
Perspectives on hiPSC-Derived Muscle Cells as Drug Discovery Models for Muscular Dystrophies

 ,
, 
Abstract
:1. Introduction
2. Preclinical Models of MD
3. hiPSCs for Drug Screening
3.1. Drug Screening for DMD
3.2. Drug Screening for LGMDs
4. hiPSCs in the Development of Disease-Modifying Therapeutic Approaches for MDs
4.1. Antisense Oligonucleotides
4.1.1. Antisense Oligonucleotides for DMD
4.1.2. Antisense Oligonucleotides for DM1
4.2. Gene Transfer
4.2.1. Gene Transfer for DMD
4.2.2. Gene Transfer for LGMDs
4.3. Genome Editing
4.3.1. Genome Editing for DMD
4.3.2. Genome Editing for Autosomal Recessive Muscular Dystrophies
4.3.3. Genome Editing for Autosomal Dominant Muscular Dystrophies
5. Challenging Issues and Future Perspectives Regarding iPSC Models and Therapeutic Screening
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef]
- Ashizawa, T.; Gagnon, C.; Groh, W.J.; Gutmann, L.; Johnson, N.E.; Meola, G.; Moxley, R.; Pandya, S.; Rogers, M.T.; Simpson, E.; et al. Consensus-based care recommendations for adults with myotonic dystrophy type 1. Neurol. Clin. Pract. 2018, 8, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoser, B.; Montagnese, F.; Bassez, G.; Fossati, B.; Gamez, J.; Heatwole, C.; Hilbert, J.; Kornblum, C.; Kostera-Pruszczyk, A.; Krahe, R.; et al. Consensus-based care recommendations for adults with myotonic dystrophy type 2. Neurol. Clin. Pract. 2019, 9, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Bockhorst, J.; Wicklund, M. Limb girdle muscular dystrophies. Neurol. Clin. 2020, 38, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Guglieri, M.; Magri, F.; Comi, G.P. Molecular etiopathogenesis of limb girdle muscular and congenital muscular dystrophies: Boundaries and contiguities. Clin. Chim. Acta 2005, 361, 54–79. [Google Scholar] [CrossRef] [PubMed]
- Magri, F.; Nigro, V.; Angelini, C.; Mongini, T.; Mora, M.; Moroni, I.; Toscano, A.; D’Angelo, M.G.; Tomelleri, G.; Siciliano, G.; et al. The italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle Nerve 2017, 55, 55–68. [Google Scholar] [CrossRef]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Gappmaier, E.; Howard, M.T.; Sampson, J.B.; Mendell, J.R.; Wall, C.; King, W.M.; et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009, 30, 1657–1666. [Google Scholar] [CrossRef] [Green Version]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Magri, F.; Govoni, A.; D’Angelo, M.G.; Del Bo, R.; Ghezzi, S.; Sandra, G.; Turconi, A.C.; Sciacco, M.; Ciscato, P.; Bordoni, A.; et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 2011, 258, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Belhasan, D.C.; Akaaboune, M. The role of the dystrophin glycoprotein complex on the neuromuscular system. Neurosci. Lett. 2020, 722, 134833. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Eteplirsen: First global approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A. Golodirsen: First approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Viltolarsen: First approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.J. Ataluren: First global approval. Drugs 2014, 74, 1709–1714. [Google Scholar] [CrossRef]
- Chemello, F.; Bassel-Duby, R.; Olson, E.N. Correction of muscular dystrophies by CRISPR gene editing. J. Clin. Investig. 2020, 130, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Datta, N.; Ghosh, P.S. Update on muscular dystrophies with focus on novel treatments and biomarkers. Curr. Neurol. Neurosci. Rep. 2020, 20, 14. [Google Scholar] [CrossRef]
