
Mechanisms of Systolic Cardiac Dysfunction in PP2A, PP5 and PP2AxPP5 Double Transgenic Mice

 ,
,
Abstract
:1. Introduction
2. Results
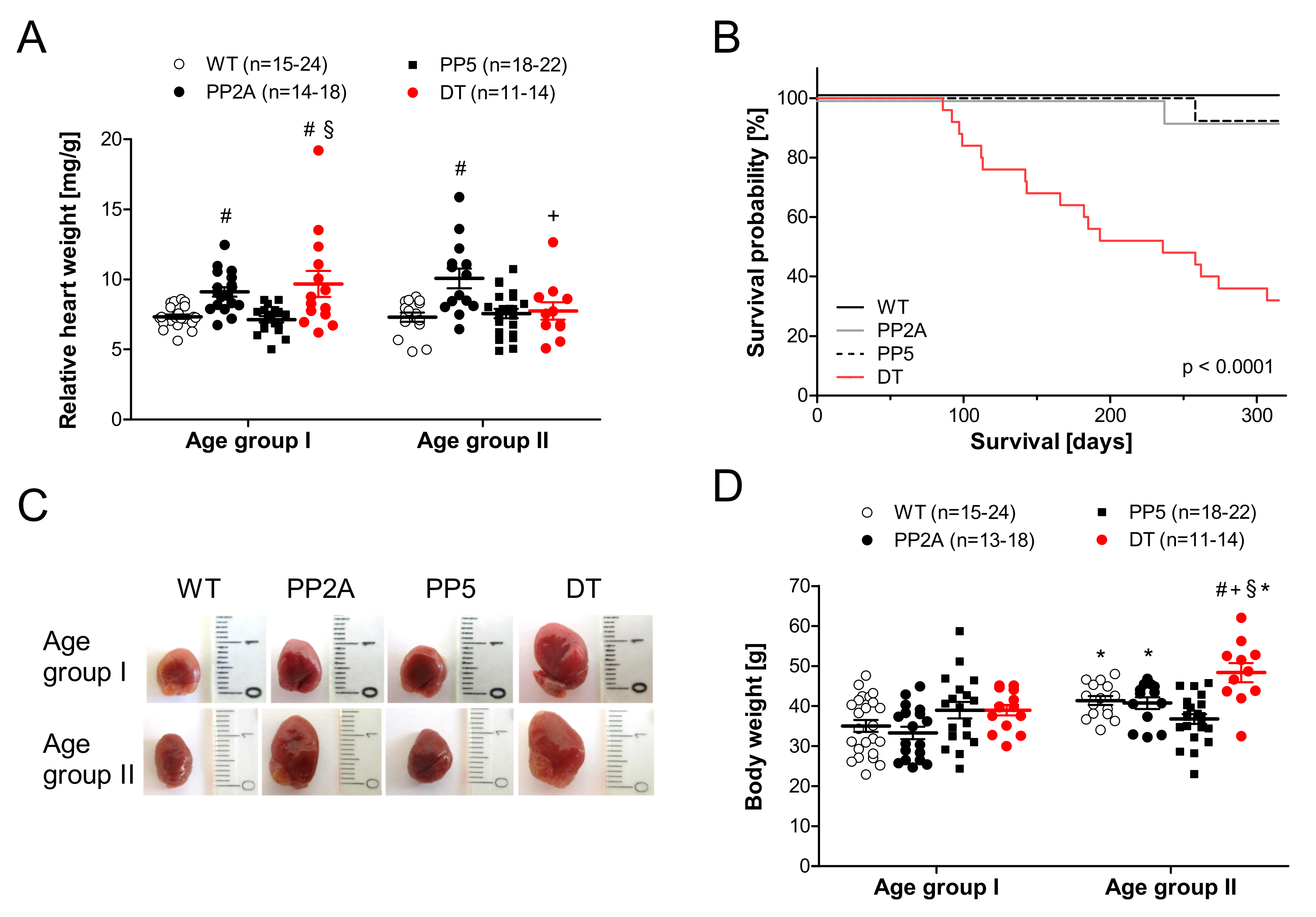
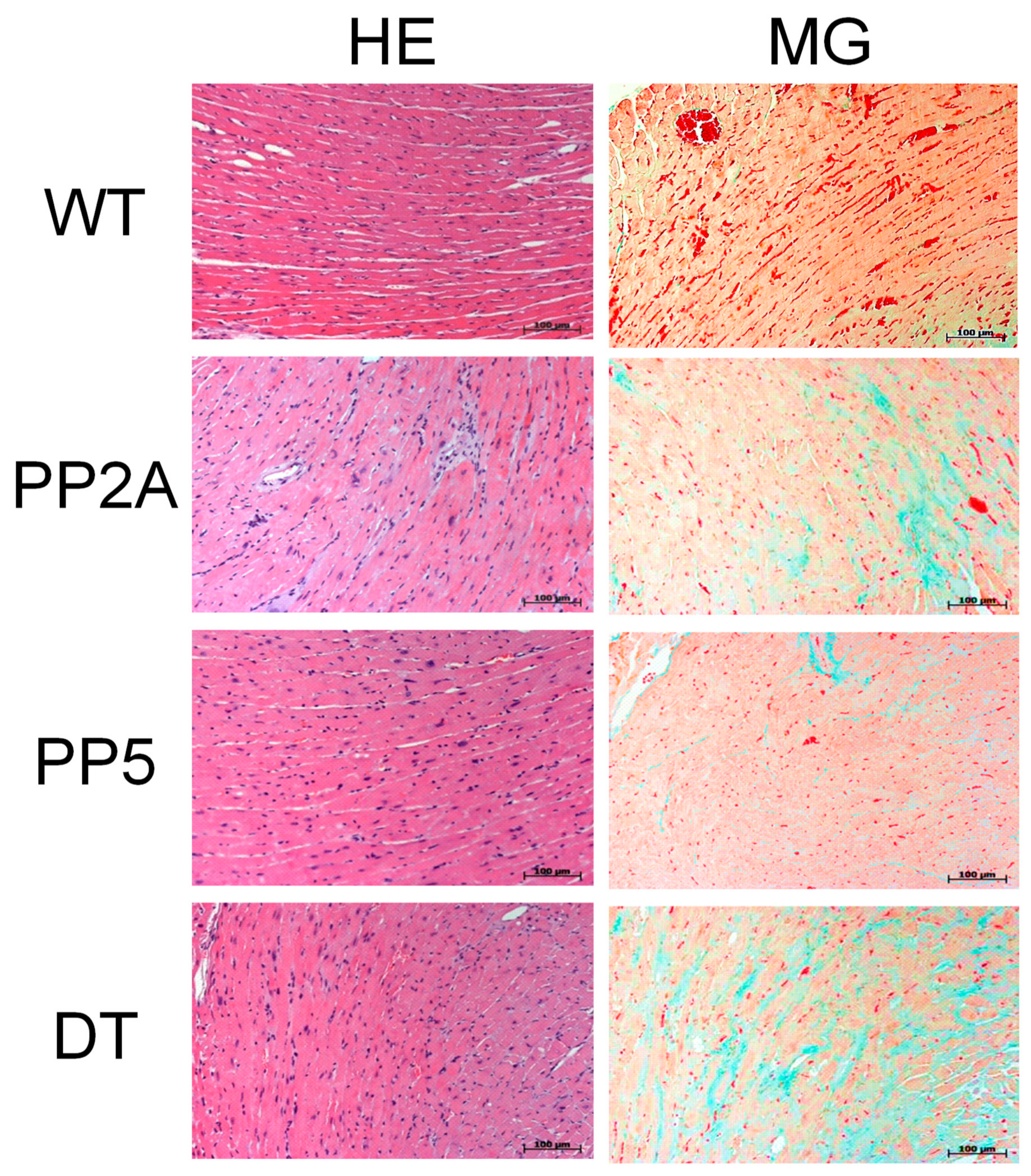
2.1. Phenotype of Transgenic Mice
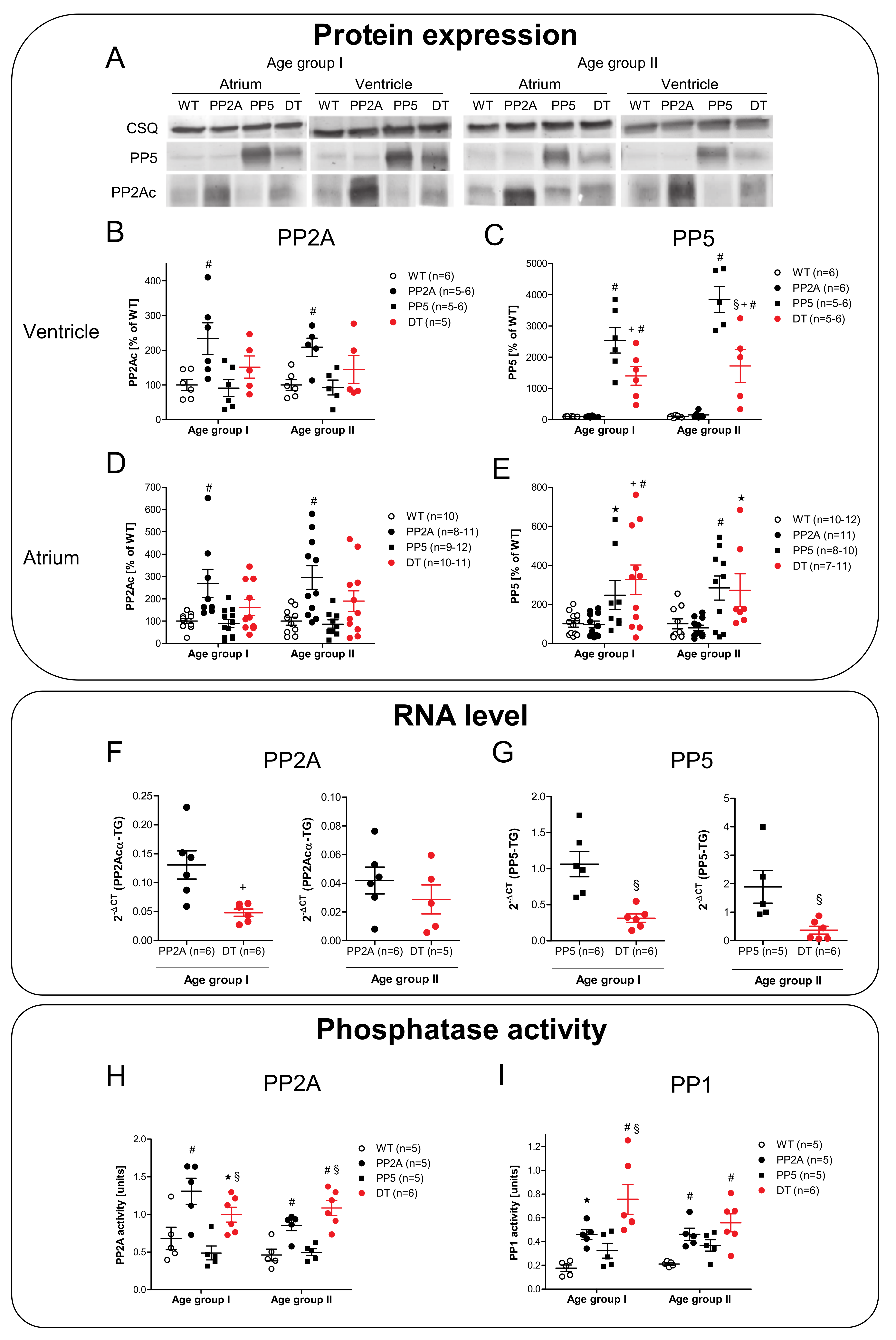
2.2. Expression of PP2A and PP5
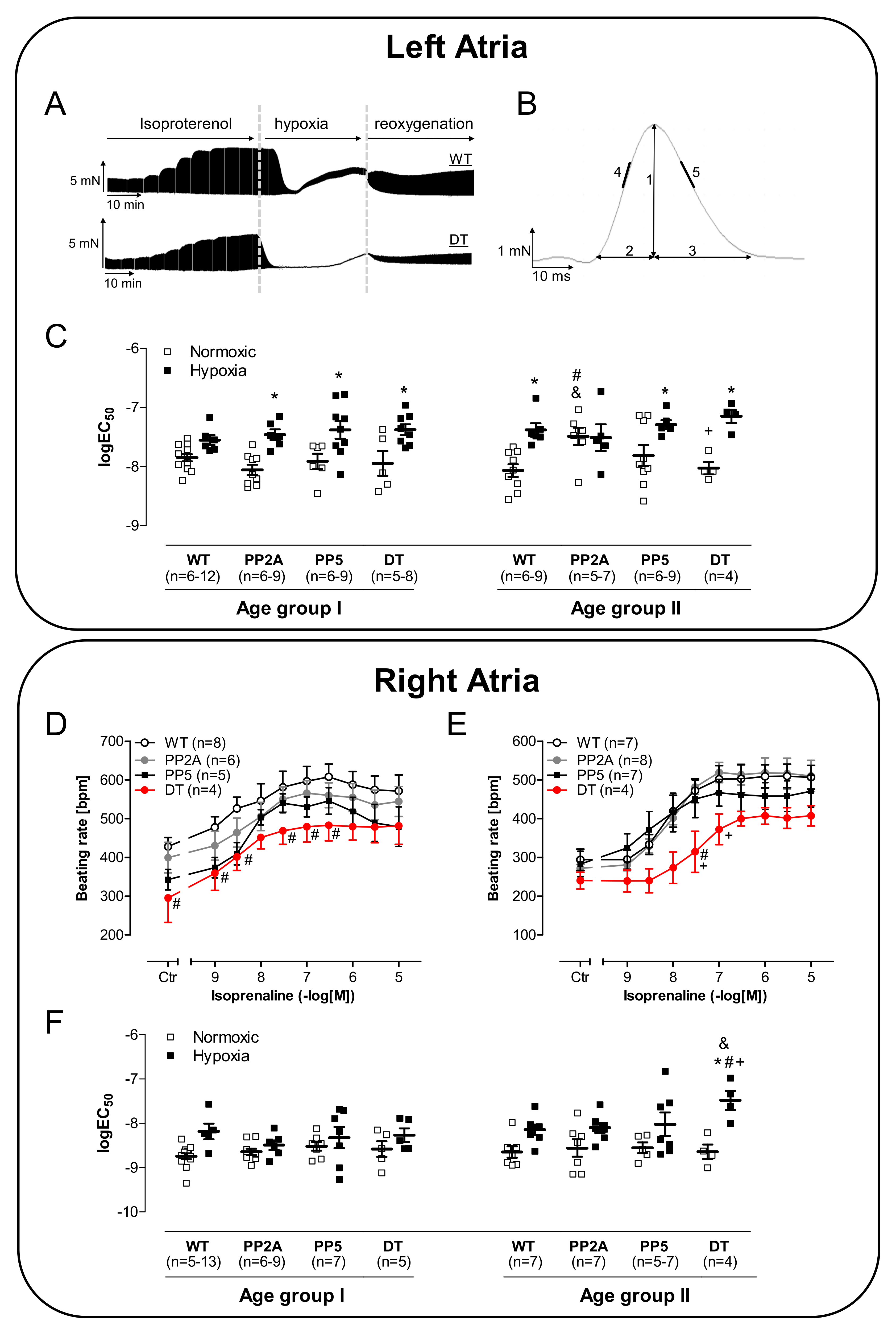
2.3. Atrial Measurements
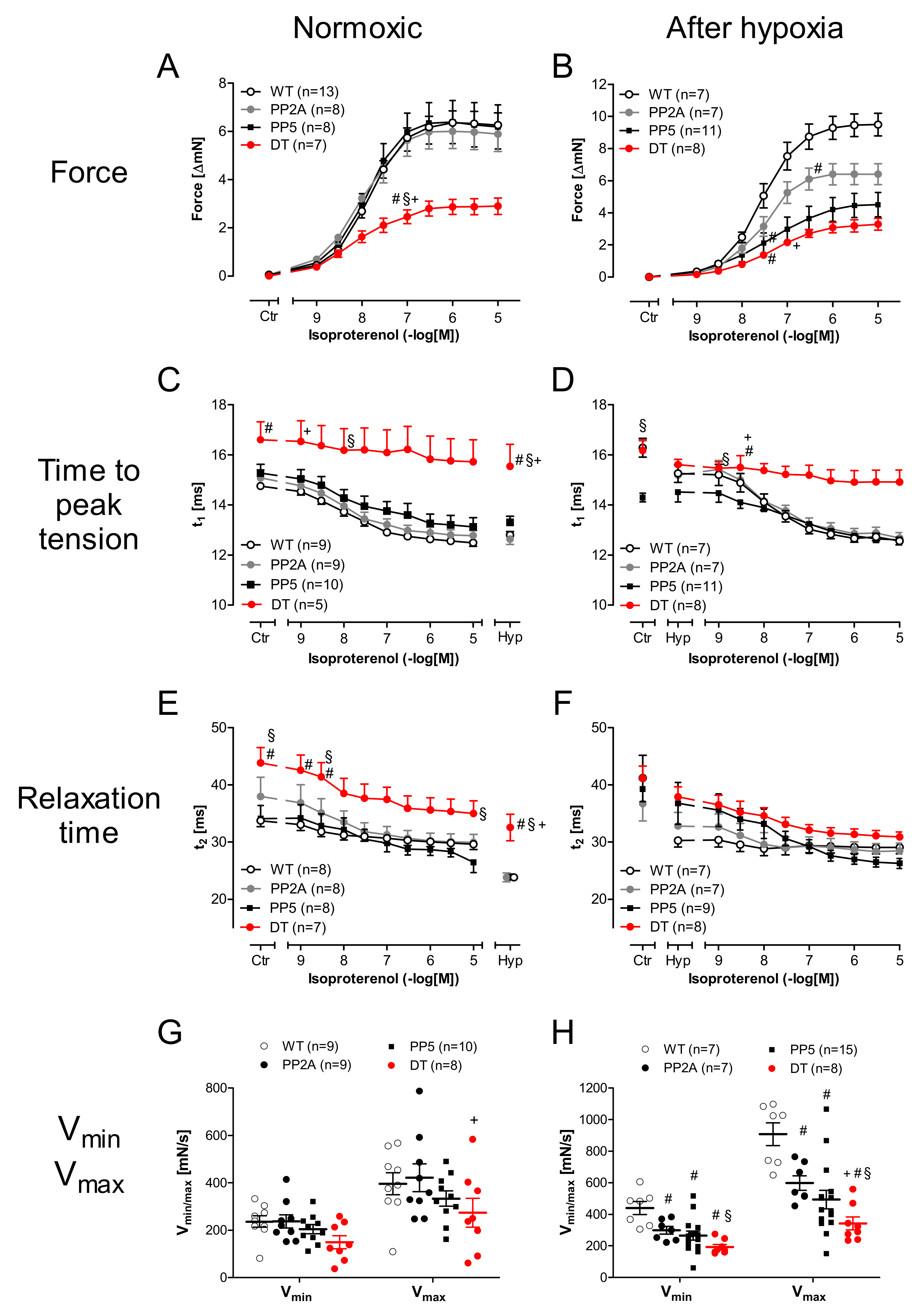
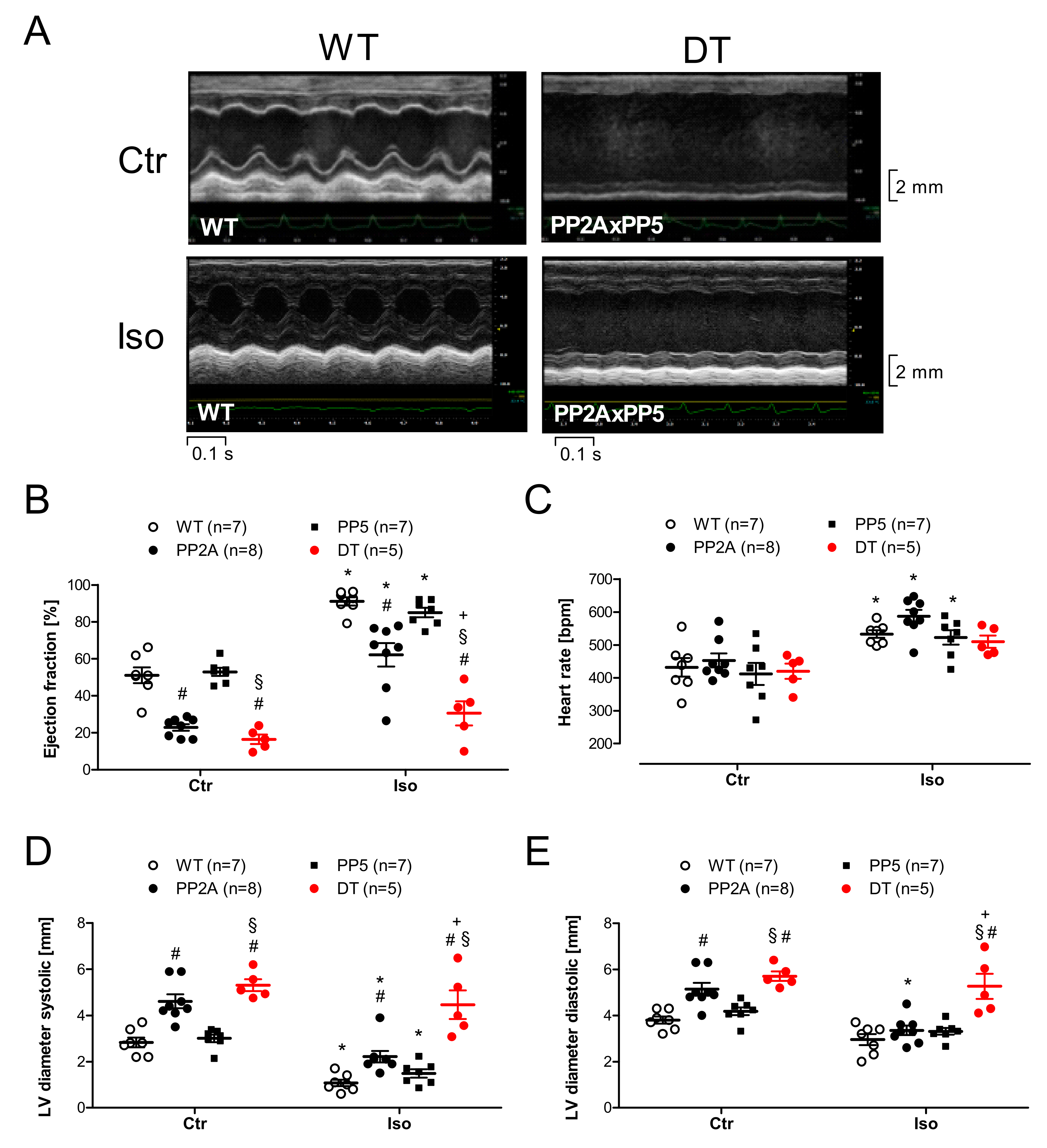
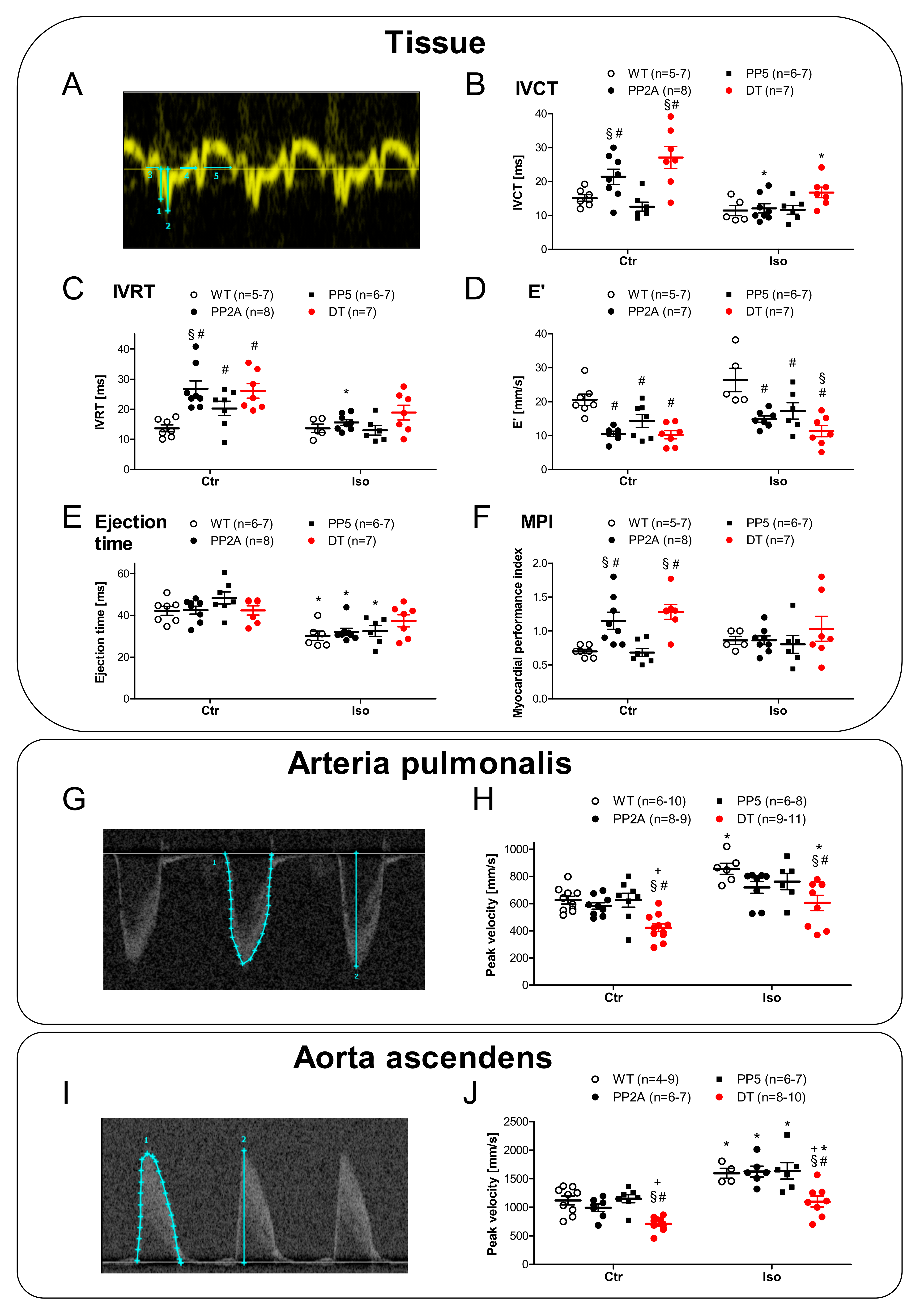
2.4. Ventricular Function
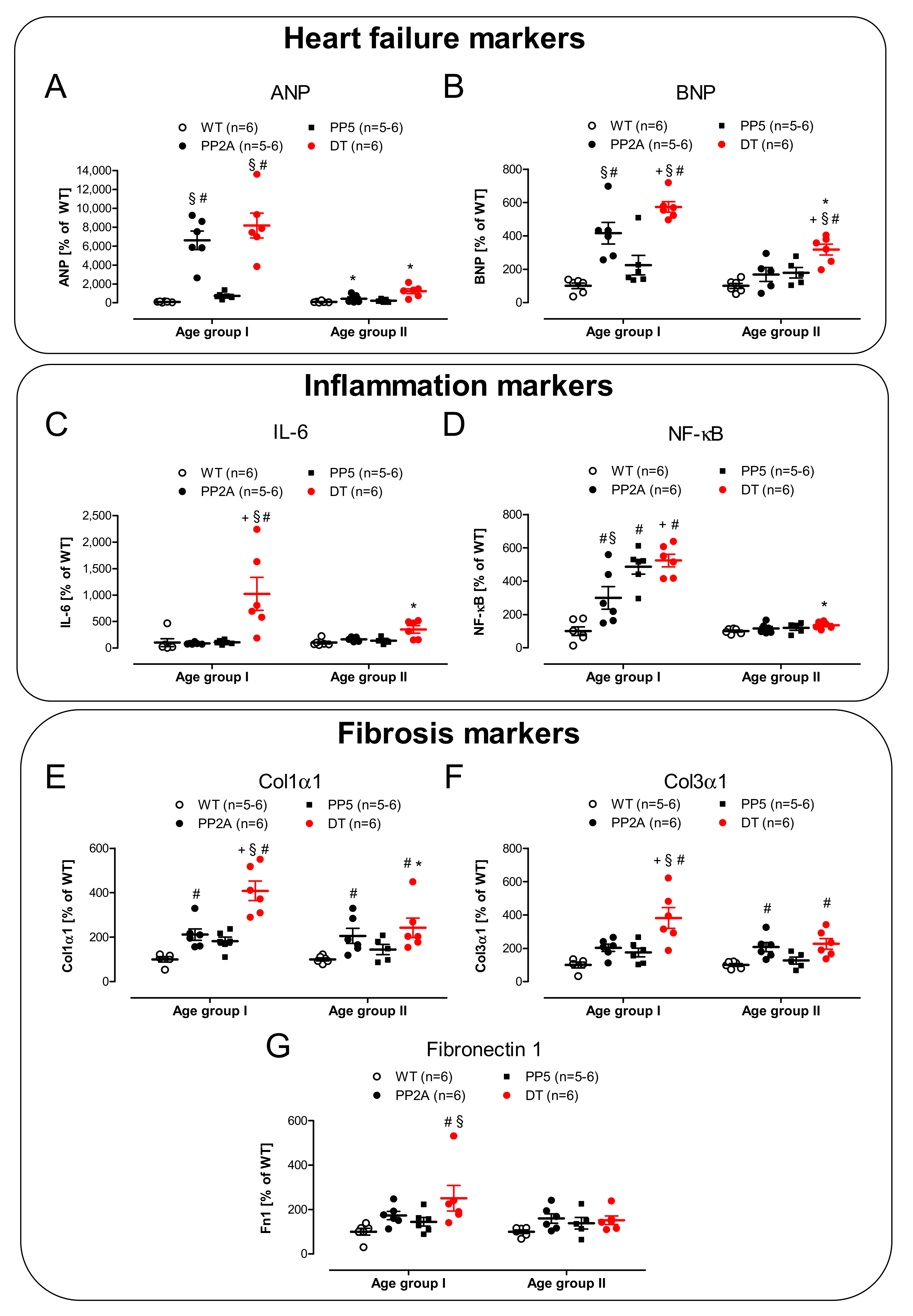
2.5. mRNA Expression
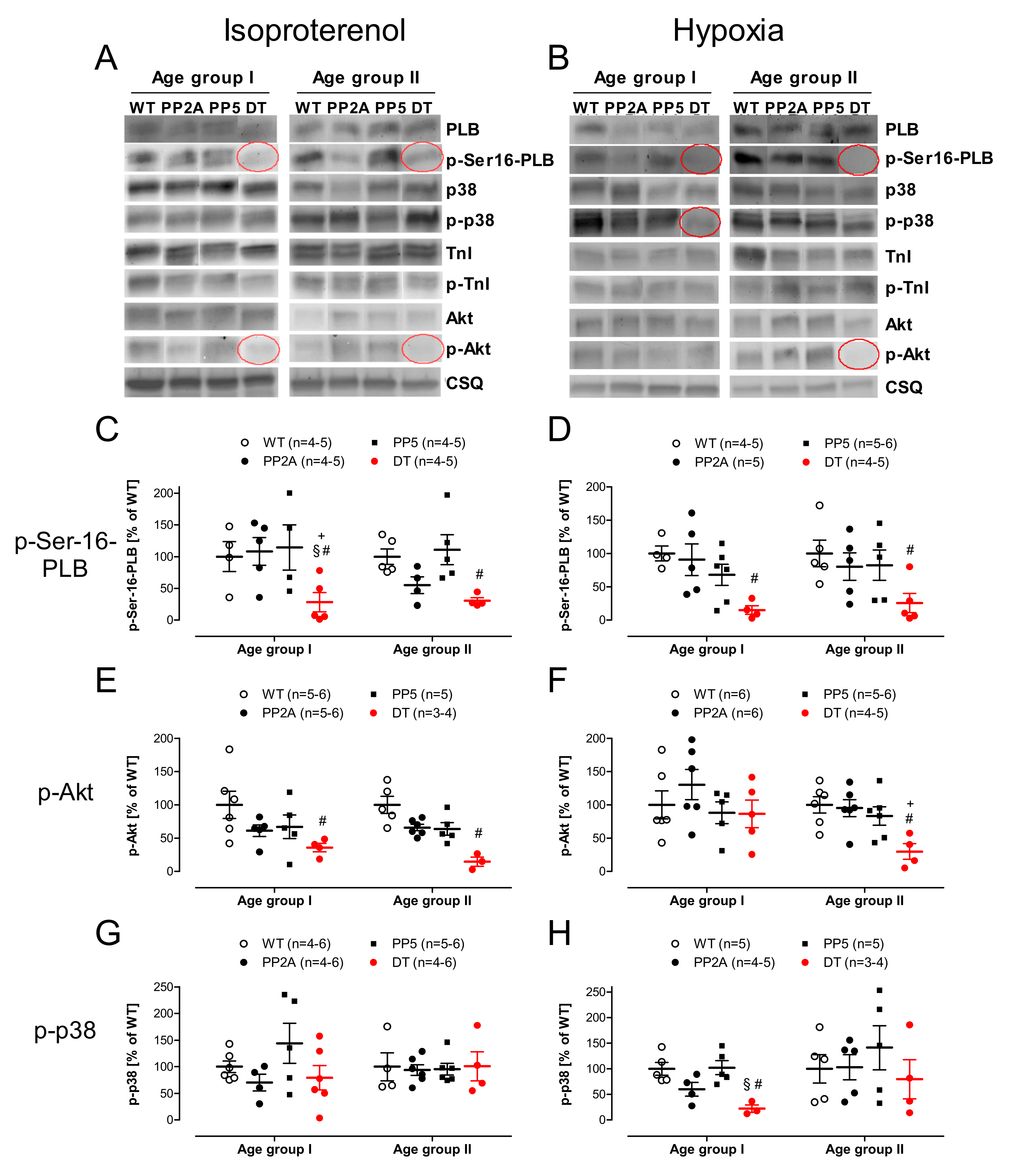
2.6. Protein Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Transgenic Mice
4.2. Contractile Studies in Mice
4.3. Echocardiography
4.4. Preparation of Homogenates
4.5. Western Blot Analysis
4.6. Protein Phosphatase Assay
4.7. Histological Analysis
4.8. Real-Time Quantitative PCR
4.9. Data Analysis
4.10. Drugs and Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brüchert, N.; Mavila, N.; Boknik, P.; Baba, H.A.; Fabritz, L.; Gergs, U.; Kirchhefer, U.; Kirchhof, P.; Matus, M.; Schmitz, W.; et al. Inhibitor-2 prevents protein phosphatase 1-induced cardiac hypertrophy and mortality. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1539–H1546. [Google Scholar] [CrossRef] [Green Version]
- DeGrande, S.T.; Little, S.C.; Nixon, D.J.; Wright, P.; Snyder, J.; Dun, W.; Murphy, N.; Kilic, A.; Higgins, R.; Binkley, P.F.; et al. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J. Biol. Chem. 2013, 288, 1032–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gergs, U.; Jahn, T.; Werner, F.; Köhler, C.; Köpp, F.; Großmann, C.; Neumann, J. Overexpression of protein phosphatase 5 in the mouse heart: Reduced contractility but increased stress tolerance—Two sides of the same coin? PLoS ONE 2019, 14, e0221289. [Google Scholar] [CrossRef]
- Herzig, S.; Neumann, J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol. Rev. 2000, 80, 173–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gergs, U.; Trapp, T.; Bushnaq, H.; Simm, A.; Silber, R.-E.; Neumann, J. Age-Dependent Protein Expression of Serine/Threonine Phosphatases and Their Inhibitors in the Human Cardiac Atrium. Adv. Med. 2019, 2019, 2675972. [Google Scholar] [CrossRef]
- Bokník, P.; Fockenbrock, M.; Herzig, S.; Knapp, J.; Linck, B.; Lüss, H.; Müller, F.U.; Müller, T.; Schmitz, W.; Schröder, F.; et al. Protein phosphatase activity is increased in a rat model of long-term beta-adrenergic stimulation. Naunyn Schmiedebergs. Arch. Pharmacol. 2000, 362, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Eschenhagen, T.; Jones, L.R.; Linck, B.; Schmitz, W.; Scholz, H.; Zimmermann, N. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J. Mol. Cell. Cardiol. 1997, 29, 265–272. [Google Scholar] [CrossRef]
- Dobrev, D.; Aguilar, M.; Heijman, J.; Guichard, J.-B.; Nattel, S. Postoperative atrial fibrillation: Mechanisms, manifestations and management. Nat. Rev. Cardiol. 2019, 16, 417–436. [Google Scholar] [CrossRef]
- Dobrev, D.; Wehrens, X.H.T. Mouse Models of Cardiac Arrhythmias. Circ. Res. 2018, 123, 332–334. [Google Scholar] [CrossRef]
- Gergs, U.; Boknik, P.; Buchwalow, I.; Fabritz, L.; Matus, M.; Justus, I.; Hanske, G.; Schmitz, W.; Neumann, J. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J. Biol. Chem. 2004, 279, 40827–40834. [Google Scholar] [CrossRef] [Green Version]
