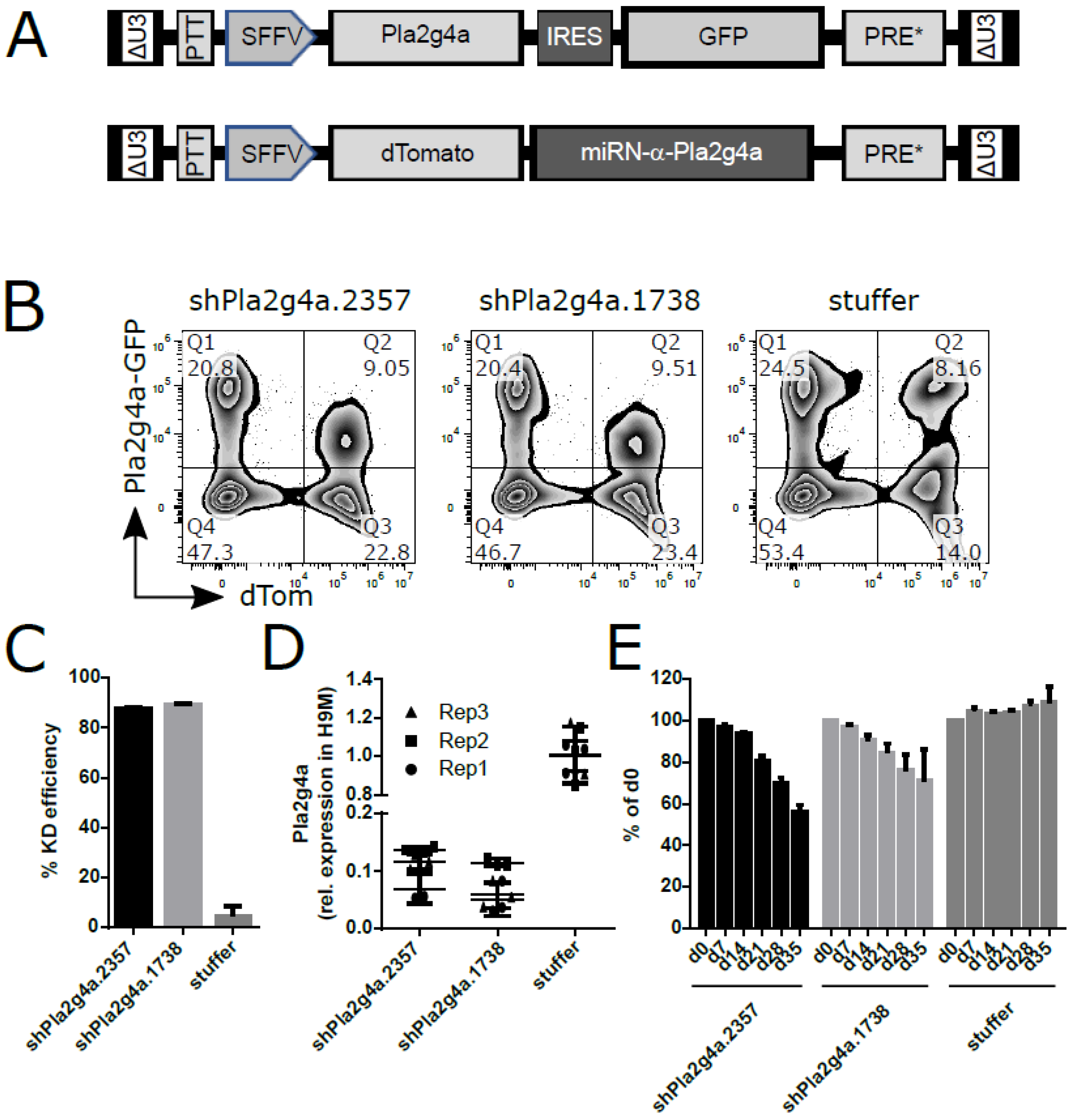
2.5. shRNA-Mediated Knockdown of Pla2g4a Recapitulates sgRNA-Induced H9M Growth Delay
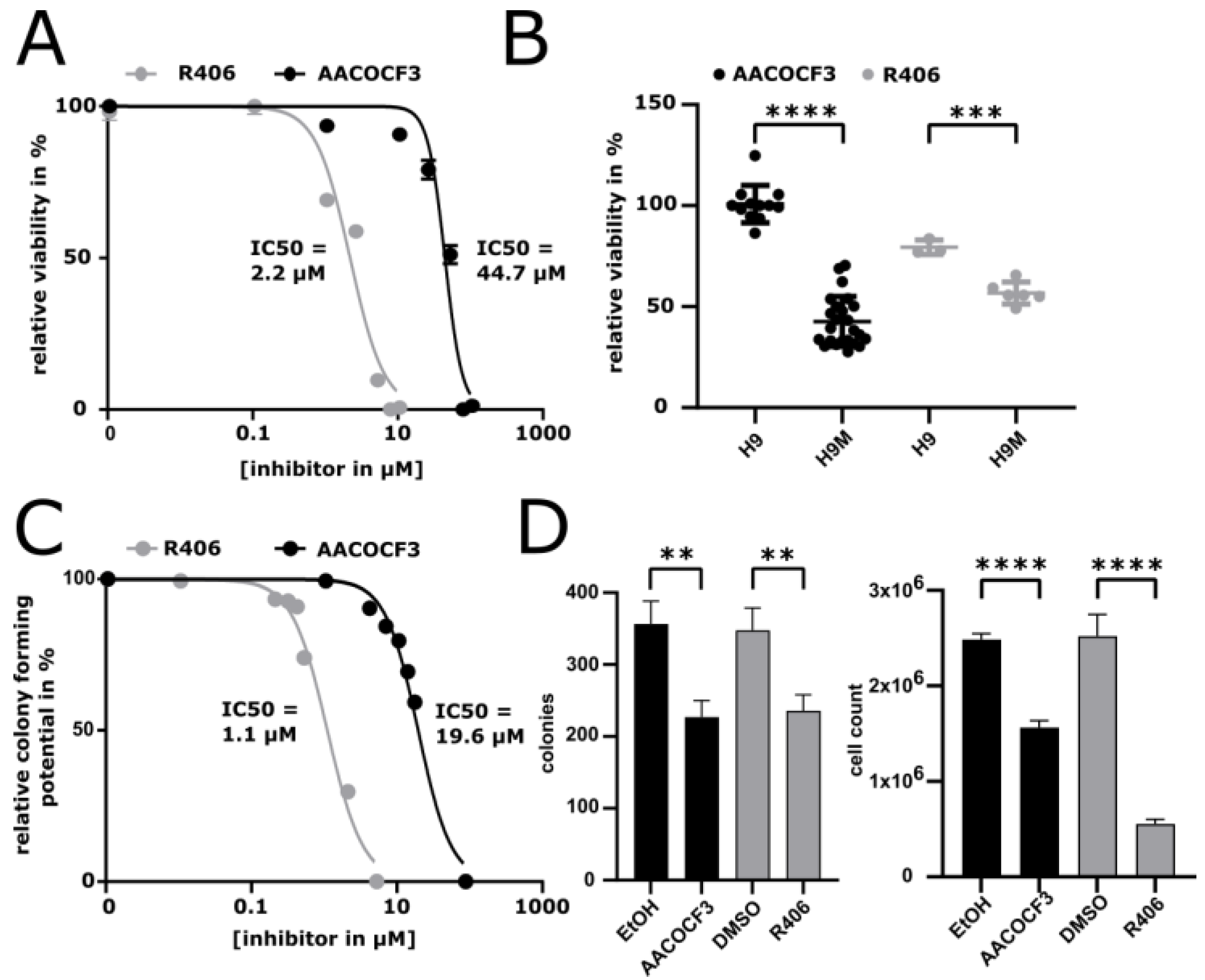
2.6. Drug-Inhibition of PLA2G4A Impairs H9M Cell Growth
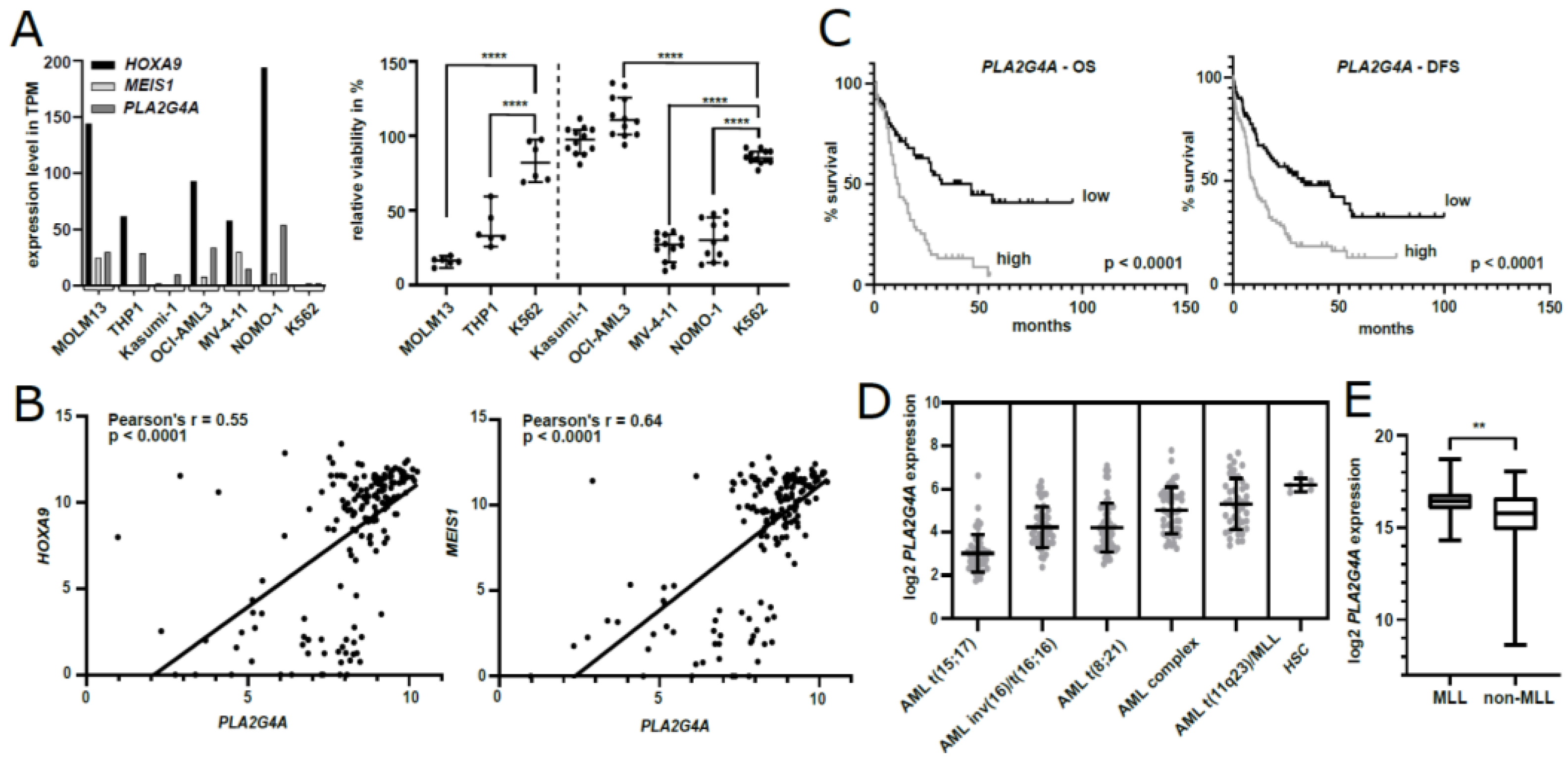
2.7. PLA2G4A Inhibition Interferes with the Growth of Human AML Cells
2.8. PLA2G4A Serves as a Prognostic Marker in Human AML
3. Discussion
4. Materials and Methods
4.1. Viral Vectors
4.2. Cell Culture
4.3. Generation of 32D-Cas9, H9 and H9M Cell Lines
4.4. Vector Production and Transduction
4.5. alamarBlue Cell Viability Assay
4.6. Colony-Forming Assay
4.7. Design and Cloning of Targeting Constructs (shRNA and sgRNA)
4.8. Flow Cytometry
4.9. Competitive Proliferation Assay
4.10. RT-qPCR
4.11. Dataset Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Thorsteinsdottir, U.; Mamo, A.; Kroon, E.; Jerome, L.; Bijl, J.; Lawrence, H.J.; Humphries, K.; Sauvageau, G. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood 2002, 99, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, H.J.; Christensen, J.; Fong, S.; Hu, Y.-L.; Weissman, I.; Sauvageau, G.; Humphries, R.K.; Largman, C. Loss of expression of the Hoxa-9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood 2005, 106, 3988–3994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineault, N.; Helgason, C.D.; Lawrence, H.J.; Humphries, R.K. Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp. Hematol. 2002, 30, 49–57. [Google Scholar] [CrossRef]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, K.R.; Knoepfler, P.S.; Sykes, D.B.; Pasillas, M.P.; Kamps, M.P. Meis1a suppresses differentiation by G-CSF and promotes proliferation by SCF: Potential mechanisms of cooperativity with Hoxa9 in myeloid leukemia. Proc. Natl. Acad. Sci. USA 2001, 98, 13120–13125. [Google Scholar] [CrossRef] [Green Version]
- Argiropoulos, B.; Palmqvist, L.; Yung, E.; Kuchenbauer, F.; Heuser, M.; Sly, L.M.; Wan, A.; Krystal, G.; Humphries, R.K. Linkage of Meis1 leukemogenic activity to multiple downstream effectors including Trib2 and Ccl3. Exp. Hematol. 2008, 36, 845–859. [Google Scholar] [CrossRef]