- Govoni, A.; Magri, F.; Brajkovic, S.; Zanetta, C.; Faravelli, I.; Corti, S.; Bresolin, N.; Comi, G.P. Ongoing therapeutic trials and outcome measures for Duchenne muscular dystrophy. Cell. Mol. Life Sci. 2013, 70, 4585–4602. [Google Scholar] [CrossRef] [PubMed]
- Scoto, M.; Finkel, R.; Mercuri, E.; Muntoni, F. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc. Health 2018, 2, 600–609. [Google Scholar] [CrossRef]
- Lawson, M.A.; Purslow, P.P. Differentiation of myoblasts in serum-free media: Effects of modified media are cell line-specific. Cells Tissues Organs 2000, 167, 130–137. [Google Scholar] [CrossRef]
- Bajaj, P.; Reddy, B.; Millet, L.; Wei, C.; Zorlutuna, P.; Bao, G.; Bashir, R. Patterning the differentiation of C2C12 skeletal myoblasts. Integr. Biol. 2011, 3, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.M.; Sather, B.K.; Burney, E.R.; Ehrlicher, S.E.; Stierwalt, H.D.; Franco, M.C.; Newsom, S.A. Robust intrinsic differences in mitochondrial respiration and H2O2 emission between L6 and C2C12 cells. Am. J. Physiol. Cell Physiol. 2019, 317, C339–C347. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Yuasa, S.; Motoda, C.; Yozu, G.; Nagai, T.; Ito, S.; Lachmann, M.; Kashimura, S.; Takei, M.; Kusumoto, D.; et al. Emerin plays a crucial role in nuclear invagination and in the nuclear calcium transient. Sci. Rep. 2017, 7, 44312. [Google Scholar] [CrossRef] [Green Version]
- Rybkin, I.I.; Markham, D.W.; Yan, Z.; Bassel-Duby, R.; Williams, R.S.; Olson, E.N. Conditional expression of SV40 T-antigen in mouse cardiomyocytes facilitates an inducible switch from proliferation to differentiation. J. Biol. Chem. 2003, 278, 15927–15934. [Google Scholar] [CrossRef] [Green Version]
- Relaix, F.; Bencze, M.; Borok, M.J.; Der Vartanian, A.; Gattazzo, F.; Mademtzoglou, D.; Perez-Diaz, S.; Prola, A.; Reyes-Fernandez, P.C.; Rotini, A.; et al. Perspectives on skeletal muscle stem cells. Nat. Commun. 2021, 12, 692. [Google Scholar] [CrossRef]
- Bossolasco, P.; Corti, S.; Strazzer, S.; Borsotti, C.; Del Bo, R.; Fortunato, F.; Salani, S.; Quirici, N.; Bertolini, F.; Gobbi, A.; et al. Skeletal muscle differentiation potential of human adult bone marrow cells. Exp. Cell Res. 2004, 295, 66–78. [Google Scholar] [CrossRef]
- Meregalli, M.; Farini, A.; Colleoni, F.; Cassinelli, L.; Torrente, Y. The role of stem cells in muscular dystrophies. Curr. Gene Ther. 2012, 12, 192–205. [Google Scholar] [CrossRef]
- Fusco-Allison, G.; Li, D.K.; Hunter, B.; Jackson, D.; Bannon, P.G.; Lal, S.; O’Sullivan, J.F. Optimizing the discovery and assessment of therapeutic targets in heart failure with preserved ejection fraction. ESC Heart Fail. 2021, ehf2.13504. [Google Scholar] [CrossRef]
- Biswas, A.; Hutchins, R. Embryonic stem cells. Stem Cells Dev. 2007, 16, 213–222. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Salani, S.; Donadoni, C.; Rizzo, F.; Bresolin, N.; Comi, G.P.; Corti, S. Generation of skeletal muscle cells from embryonic and induced pluripotent stem cells as an in vitro model and for therapy of muscular dystrophies. J. Cell. Mol. Med. 2012, 16, 1353–1364. [Google Scholar] [CrossRef]