- Gergs, U.; Boknik, P.; Buchwalow, I.B.; Fabritz, L.; Gründker, N.; Kucerova, D.; Matus, M.; Werner, F.; Schmitz, W.; Neumann, J. Modulation of cardiac contractility by serine/threonine protein phosphatase type 5. Int. J. Cardiol. 2012, 154, 116–121. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Lu, J.-R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A Calcineurin-Dependent Transcriptional Pathway for Cardiac Hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Hoehn, M.; Zhang, Y.; Xu, J.; Gergs, U.; Boknik, P.; Werdan, K.; Neumann, J.; Ebelt, H. Overexpression of protein phosphatase 2A in a murine model of chronic myocardial infarction leads to increased adverse remodeling but restores the regulation of β-catenin by glycogen synthase kinase 3β. Int. J. Cardiol. 2015, 183, 39–46. [Google Scholar] [CrossRef]
- Morita, K.; Saitoh, M.; Tobiume, K.; Matsuura, H.; Enomoto, S.; Nishitoh, H.; Ichijo, H. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 2001, 20, 6028–6036. [Google Scholar] [CrossRef] [Green Version]
- Schulz, N.; Gergs, U.; Neumann, J. Heart specific overexpression of PP2A leads to impaired contractility but protects against ischemia. In Proceedings of the 48th Spring Meeting of the German Society for Experimental and Clinical Pharmacology and Toxicology, Mainz, Germany, 13–15 March 2007; Springer: New York, NY, USA, 2007; p. 62. [Google Scholar]
- Schulz, N.; Gergs, U.; Loppnow, H.; Neumann, J. Increased expression of PP2A protects against lipopolysaccharide-induced stress. In Proceedings of the 49th Annual Meeting of the German Society for Experimental and Clinical Pharmacology and Toxicology, Mainz, Germany, 11–13 March 2008; Springer: New York, NY, USA, 2008; p. 56. [Google Scholar]
- Bartel, S.; Stein, B.; Eschenhagen, T.; Mende, U.; Neumann, J.; Schmitz, W.; Krause, E.G.; Karczewski, P.; Scholz, H. Protein phosphorylation in isolated trabeculae from nonfailing and failing human hearts. Mol. Cell. Biochem. 1996, 157, 171–179. [Google Scholar] [CrossRef]
- Neumann, J.; Boknik, P.; Herzig, S.; Schmitz, W.; Scholz, H.; Gupta, R.C.; Watanabe, A.M. Evidence for physiological functions of protein phosphatases in the heart: Evaluation with okadaic acid. Am. J. Physiol. 1993, 265, H257–H266. [Google Scholar] [CrossRef]
- Neumann, J.; Bokník, P.; Herzig, S.; Schmitz, W.; Scholz, H.; Wiechen, K.; Zimmermann, N. Biochemical and electrophysiological mechanisms of the positive inotropic effect of calyculin A, a protein phosphatase inhibitor. J. Pharmacol. Exp. Ther. 1994, 271, 535–541. [Google Scholar]
- Neumann, J.; Herzig, S.; Boknik, P.; Apel, M.; Kaspareit, G.; Schmitz, W.; Scholz, H.; Tepel, M.; Zimmermann, N. On the cardiac contractile, biochemical and electrophysiological effects of cantharidin, a phosphatase inhibitor. J. Pharmacol. Exp. Ther. 1995, 274, 530–539. [Google Scholar]
- El-Armouche, A.; Pamminger, T.; Ditz, D.; Zolk, O.; Eschenhagen, T. Decreased protein and phosphorylation level of the protein phosphatase inhibitor-1 in failing human hearts. Cardiovasc. Res. 2004, 61, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Grote-Wessels, S.; Baba, H.A.; Boknik, P.; El-Armouche, A.; Fabritz, L.; Gillmann, H.