- Argiropoulos, B.; Humphries, R.K. Hox genes in hematopoiesis and leukemogenesis. Oncogene 2007, 26, 6766–6776. [Google Scholar] [CrossRef] [Green Version]
- Collins, C.T.; Hess, J.L. Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Curr. Opin. Hematol. 2016, 23, 354–361. [Google Scholar] [CrossRef]
- Staffas, A.; Arabanian, L.S.; Wei, S.Y.; Jansson, A.; Stahlman, S.; Johansson, P.; Fogelstrand, L.; Cammenga, J.; Kuchenbauer, F.; Palmqvist, L. Upregulation of Flt3 is a passive event in Hoxa9/Meis1-induced acute myeloid leukemia in mice. Oncogene 2020, 36, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Doebele, C.; Comoglio, F.; Berg, T.; Beck, J.; Bohnenberger, H.; Alexe, G.; Corso, J.; Strobel, P.; Wachter, A.; et al. Hoxa9 and Meis1 Cooperatively Induce Addiction to Syk Signaling by Suppressing miR-146a in Acute Myeloid Leukemia. Cancer Cell 2017, 31, 549–562.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/ b-Catenin Pathway Is Required for the Development of Leukemia Stem Cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [Green Version]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W.E. β-Catenin Mediates the Establishment and Drug Resistance of MLL Leukemic Stem Cells. Cancer Cell 2010, 18, 606–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ysebaert, L.; Chicanne, G.; Demur, C.; De Toni, F.; Prade-Houdellier, N.; Ruidavets, J.-B.; Mansat-De Mas, V.; Rigal-Huguet, F.; Laurent, G.; Payrastre, B.; et al. Expression of β-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia 2006, 20, 1211–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, E.A.; Golding, M.C.; Srivastava, P.; Povinelli, B.J.; James, S.R.; Ford, L.A.; Wetzler, M.; Wang, E.S.; Nemeth, M.J. Pharmacological targeting of beta-catenin in normal karyotype acute myeloid leukemia blasts. Haematologica 2015, 100, e49–e52. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Wu, C.; Wang, Y.; Qi, R.; Bhavanasi, D.; Zuo, Z.; Dos Santos, C.; Chen, S.; Chen, Y.; Zheng, H.; et al. A Genome-Wide CRISPR Screen Identifies Genes Critical for Resistance to FLT3 Inhibitor AC220. Cancer Res. 2017, 77, 4402–4413. [Google Scholar] [CrossRef] [Green Version]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wang, E.; Milazzo, J.P.; Wang, Z.; Kinney, J.B.; Vakoc, C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Li, B.; Millman, S.E.; Chen, C.; Li, X.; Morris, J.P.; Mayle, A.; Ho, Y.