- Piga, D.; Salani, S.; Magri, F.; Brusa, R.; Mauri, E.; Comi, G.P.; Bresolin, N.; Corti, S. Human induced pluripotent stem cell models for the study and treatment of Duchenne and Becker muscular dystrophies. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419833478. [Google Scholar] [CrossRef] [Green Version]
- Talkhabi, M.; Aghdami, N.; Baharvand, H. Human cardiomyocyte generation from pluripotent stem cells: A state-of-art. Life Sci. 2016, 145, 98–113. [Google Scholar] [CrossRef]
- Brandão, K.O.; Tabel, V.A.; Atsma, D.E.; Mummery, C.L.; Davis, R.P. Human pluripotent stem cell models of cardiac disease: From mechanisms to therapies. Dis. Model. Mech. 2017, 10, 1039–1059. [Google Scholar] [CrossRef] [Green Version]
- Abujarour, R.; Bennett, M.; Valamehr, B.; Lee, T.T.; Robinson, M.; Robbins, D.; Le, T.; Lai, K.; Flynn, P. Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl. Med. 2014, 3, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Lim, H.; Estrellas, K.; Mula, J.; Cohen, T.V.; Zhang, Y.; Donnelly, C.J.; Richard, J.-P.; Kim, Y.J.; Kim, H.; et al. Concordant but varied phenotypes among duchenne muscular dystrophy patient-specific myoblasts derived using a human iPSC-based model. Cell Rep. 2016, 15, 2301–2312. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Li, Y.; Han, L.; Kaplan, A.D.; Ao, Y.; Kalra, S.; Bett, G.C.L.; Rasmusson, R.L.; Denning, C.; Yang, L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with duchenne muscular dystrophy. DMM Dis. Model. Mech. 2015, 8, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzal, M.Z.; Reiter, M.; Gastonguay, C.; McGivern, J.V.; Guan, X.; Ge, Z.D.; Mack, D.L.; Childers, M.K.; Ebert, A.D.; Strande, J.L. Nicorandil, a Nitric Oxide Donor and ATP-Sensitive Potassium Channel Opener, Protects Against Dystrophin-Deficient Cardiomyopathy. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Choi, I.Y.; Rovira Gonzalez, Y.I.; Andersen, P.; Talbot, C.C.; Iyer, S.R.; Lovering, R.M.; Wagner, K.R.; Lee, G. Duchenne muscular dystrophy hiPSC-derived myoblast drug screen identifies compounds that ameliorate disease in mdx mice. JCI Insight 2020, 5, e134287. [Google Scholar] [CrossRef]
- Kokubu, Y.; Nagino, T.; Sasa, K.; Oikawa, T.; Miyake, K.; Kume, A.; Fukuda, M.; Fuse, H.; Tozawa, R.; Sakurai, H. Phenotypic drug screening for dysferlinopathy using patient-derived induced pluripotent stem cells. Stem Cells Transl. Med. 2019, 8, 1017–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchimura, T.; Otomo, J.; Sato, M.; Sakurai, H. A human iPS cell myogenic differentiation system permitting high-throughput drug screening. Stem Cell Res. 2017, 25, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Dick, E.; Kalra, S.; Anderson, D.; George, V.; Ritso, M.; Laval, S.H.; Barresi, R.; Aartsma-Rus, A.; Lochmüller, H.; Denning, C. Exon skipping and gene transfer restore dystrophin expression in human induced pluripotent stem cells-cardiomyocytes harboring DMD mutations. Stem Cells Dev. 2013, 22, 2714–2724. [Google Scholar] [CrossRef] [Green Version]
- Shoji, E.; Sakurai, H.; Nishino, T.; Nakahata, T.; Heike, T.; Awaya, T.; Fujii, N.; Manabe, Y.; Matsuo, M.; Sehara-Fujisawa, A. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 2015, 5, 12831. [Google Scholar] [CrossRef] [Green Version]
- Mondragon-Gonzalez, R.; Perlingeiro, R.C.R. Recapitulating muscle disease phenotypes with myotonic dystrophy 1 induced pluripotent stem cells: A tool for disease modeling and drug discovery. Dis. Models Mech. 2018, 11, dmm034728. [Google Scholar] [CrossRef] [Green Version]