-J.; Kucerova, D.; Matus, M.; Müller, F.U.; Neumann, J.; et al. Inhibition of protein phosphatase 1 by inhibitor-2 exacerbates progression of cardiac failure in a model with pressure overload. Cardiovasc. Res. 2008, 79, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Kirchhefer, U.; Baba, H.A.; Bokník, P.; Breeden, K.M.; Mavila, N.; Brüchert, N.; Justus, I.; Matus, M.; Schmitz, W.; DePaoli-Roach, A.A.; et al. Enhanced cardiac function in mice overexpressing protein phosphatase Inhibitor-2. Cardiovasc. Res. 2005, 68, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Krause, T.; Grote-Wessels, S.; Balzer, F.; Boknik, P.; Gergs, U.; Kirchhefer, U.; Buchwalow, I.B.; Müller, F.U.; Schmitz, W.; Neumann, J. Successful overexpression of wild-type inhibitor-2 of PP1 in cardiovascular cells. Naunyn Schmiedebergs. Arch. Pharmacol. 2018, 391, 859–873. [Google Scholar] [CrossRef]
- Pathak, A.; del Monte, F.; Zhao, W.; Schultz, J.-E.; Lorenz, J.N.; Bodi, I.; Weiser, D.; Hahn, H.; Carr, A.N.; Syed, F.; et al. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ. Res. 2005, 96, 756–766. [Google Scholar] [CrossRef] [Green Version]
- Brautigan, D.L.; Shenolikar, S. Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates. Annu. Rev. Biochem. 2018, 87, 921–964. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [Green Version]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Lubbers, E.R.; Mohler, P.J. Roles and regulation of protein phosphatase 2A (PP2A) in the heart. J. Mol. Cell. Cardiol. 2016, 101, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Davare, M.A.; Horne, M.C.; Hell, J.W. Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J. Biol. Chem. 2000, 275, 39710–39717. [Google Scholar] [CrossRef] [Green Version]
- Bhasin, N.; Cunha, S.R.; Mudannayake, M.; Gigena, M.S.; Rogers, T.B.; Mohler, P.J. Molecular basis for PP2A regulatory subunit B56alpha targeting in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H109–H119. [Google Scholar] [CrossRef] [Green Version]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Gaburjakova, M.; Gaburjakova, J.; Yang, Y.M.; Rosemblit, N.; Marks, A.R. Phosphorylation-dependent regulation of ryanodine receptors: A novel role for leucine/isoleucine zippers. J. Cell Biol. 2001, 153, 699–708. [Google Scholar] [CrossRef]
- Reiken, S.; Gaburjakova, M.; Guatimosim, S.; Gomez, A.M.; D’Armiento, J.; Burkhoff, D.; Wang, J.; Vassort, G.; Lederer, W.J.; Marks, A.R. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J. Biol. Chem. 2003, 278, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDougall, L.K.; Jones, L.R.; Cohen, P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur. J. Biochem. 1991, 196, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Maas, R.; Bokník, P.; Jones, L.R.; Zimmermann, N.; Scholz, H. Pharmacological characterization of protein phosphatase activities in preparations from failing human hearts. J. Pharmacol. Exp. Ther. 1999, 289, 188–193. [Google Scholar] [PubMed]
- Weber, S.; Meyer-Roxlau, S.; Wagner, M.; Dobrev, D.; El-Armouche, A. Counteracting Protein Kinase Activity in the Heart: The Multiple Roles of Protein Phosphatases. Front. Pharmacol. 2015, 6, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jideama, N.M.; Crawford, B.H.; Hussain, A.K.M.A.; Raynor, R.L. Dephosphorylation specificities of protein phosphatase for cardiac troponin I, troponin T, and sites within troponin T. Int. J. Biol. Sci. 2006, 2, 1–9. [Google Scholar] [CrossRef]