-J.; Loizou, E.; Liu, H.; et al. Vitamin B6 Addiction in Acute Myeloid Leukemia. Cancer Cell 2020, 37, 71–84.e7. [Google Scholar] [CrossRef]
- Maetzig, T.; Ruschmann, J.; Lai, C.K.; Ngom, M.; Imren, S.; Rosten, P.; Norddahl, G.L.; von Krosigk, N.; Sanchez Milde, L.; May, C.; et al. A Lentiviral Fluorescent Genetic Barcoding System for Flow Cytometry-Based Multiplex Tracking. Mol. Ther. 2017, 25, 606–620. [Google Scholar] [CrossRef] [Green Version]
- Maetzig, T.; Ruschmann, J.; Sanchez Milde, L.; Lai, C.K.; von Krosigk, N.; Humphries, R.K. Lentiviral Fluorescent Genetic Barcoding for Multiplex Fate Tracking of Leukemic Cells. Mol. Ther. Methods Clin. Dev. 2017, 6, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Mohme, M.; Maire, C.L.; Riecken, K.; Zapf, S.; Aranyossy, T.; Westphal, M.; Lamszus, K.; Fehse, B. Optical Barcoding for Single-Clone Tracking to Study Tumor Heterogeneity. Mol. Ther. 2017, 25, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Smurthwaite, C.A.; Hilton, B.J.; O’Hanlon, R.; Stolp, Z.D.; Hancock, B.M.; Abbadessa, D.; Stotland, A.; Sklar, L.A.; Wolkowicz, R. Fluorescent genetic barcoding in mammalian cells for enhanced multiplexing capabilities in flow cytometry. Cytom. A 2014, 85, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Maetzig, T.; Morgan, M.; Schambach, A. Fluorescent genetic barcoding for cellular multiplex analyses. Exp. Hematol. 2018, 67, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Miller, P.G.; Al-Shahrour, F.; Hartwell, K.A.; Chu, L.P.; Jaras, M.; Puram, R.V.; Puissant, A.; Callahan, K.P.; Ashton, J.; McConkey, M.E.; et al. In Vivo RNAi screening identifies a leukemia-specific dependence on integrin beta 3 signaling. Cancer Cell 2013, 24, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oellerich, T.; Oellerich, M.F.; Engelke, M.; Münch, S.; Mohr, S.; Nimz, M.; Hsiao, H.-H.; Corso, J.; Zhang, J.; Bohnenberger, H.; et al. β2 integrin–derived signals induce cell survival and proliferation of AML blasts by activating a Syk/STAT signaling axis. Blood 2013, 121, 3889–3899. [Google Scholar] [CrossRef] [Green Version]