- Kazuki, Y.; Hiratsuka, M.; Takiguchi, M.; Osaki, M.; Kajitani, N.; Hoshiya, H.; Hiramatsu, K.; Yoshino, T.; Kazuki, K.; Ishihara, C.; et al. Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol. Ther. 2010, 18, 386–393. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Gerli, M.F.M.; Perani, L.; Benedetti, S.; Ungaro, F.; Cassano, M.; Antonini, S.; Tagliafico, E.; Artusi, V.; Longa, E.; et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Transl. Med. 2012, 4, 140ra89. [Google Scholar] [CrossRef]
- Tanaka, A.; Woltjen, K.; Miyake, K.; Hotta, A.; Ikeya, M.; Yamamoto, T.; Nishino, T.; Shoji, E.; Sehara-Fujisawa, A.; Manabe, Y.; et al. Efficient and reproducible myogenic differentiation from human iPS cells: Prospects for modeling Miyoshi Myopathy in vitro. PLoS ONE 2013, 8, e61540. [Google Scholar] [CrossRef]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3, e1602814. [Google Scholar] [CrossRef] [Green Version]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.-H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2, e95918. [Google Scholar] [CrossRef]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.-L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef] [Green Version]
- Ifuku, M.; Iwabuchi, K.A.; Tanaka, M.; Lung, M.S.Y.; Hotta, A. Restoration of dystrophin protein expression by exon skipping utilizing CRISPR-Cas9 in myoblasts derived from DMD patient iPS cells. Methods Mol. Biol. 2018, 1828, 191–217. [Google Scholar] [CrossRef]
- Min, Y.-L.; Bassel-Duby, R.; Olson, E.N. CRISPR Correction of Duchenne Muscular Dystrophy. Annu. Rev. Med. 2019, 70, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Morisaka, H.; Yoshimi, K.; Okuzaki, Y.; Gee, P.; Kunihiro, Y.; Sonpho, E.; Xu, H.; Sasakawa, N.; Naito, Y.; Nakada, S.; et al. CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat. Commun. 2019, 10, 5302. [Google Scholar] [CrossRef]
- Moretti, A.; Fonteyne, L.; Giesert, F.; Hoppmann, P.; Meier, A.B.; Bozoglu, T.; Baehr, A.; Schneider, C.M.; Sinnecker, D.; Klett, K.; et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020, 26, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Gee, P.; Lung, M.S.Y.; Okuzaki, Y.; Sasakawa, N.; Iguchi, T.; Makita, Y.; Hozumi, H.; Miura, Y.; Yang, L.F.; Iwasaki, M.; et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat. Commun. 2020, 11, 1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Ma, Y.; Huang, T.; Chen, Y.; Peng, Y.; Li, B.; Li, J.; Zhang, Y.; Song, B.; Sun, X.; et al. Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol. Cell 2018, 72, 380–394.e7. [Google Scholar] [CrossRef] [Green Version]
- Chemello, F.; Chai, A.C.; Li, H.; Rodriguez-Caycedo, C.; Sanchez-Ortiz, E.; Atmanli, A.; Mireault, A.A.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv. 2021, 7, eabg4910. [Google Scholar] [CrossRef]