- Kuster, D.W.D.; Bawazeer, A.C.; Zaremba, R.; Goebel, M.; Boontje, N.M.; van der Velden, J. Cardiac myosin binding protein C phosphorylation in cardiac disease. J. Muscle Res. Cell Motil. 2012, 33, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Mumby, M.C.; Walter, G. Protein serine/threonine phosphatases: Structure, regulation, and functions in cell growth. Physiol. Rev. 1993, 73, 673–699. [Google Scholar] [CrossRef]
- Solaro, R.J.; Kobayashi, T. Protein phosphorylation and signal transduction in cardiac thin filaments. J. Biol. Chem. 2011, 286, 9935–9940. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.; Sun, Q.; Li, Y.; Zhang, L.; Zhang, P.; Wang, X.; Tian, C.; Li, Q.; Song, J.; Liu, H.; et al. CKIP-1 inhibits cardiac hypertrophy by regulating class II histone deacetylase phosphorylation through recruiting PP2A. Circulation 2012, 126, 3028–3040. [Google Scholar] [CrossRef] [Green Version]
- Paroni, G.; Cernotta, N.; Dello Russo, C.; Gallinari, P.; Pallaoro, M.; Foti, C.; Talamo, F.; Orsatti, L.; Steinkühler, C.; Brancolini, C. PP2A regulates HDAC4 nuclear import. Mol. Biol. Cell 2008, 19, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Swingle, M.R.; Honkanen, R.E.; Ciszak, E.M. Structural basis for the catalytic activity of human serine/threonine protein phosphatase-5. J. Biol. Chem. 2004, 279, 33992–33999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blatch, G.L.; Lässle, M. The tetratricopeptide repeat: A structural motif mediating protein-protein interactions. Bioessays 1999, 21, 932–939. [Google Scholar] [CrossRef]
- Lamb, J.R.; Tugendreich, S.; Hieter, P. Tetratrico peptide repeat interactions: To TPR or not to TPR? Trends Biochem. Sci. 1995, 20, 257–259. [Google Scholar] [CrossRef]
- Hinds, T.D.; Sánchez, E.R. Protein phosphatase 5. Int. J. Biochem. Cell Biol. 2008, 40, 2358–2362. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, A.M.; Galigniana, M.D.; Chen, M.S.; Owens-Grillo, J.K.; Chinkers, M.; Pratt, W.B. Protein phosphatase 5 is a major component of glucocorticoid receptor.hsp90 complexes with properties of an FK506-binding immunophilin. J. Biol. Chem. 1997, 272, 16224–16230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, T.H.; Ning, Y.-M.; Sánchez, E.R. Differential control of glucocorticoid receptor hormone-binding function by tetratricopeptide repeat (TPR) proteins and the immunosuppressive ligand FK506. Biochemistry 2005, 44, 2030–2038. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, A.J.; Russell, L.C.; Whitt, S.R.; Chinkers, M. Overlapping sites of tetratricopeptide repeat protein binding and chaperone activity in heat shock protein 90. J. Biol. Chem. 2000, 275, 17857–17862. [Google Scholar] [CrossRef] [Green Version]
- Conde, R.; Xavier, J.; McLoughlin, C.; Chinkers, M.; Ovsenek, N. Protein phosphatase 5 is a negative modulator of heat shock factor 1. J. Biol. Chem. 2005, 280, 28989–28996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, F.; Umeda, Y.; Shimamoto, S.; Tsuchiya, M.; Tokumitsu, H.; Tokuda, M.; Kobayashi, R. S100 proteins modulate protein phosphatase 5 function: A link between CA2+ signal transduction and protein dephosphorylation. J. Biol. Chem. 2012, 287, 13787–13798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbalzano, E.; Mandraffino, G.; Casciaro, M.; Quartuccio, S.; Saitta, A.; Gangemi, S. Pathophysiological mechanism and therapeutic role of S100 proteins in cardiac failure: A systematic review. Heart Fail. Rev. 2016, 21, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lu, Y.; Li, Y.; Xiao, L.; Xing, Y.; Li, Y.; Wu, L. Role of S100A1 in hypoxia-induced inflammatory response in cardiomyocytes via TLR4/ROS/NF-κB pathway. J. Pharm. Pharmacol. 