- Stemmer, M.; Thumberger, T.; del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS ONE 2015, 10, e0124633. [Google Scholar] [CrossRef] [Green Version]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; McConkey, M.E.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Maetzig, T.; Galla, M.; Brugman, M.H.; Loew, R.; Baum, C.; Schambach, A. Mechanisms controlling titer and expression of bidirectional lentiviral and gammaretroviral vectors. Gene Ther. 2010, 17, 400–411. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Adams, F.F.; Heckl, D.; Hoffmann, T.; Talbot, S.R.; Kloos, A.; Thol, F.; Heuser, M.; Zuber, J.; Schambach, A.; Schwarzer, A. An optimized lentiviral vector system for conditional RNAi and efficient cloning of microRNA embedded short hairpin RNA libraries. Biomaterials 2017, 139, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, X.; Zhang, L.; Geng, D.; Wang, Y.; Sun, D.; Sui, P.; Zhao, X.; Xin, C.; Jiang, J.; et al. Inhibition of PLA2G4A Reduces the Expression of Lung Cancer-Related Cytokines. DNA Cell Biol. 2018, 16, 1076–1081. [Google Scholar] [CrossRef]
- Loweth, A.C.; Scarpello, J.H.B.; Morgan, N.G. A Specific Inhibitor of Cytosolic Phospholipase A2 Activity, AACOCF3, Inhibits Glucose-Induced Insulin Secretion from Isolated Rat Islets. Biochem. Biophys. Res. Commun. 1996, 218, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Gowda, R.; Dinavahi, S.S.; Iyer, S.; Banerjee, S.; Neves, R.I.; Pameijer, C.R.; Robertson, G.P. Nanoliposomal delivery of cytosolic phospholipase A2 inhibitor arachidonyl trimethyl ketone for melanoma treatment. Nanomedicine 2018, 14, 863–873. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Moreno, P.; Manning, J.; Fuentes, A.M.-P.; George, N.; Fexova, S.; Fonseca, N.A.; Füllgrabe, A.; Green, M.; Huang, N.; et al. Expression Atlas update: From tissues to single cells. Nucleic Acids Res. 2019, 48, D77–D83. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; Zhou, M.; Zeng, M.; Han, L. PLA2G4A Is a Potential Biomarker Predicting Shorter Overall Survival in Patients with Non-M3/ NPM1 Wildtype Acute Myeloid Leukemia. DNA Cell Biol. 2020, 39, 700–708. [Google Scholar] [CrossRef]