- Turan, S.; Farruggio, A.P.; Srifa, W.; Day, J.W.; Calos, M.P. Precise correction of disease mutations in induced pluripotent stem cells derived from patients with limb girdle muscular dystrophy. Mol. Ther. 2016, 24, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, S.; Dhoke, N.R.; Kiley, J.; Mateos-Aierdi, A.J.; Tungtur, S.; Mondragon-Gonzalez, R.; Killeen, G.; Oliveira, V.K.P.; López de Munain, A.; Perlingeiro, R.C.R. Gene correction of LGMD2A patient-specific iPSCs for the development of targeted autologous cell therapy. Mol. Ther. 2019, 27, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Terada, N.; Ashizawa, T. Human iPSC models to study orphan diseases: Muscular dystrophies. Curr. Stem Cell Rep. 2018, 4, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, C.; Cappella, M.; Valaperta, R.; Cardani, R.; Meola, G.; Martelli, F.; Cardinali, B.; Falcone, G. CRISPR/Cas9-mediated deletion of CTG expansions recovers normal phenotype in myogenic cells derived from myotonic dystrophy 1 patients. Mol. Ther. Nucleic Acids 2017, 9, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Agtmaal, E.L.; André, L.M.; Willemse, M.; Cumming, S.A.; van Kessel, I.D.G.; van den Broek, W.J.A.A.; Gourdon, G.; Furling, D.; Mouly, V.; Monckton, D.G.; et al. CRISPR/Cas9-induced (CTG⋅CAG)n repeat instability in the myotonic dystrophy type 1 locus: Implications for therapeutic genome editing. Mol. Ther. 2017, 25, 24–43. [Google Scholar] [CrossRef] [Green Version]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef] [Green Version]
- Parente, V.; Corti, S. Advances in spinal muscular atrophy therapeutics. Ther. Adv. Neurol. Disord. 2018, 11, 175628561875450. [Google Scholar] [CrossRef] [PubMed]
- Duan, D. Systemic AAV micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with duchenne muscular dystrophy: A nonrandomized controlled trial. JAMA Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E. V Annotation and Classification of CRISPR-Cas Systems. Methods Mol. Biol. 2015, 1311, 47–75. [Google Scholar] [CrossRef] [Green Version]
- Asher, D.R.; Thapa, K.; Dharia, S.D.; Khan, N.; Potter, R.A.; Rodino-Klapac, L.R.; Mendell, J.R. Clinical development on the frontier: Gene therapy for duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2020, 20, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, G.; Gao, Y.; Jin, S.; Subramony, S.H.; Terada, N.; Ranum, L.P.W.; Swanson, M.S.; Ashizawa, T. Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 2015, 33, 1829–1838. [Google Scholar] [CrossRef] [Green Version]
- Corti, S.; Faravelli, I.; Cardano, M.; Conti, L. Human pluripotent stem cells as tools for neurodegenerative and neurodevelopmental disease modeling and drug discovery. Expert Opin. Drug Discov. 2015, 10, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Huo, H.; Loh, Y.-H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faravelli, I.; Costamagna, G.; Tamanini, S.; Corti, S. Back to the origins: Human brain organoids to investigate neurodegeneration. Brain Res. 2020, 1727, 146561. [Google Scholar] [CrossRef]
- Agrawal, G.; Aung, A.; Varghese, S. Skeletal muscle-on-a-chip: An in vitro model to evaluate tissue formation and injury. Lab Chip 2017, 17, 3447–3461. [Google Scholar] [CrossRef]
- Abati, E.; Bresolin, N.; Comi, G.P.; Corti, S. Preconditioning and cellular engineering to increase the survival of transplanted neural stem cells for motor neuron disease therapy. Mol. Neurobiol. 2018, 56, 3356–3367. [Google Scholar] [CrossRef]



{kind=link}
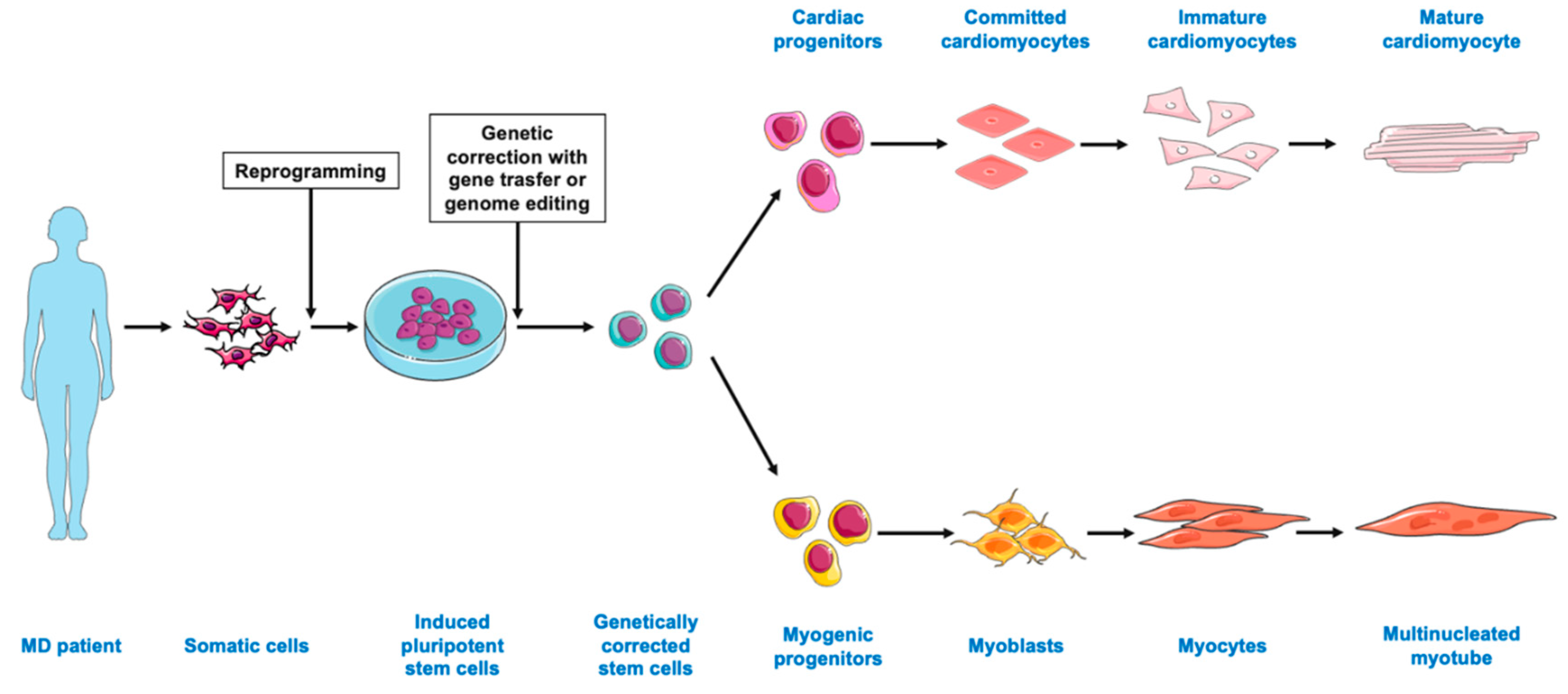{kind=link}
| Disease | Cell Type Obtained | Differentiation Strategy | Screened Molecule(s) | Results | References |
|---|---|---|---|---|---|
| DMD, BMD | Myotubes | Overexpression of the myogenic regulatory factor MyoD | Insulin-like growth factor (IGF1) and wingless-type protein 7a (Wnt7a) | Increase in myotube diameter | [37] |