2015, 67, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Krysiak, J.; Unger, A.; Beckendorf, L.; Hamdani, N.; von Frieling-Salewsky, M.; Redfield, M.M.; Dos Remedios, C.G.; Sheikh, F.; Gergs, U.; Boknik, P.; et al. Protein phosphatase 5 regulates titin phosphorylation and function at a sarcomere-associated mechanosensor complex in cardiomyocytes. Nat. Commun. 2018, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Dörner, M.; Gergs, U.; Neumann, J. Influence of aging on cardiac hypoxia in PP2A overexpressing mice. In Proceedings of the 84th Annual Meeting of the German Society for Experimental and Clinical Pharmacology and Toxicology (DGPT) and the 20th Annual Meeting of the Association of the Clinical Pharmacology Germany (VKliPha) with contribution of the Arbeitsgemeinschaft für Angewandte Humanpharmakologie e. V. (AGAH), Stuttgart, Germany, 25–28 February 2019; Springer: New York, NY, USA, 2019; p. 30. [Google Scholar]
- Dörner, M.; Gergs, U.; Neumann, J. Cardiac function in young and old PP2AxPP5 overexpressing mice. In Proceedings of the 85th Annual Meeting of the German Society for Experimental and Clinical Pharmacology and Toxicology (DGPT) and the 21th Annual Meeting of the Association of the Clinical Pharmacology Germany (VKliPha) with contribution of the Arbeitsgemeinschaft für Angewandte Humanpharmakologie e. V. (AGAH), Stuttgart, Germany, 25–28 February 2019; Springer: New York, NY, USA, 2019; pp. 41–42. [Google Scholar]
- Dörner, M.; Köpp, F.; Gergs, U.; Neumann, J. Cardiac regulation and systolic function of PP2A and PP5 in PP2AxPP5 double-transgenic mice. Clin. Res. Cardiol. 2019, 108, P1241. [Google Scholar]
- Köpp, F.; Dörner, M.; Runte, J.; Gergs, U.; Neumann, J. Mechanisms of cardiac hypertrophy in PP2AxPP5 double transgenic mice. In Proceedings of the 84th Annual Meeting of the German Society for Experimental and Clinical Pharmacology and Toxicology (DGPT) and the 20th Annual Meeting of the Association of the Clinical Pharmacology Germany (VKliPha) With contribution of the Arbeitsgemeinschaft für Angewandte Humanpharmakologie e. V. (AGAH), Stuttgart, Germany, 25–28 February 2019; Springer: New York, NY, USA, 2019; p. 29. [Google Scholar]
- Neumann, J.; Köpp, F.; Dörner, M.; Gergs, U. Cardiac dysfunction in mice overexpressing both PP2A and PP5. In Proceedings of the FEBS Advanced Courses, Europhosphatase: From Molecular Mechanisms to System-Wide Responses, Debrecen, Hungary, 11–16 June 2019; p. 27, ISBN 978-615-5270-54-3. [Google Scholar]
- Kuhn, M. Molecular physiology of natriuretic peptide signalling. Basic Res. Cardiol. 2004, 99, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Endothelial actions of atrial and B-type natriuretic peptides. Br. J. Pharmacol. 2012, 166, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Yoder, A.R.; Flora, D.R.; Antos, L.K.; Dickey, D.M. Natriuretic peptides: Their structures, receptors, physiologic functions and therapeutic applications. Handb. Exp. Pharmacol. 2009, 341–366. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K.; Nakao, K. Regulation and significance of atrial and brain natriuretic peptides as cardiac hormones. Endocr. J. 2010, 57, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K. The natriuretic peptide system in heart failure: Diagnostic and therapeutic implications. Pharmacol. Ther. 2021, 227, 107863. [Google Scholar] [CrossRef]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of Inflammation in Heart Failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef]