- Sha, K.; Lu, Y.; Zhang, P.; Pei, R.; Shi, X.; Fan, Z.; Chen, L. Identifying a novel 5-gene signature predicting clinical outcomes in acute myeloid leukemia. Clin. Transl. Oncol. 2020, 23, 648–656. [Google Scholar] [CrossRef]
- Bagger, F.O.; Kinalis, S.; Rapin, N. BloodSpot: A database of healthy and malignant haematopoiesis updated with purified and single cell mRNA sequencing profiles. Nucleic Acids Res. 2019, 47, D881–D885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Narumiya, S. Prostaglandins and chronic inflammation. Trends Pharmacol. Sci. 2012, 33, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilley, S.L.; Coffman, T.M.; Koller, B.H. Mixed messages: Modulation of inflammation and immune responses by prostaglandins and thromboxanes. J. Clin. Investig. 2001, 108, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, M.; Rosenberg, D.W. Roles of cPLA2alpha and arachidonic acid in cancer. Biochim. Biophys Acta 2006, 1761, 1335–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; DuBois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Sun, T.-Q.; Lu, B.; Feng, J.-J.; Reinhard, C.; Jan, Y.N.; Fantl, W.J.; Williams, L.T. PAR-1 is a Dishevelled-associated kinase and a positive regulator of Wnt signalling. Nat. Cell Biol. 2001, 3, 628–636. [Google Scholar] [CrossRef]
- De Veiga, S.B.; Carneiro-Lobo, T.C.; Coelho, C.J.B.P.; Carvalho, S.M.F.; Maia, R.C.; Vasconcelos, F.C.; Abdelhay, E.; Mencalha, A.L.; Ferreira, A.F.; Castro, F.A.; et al. Increased expression of protease-activated receptor 1 (PAR-1) in human leukemias. Blood Cells Mol. Dis. 2011, 46, 230–234. [Google Scholar] [CrossRef]
- Heidel, F.H.; Bullinger, L.; Feng, Z.; Wang, Z.; Neff, T.A.; Stein, L.; Kalaitzidis, D.; Lane, S.W.; Armstrong, S.A. Genetic and Pharmacologic Inhibition of β-Catenin Targets Imatinib-Resistant Leukemia Stem Cells in CML. Cell Stem Cell 2012, 10, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Sitwala, K.; Bronstein, J.; Sanders, D.; Dandekar, M.; Collins, C.; Robertson, G.; MacDonald, J.; Cezard, T.; Bilenky, M.; et al. Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood 2012, 119, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Fonteh, A.N. Differential effects of arachidonoyl trifluoromethyl ketone on arachidonic acid release and lipid mediator biosynthesis by human neutrophils: Evidence for different arachidonate pools. Eur. J. Biochem. 2002, 269, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Leis, H.J.; Windischhofer, W. Inhibition of cyclooxygenases 1 and 2 by the phospholipase-blocker, arachidonyl trifluoromethyl ketone: COX-inhibition by ATK. Br. J. Pharmacol. 2008, 155, 731–737. [Google Scholar] [CrossRef]
- Gibbs, K.D.; Jager, A.; Crespo, O.; Goltsev, Y.; Trejo, A.; Richard, C.E.; Nolan, G.P. Decoupling of tumor-initiating activity from stable immunophenotype in HoxA9-Meis1-driven AML. Cell Stem Cell 2012, 10, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomoo, T.; Nakatsuka, T.; Katayama, T.; Hayashi, Y.; Fujieda, Y.; Terakawa, M.; Nagahira, K. Design, Synthesis, and Biological Evaluation of 3-(1-Aryl-1 H -indol-5-yl)propanoic Acids as New Indole-Based Cytosolic Phospholipase A2 α Inhibitors. J. Med. Chem. 2014, 57, 7244–7262. [Google Scholar] [CrossRef]
- Kanai, S.; Ishihara, K.; Kawashita, E.; Tomoo, T.; Nagahira, K.; Hayashi, Y.; Akiba, S. ASB14780, an Orally Active Inhibitor of Group IVA Phospholipase A2, Is a Pharmacotherapeutic Candidate for Nonalcoholic Fatty Liver Disease. J. Pharm. Exp. Ther. 2016, 356, 604–614. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673.e11. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.-H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 2018, 34, 499–512.e9. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Demetris, A.J.; Michalopoulos, G.; Shelhamer, J.H.; Wu, T. 85-kDa cPLA2 plays a critical role in PPAR-mediated gene transcription in human hepatoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G586–G597. [Google Scholar] [CrossRef]