| DMD | Myoblasts | Exogenous growth factor-based protocol | Pharmacological “dual-SMAD” inhibition (LDN193189 and SB431542) | Reversal of increased nuclear localization of pSMAD protein and expression of IL-6 and -8 and Col3; rescue of myoblasts fusion defects | [38] |
| DMD | Cardiomyocytes | Exogenous growth factor-based protocol | Poloxamer 188 (P188) | Decreased resting cytosolic Ca2+ level, repressed caspase-3 activation | [39] |
| DMD | Cardiomyocytes | Exogenous growth factor-based protocol | Nicorandil | Expression of SOD2 and decreased mitochondrial ROS production | [40] |
| DMD | Myoblasts | Exogenous growth factor-based protocol with FACS purification of NCAM+/HNK1− cells | Ginsenoside Rd, and fenofibrate | Enhancement of myogenic fusion | [41] |
| MM | Myocytes | Exogenous growth factor-based protocol | Nocodazole | Increased dysferlin level and rescue of membrane resealing properties after injury | [42] |
| Intervention | Cell Type | Outcome | References |
|---|---|---|---|
| Antisense oligonucleotides (ASOs) | |||
| ASO-mediated DMD exon 51 skipping | DMD iPSC-derived CMs | Restoration of dystrophin expression in 30% of CMs. | [44] |
| ASO-mediated DMD exon 45 skipping | DMD iPSC-derived myotubes | Restoration of dystrophin expression, decrease in Ca2+ overflow and reduction in CK secretion. | [45] |
| 2′-OMe-PT-(CAG)7-ASO to abolish RNA foci | DM1 iPSC-derived myogenic cells | Reduction in the number of nuclei containing RNA foci and increased inclusion of BIN1 exon 11. | [46] |
| Gene transfer | |||
| Human artificial chromosome vector carrying a genomic dystrophin sequence (HAC-DYS) | DMD iPSCs | Restoration of dystrophin expression in muscle-like tissues upon differentiation of iPSCs into teratomas. | [47] |
| Human artificial chromosome vector carrying a genomic dystrophin sequence (HAC-DYS) | DMD iPSCs | Restoration of dystrophin expression and reversal of the pathologic phenotype (reduced nuclear localization of phosphorylated SMAD and reduced IL-6, IL-8 and collagen 3 expression compared to those in untreated cells). | [38] |
| Lentiviral vector-encoding human α-sarcoglycan cDNA | LGMD R3 iPSCs | Restoration of alpha-SG expression, functional amelioration upon transplantation into α-SG-null mice. | [48] |
| Plasmid vector encoding human dysferlin cDNA | MM iPSCs | Restoration of dysferlin expression and rescue of defective membrane repair. | [49] |
| CRISPR/Cas9 | |||
| TALEN and CRISPR/Cas9 | DMD iPSCs | Comparison of three correction methods: exon skipping, frameshifting, and exon knock-in; exon skipping was the most effective. | [50] |
| CRISPR/Cas9 | iPSC-derived skeletal muscle fibers and CMs | Restoration of dystrophin by the CRISPR/Cas 9-mediated skipping of exons 45–55. Method applicable for 60% of DMD patient mutations. | [51] |
| CRISPR/Cpf1 | iPSC-derived CMs | Cpf1-mediated genome editing restored dystrophin expression in CMs by skipping out-of-frame DMD exons or by correcting nonsense mutations. Reversal of dystrophy hallmarks after germline correction in a mouse model. | [52] |
| CRISPR/Cas9 | DMD iPSC-derived CMs | Correction of dystrophin actin-binding domain mutations with expression of truncated dystrophin and rescue of CM features (contractility and calcium currents). | [53] |