- Bokník, P.; Khorchidi, S.; Bodor, G.S.; Huke, S.; Knapp, J.; Linck, B.; Lüss, H.; Müller, F.U.; Schmitz, W.; Neumann, J. Role of protein phosphatases in regulation of cardiac inotropy and relaxation. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H786–H794. [Google Scholar] [CrossRef] [PubMed]
- Tei, C.; Nishimura, R.A.; Seward, J.B.; Tajik, A.J. Noninvasive Doppler-derived myocardial performance index: Correlation with simultaneous measurements of cardiac catheterization measurements. J. Am. Soc. Echocardiogr. 1997, 10, 169–178. [Google Scholar] [CrossRef]
- Truttmann, A.C.; Ashraf, Q.; Mishra, O.P.; Delivoria-Papadopoulos, M. Effect of hypoxia on protein phosphatase 2A activity, subcellular distribution and expression in cerebral cortex of newborn piglets. Neuroscience 2004, 127, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Elgenaidi, I.S.; Spiers, J.P. Hypoxia modulates protein phosphatase 2A through HIF-1α dependent and independent mechanisms in human aortic smooth muscle cells and ventricular cardiomyocytes. Br. J. Pharmacol. 2019, 176, 1745–1763. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.A.; Payne, E.S.; Csortos, C.; DePaoli-Roach, A.A.; Lefkowitz, R.J. The G-protein-coupled receptor phosphatase: A protein phosphatase type 2A with a distinct subcellular distribution and substrate specificity. Proc. Natl. Acad. Sci. USA 1995, 92, 8343–8347. [Google Scholar] [CrossRef] [Green Version]
- Bristow, M.R.; Ginsburg, R.; Minobe, W.; Cubicciotti, R.S.; Sageman, W.S.; Lurie, K.; Billingham, M.E.; Harrison, D.C.; Stinson, E.B. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 1982, 307, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.J. Adrenergic signaling in heart failure: A balance of toxic and protective effects. Pflug. Arch. 2014, 466, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Mika, D. cAMP/PKA signaling compartmentalization in cardiomyocytes: Lessons from FRET-based biosensors. J. Mol. Cell. Cardiol. 2019, 131, 112–121. [Google Scholar] [CrossRef]
- Gergs, U.; Baumann, M.; Böckler, A.; Buchwalow, I.B.; Ebelt, H.; Fabritz, L.; Hauptmann, S.; Keller, N.; Kirchhof, P.; Klöckner, U.; et al. Cardiac overexpression of the human 5-HT4 receptor in mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H788–H798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gergs, U.; Gerigk, T.; Wittschier, J.; Schmidbaur, C.T.; Röttger, C.; Mahnkopf, M.; Edler, H.; Wache, H.; Neumann, J. Influence of Serotonin 5-HT4 Receptors on Responses to Cardiac Stressors in Transgenic Mouse Models. Biomedicines 2021, 9, 569. [Google Scholar] [CrossRef] [PubMed]
- Lubert, E.J.; Hong, Y.; Sarge, K.D. Interaction between Protein Phosphatase 5 and the A subunit of Protein Phosphatase 2A. J. Biol. Chem. 2001, 276, 38582–38587. [Google Scholar] [CrossRef] [Green Version]
- Katayama, K.; Yamaguchi, M.; Noguchi, K.; Sugimoto, Y. Protein phosphatase complex PP5/PPP2R3C dephosphorylates P-glycoprotein/ABCB1 and down-regulates the expression and function. Cancer Lett. 2014, 345, 124–131. [Google Scholar] [CrossRef]