- Heasley, L.E.; Thaler, S.; Nicks, M.; Price, B.; Skorecki, K.; Nemenoff, R.A. Induction of Cytosolic Phospholipase A2 by Oncogenic Ras in Human Non-small Cell Lung Cancer. J. Biol. Chem. 1997, 272, 14501–14504. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Han, C.; Lunz, J.G.; Michalopoulos, G.; Shelhamer, J.H.; Demetris, A.J. Involvement of 85-kd cytosolic phospholipase A2 and cyclooxygenase-2 in the proliferation of human cholangiocarcinoma cells. Hepatology 2002, 36, 363–373. [Google Scholar] [CrossRef]
- Lieske, A.; Ha, T.C.; Schambach, A.; Maetzig, T. An improved lentiviral fluorescent genetic barcoding approach distinguishes hematopoietic stem cell properties in multiplexed in vivo experiments. Hum. Gene Ther. 2021. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Rapin, N.; Bagger, F.O.; Jendholm, J.; Mora-Jensen, H.; Krogh, A.; Kohlmann, A.; Thiede, C.; Borregaard, N.; Bullinger, L.; Winther, O.; et al. Comparing cancer vs normal gene expression profiles identifies new disease entities and common transcriptional programs in AML patients. Blood 2014, 123, 894–904. [Google Scholar] [CrossRef] [Green Version]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, J.J.; Lieske, A.; Dörpmund, N.; Klatt, D.; Hoffmann, D.; Kleppa, M.-J.; Kustikova, O.S.; Stahlhut, M.; Schwarzer, A.; Schambach, A.; et al. A Multiplex CRISPR-Screen Identifies PLA2G4A as Prognostic Marker and Druggable Target for HOXA9 and MEIS1 Dependent AML. Int. J. Mol. Sci. 2021, 22, 9411. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179411
Hassan JJ, Lieske A, Dörpmund N, Klatt D, Hoffmann D, Kleppa M-J, Kustikova OS, Stahlhut M, Schwarzer A, Schambach A, et al. A Multiplex CRISPR-Screen Identifies PLA2G4A as Prognostic Marker and Druggable Target for HOXA9 and MEIS1 Dependent AML. International Journal of Molecular Sciences. 2021; 22(17):9411. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179411
Chicago/Turabian StyleHassan, Jacob Jalil, Anna Lieske, Nicole Dörpmund, Denise Klatt, Dirk Hoffmann, Marc-Jens Kleppa, Olga S. Kustikova, Maike Stahlhut, Adrian Schwarzer, Axel Schambach, and et al. 2021. "A Multiplex CRISPR-Screen Identifies PLA2G4A as Prognostic Marker and Druggable Target for HOXA9 and MEIS1 Dependent AML" International Journal of Molecular Sciences 22, no. 17: 9411. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179411