| CRISPR/Cas9 with sgRNAs | DMD iPSC-derived 3D-engineered heart muscle | Correction of exon deletion, pseudoexons, point mutations and large duplication mutations; restoration of dystrophin expression and the corresponding mechanical force of contraction. | [54] |
| CRISPR/Cas9 | DMD iPSC-derived myoblasts | Skipping of exon 45 and restoration of dystrophin expression. | [55] |
| CRISPR/Cas9 | DMD iPSC-derived CMs | Correction of exon 44 deletion mutations using AAV9 encoding Cas9 and single-guide RNAs, leading to dystrophin restoration. | [56] |
| CRISPR/Cas3 | DMD iPSC-derived myoblasts | Correction of exon 45 by Cas3; skipping and restoration of dystrophin was achieved, but the editing efficiency was lower than that of CRISPR/Cas9. | [57] |
| CRISPR/Cas9 | DMD iPSC-derived myotubes and CMs | Correction of exon 52 deletion mutations using AAV9 intein-split Cas9 and a pair of guide RNAs, leading to dystrophin restoration and phenotypic rescue. | [58] |
| CRISPR/Cas9 | DMD iPSC-derived myoblasts | Use of extracellular nanovesicles (NanoMEDIC) to deliver the CRISPR/Cas9 protein with a high transfection rate in different cell types and 90% of exon 45 being skipped in CMs. | [59] |
| CRISPR/Cas9-cytidine deaminase | DMD iPSCs | Use of a CRISPR-guided cytidine deaminase to induce exon 50 skipping and thus restore the ORF and dystrophin function. | [60] |
| CRISPR/Cas9 | DMD iPSC-derived CMs | Use of an adenine base editor (ABE) to modify splice donor sites on the DMD gene, leading to skipping of the stop signal and correction of exon 51 deletions. | [61] |
| CRISPR/Cas9 | LGMD R2 iPSCs and LGMD R3 iPSCs | Correction of point mutations leading to proper expression of dysferlin and α-SG proteins. | [62] |
| CRISPR/Cas9 | LGMD R1 iPSC-derived myotubes | Correction of point mutations leading to proper expression of Calpain 3. | [63] |
| CRISPR/Cas9 | DM1 iPSCs | Elimination of CTG pathological repeats. | [64] |
| CRISPR/Cas9 | DM1 iPSC-derived myogenic cells | Removal of repeat expansion, thereby preventing nuclear focus formation and splicing alterations. | [65] |
| CRISPR/Cas9 | DM1 iPSC-derived myoblasts | Removal of repeat expansion, thereby restoring myogenic capacity and nucleocytoplasmic distribution and preventing nuclear focus formation. | [66] |
| CRISPR/Cas9 | DM1 iPSC-derived myogenic cells and myoblasts | Removal of repeat expansion, thereby preventing nuclear focus formation and splicing alterations. | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abati, E.; Sclarandi, E.; Comi, G.P.; Parente, V.; Corti, S. Perspectives on hiPSC-Derived Muscle Cells as Drug Discovery Models for Muscular Dystrophies. Int. J. Mol. Sci. 2021, 22, 9630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179630
Abati E, Sclarandi E, Comi GP, Parente V, Corti S. Perspectives on hiPSC-Derived Muscle Cells as Drug Discovery Models for Muscular Dystrophies. International Journal of Molecular Sciences. 2021; 22(17):9630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179630
Chicago/Turabian StyleAbati, Elena, Emanuele Sclarandi, Giacomo Pietro Comi, Valeria Parente, and Stefania Corti. 2021. "Perspectives on hiPSC-Derived Muscle Cells as Drug Discovery Models for Muscular Dystrophies" International Journal of Molecular Sciences 22, no. 17: 9630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179630