- Chiang, C.-W.; Liu, W.-K.; Chiang, C.-W.; Chou, C.-K. Phosphorylation-dependent association of the G4-1/G5PR regulatory subunit with IKKβ negatively modulates NF-κB activation through recruitment of protein phosphatase 5. Biochem. J. 2011, 433, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Kono, Y.; Maeda, K.; Kuwahara, K.; Yamamoto, H.; Miyamoto, E.; Yonezawa, K.; Takagi, K.; Sakaguchi, N. MCM3-binding GANP DNA-primase is associated with a novel phosphatase component G5PR. Genes Cells 2002, 7, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Urban, G.; Scammell, J.G.; Dean, N.M.; McLean, T.K.; Aragon, I.; Honkanen, R.E. Ser/Thr Protein Phosphatase Type 5 (PP5) Is a Negative Regulator of Glucocorticoid Receptor-Mediated Growth Arrest. Biochemistry 1999, 38, 8849–8857. [Google Scholar] [CrossRef] [PubMed]
- Messner, D.J.; Romeo, C.; Boynton, A.; Rossie, S. Inhibition of PP2A, but not PP5, mediates p53 activation by low levels of okadaic acid in rat liver epithelial cells. J. Cell. Biochem. 2006, 99, 241–255. [Google Scholar] [CrossRef]
- Schwinger, R.H.; Münch, G.; Bölck, B.; Karczewski, P.; Krause, E.G.; Erdmann, E. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J. Mol. Cell. Cardiol. 1999, 31, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.C.; Mishra, S.; Rastogi, S.; Imai, M.; Habib, O.; Sabbah, H.N. Cardiac SR-coupled PP1 activity and expression are increased and inhibitor 1 protein expression is decreased in failing hearts. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2373–H2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.; Gupta, R.C.; Tiwari, N.; Sharov, V.G.; Sabbah, H.N. Molecular mechanisms of reduced sarcoplasmic reticulum Ca2+ uptake in human failing left ventricular myocardium. J. Heart Lung Transplant. 2002, 21, 366–373. [Google Scholar] [CrossRef]
- Carr, A.N.; Schmidt, A.G.; Suzuki, Y.; del Monte, F.; Sato, Y.; Lanner, C.; Breeden, K.; Jing, S.-L.; Allen, P.B.; Greengard, P.; et al. Type 1 phosphatase, a negative regulator of cardiac function. Mol. Cell. Biol. 2002, 22, 4124–4135. [Google Scholar] [CrossRef] [Green Version]
- Vasan, R.S.; Urbina, E.M.; Jin, L.; Xanthakis, V. Prognostic Significance of Echocardiographic Measures of Cardiac Remodeling in the Community. Curr. Cardiol. Rep. 2021, 23, 86. [Google Scholar] [CrossRef]
- Packer, M. Differential Pathophysiological Mechanisms in Heart Failure With a Reduced or Preserved Ejection Fraction in Diabetes. JACC Heart Fail. 2021, 9, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Gergs, U.; Böckler, A.; Ebelt, H.; Hauptmann, S.; Keller, N.; Otto, V.; Pönicke, K.; Schmitz, W.; Neumann, J. Human 5-HT₄receptor stimulation in atria of transgenic mice. Naunyn Schmiedebergs. Arch. Pharmacol. 2013, 386, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Kirchhefer, U.; Baba, H.A.; Hanske, G.; Jones, L.R.; Kirchhof, P.; Schmitz, W.; Neumann, J. Age-dependent biochemical and contractile properties in atrium of transgenic mice overexpressing junctin. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2216–H2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, J.; Boknik, P.; Matherne, G.P.; Lankford, A.; Schmitz, W. Pertussis toxin sensitive and insensitive effects of adenosine and carbachol in murine atria overexpressing A(1)-adenosine receptors. Br. J. Pharmacol. 2003, 138, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gergs, U.; Fahrion, C.M.; Bock, P.; Fischer, M.; Wache, H.; Hauptmann, S.; Schmitz, W.; Neumann, J. Evidence for a functional role of calsequestrin 2 in mouse atrium. Acta Physiol. 2017, 219, 669–682. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Kirchhefer, U.; Heinick, A.; König, S.; Kristensen, T.; Müller, F.U.; Seidl, M.D.; Boknik, P. Protein phosphatase 2A is regulated by protein kinase Cα (PKCα)-dependent phosphorylation of its targeting subunit B56α at Ser41. J. Biol. Chem. 2014, 289, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Huang, Y.-L.; Lin, N.-Y.; Chen, H.-C.; Chiu, W.-C.; Chang, C.-J. Differential regulation of ARE-mediated TNFalpha and IL-1beta mRNA stability by lipopolysaccharide in RAW264.7 cells. Biochem. Biophys. Res. Commun. 2006, 346, 160–168. [Google Scholar] [CrossRef]
- Furlow, J.D.; Watson, M.L.; Waddell, D.S.; Neff, E.S.; Baehr, L.M.; Ross, A.P.; Bodine, S.C. Altered gene expression patterns in muscle ring finger 1 null mice during denervation- and dexamethasone-induced muscle atrophy. Physiol. Genom. 2013, 45, 1168–1185. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Chen, Y.; Chiang, S.K.S.; Tso, M.O.M. NF-kappaB activation in light-induced retinal degeneration in a mouse model. Invest. Ophthalmol. Vis. Sci. 2002, 43, 2834–2840. [Google Scholar] [PubMed]
- Yamamoto, H.; Omelchenko, I.; Shi, X.; Nuttall, A.L. The influence of NF-kappaB signal-transduction pathways on the murine inner ear by acoustic overstimulation. J. Neurosci. Res. 2009, 87, 1832–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age Group I | Age Group II | |||||||
|---|---|---|---|---|---|---|---|---|
| WT | PP2A | PP5 | DT | WT | PP2A | PP5 | DT | |
| n | 24 | 18 | 20 | 13 | 21 | 19 | 22 | 8 |
| mLung [mg] | 220 ± 9 | 214 ± 8 | 213 ± 5 | 262 ± 17 #,+,§ | 290 ± 7 * | 311 ± 10 * | 245 ± 8 #,* | 273 ± 22 |
| n | 24 | 18 | 20 | 13 | 21 | 19 | 22 | 8 |
| mLiver [mg] | 1878 ± 83 | 1694 ± 99 | 1752 ± 85 | 2025 ± 99 | 2310 ± 88 * | 2468 ± 162 * | 1846 ± 82 # | 2695 ± 227 §,* |
| n | 11 | 12 | 19 | 13 | 8 | 8 | 22 | 13 |
| mKidney [mg] | 566 ± 47 | 523 ± 38 | 511 ± 39 | 501 ± 29 | 587 ± 53 | 527 ± 48 | 518 ± 26 | 573 ± 44 |
| WT (n = 7) | PP2A (n = 8) | PP5 (n = 7) | DT (n = 5) | |||||
|---|---|---|---|---|---|---|---|---|
| EF [%] | Age Group I | Age Group II | Age Group I | Age Group II | Age Group I | Age Group II | Age Group I | Age Group II |
| Basal | 51.1 ± 4.31 | 51.5 ± 3.35 | 22.6 ± 1.60 # | 24.9 ± 2.31 # | 51.5 ± 2.4 | 48.6 ± 3.99 | 19.0 ± 3.2 #,§ | 19.8 ± 3.2 #,§ |
| Iso | 91.1 ± 2.26 * | 92.4 ± 1.28 * | 62.1 ± 6.33 #,* | 61.0 ± 4.21 #,* | 85.0 ± 2.56 * | 79.0 ± 4.53 * | 30.5 ± 6.55 #,+,§ | 32.2 ± 5.10 #,+,§ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dörner, M.-F.; Boknik, P.; Köpp, F.; Buchwalow, I.B.; Neumann, J.; Gergs, U. Mechanisms of Systolic Cardiac Dysfunction in PP2A, PP5 and PP2AxPP5 Double Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 9448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179448
Dörner M-F, Boknik P, Köpp F, Buchwalow IB, Neumann J, Gergs U. Mechanisms of Systolic Cardiac Dysfunction in PP2A, PP5 and PP2AxPP5 Double Transgenic Mice. International Journal of Molecular Sciences. 2021; 22(17):9448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179448
Chicago/Turabian StyleDörner, Mara-Francine, Peter Boknik, Friedrich Köpp, Igor B. Buchwalow, Joachim Neumann, and Ulrich Gergs. 2021. "Mechanisms of Systolic Cardiac Dysfunction in PP2A, PP5 and PP2AxPP5 Double Transgenic Mice" International Journal of Molecular Sciences 22, no. 17: 9448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179448