
Cyclosporine A and Tacrolimus Induce Functional Impairment and Inflammatory Reactions in Endothelial Progenitor Cells

 and
and
Abstract
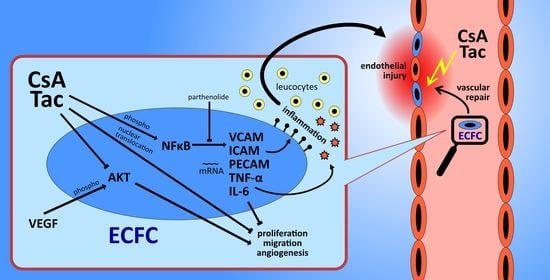
:
1. Introduction
2. Results
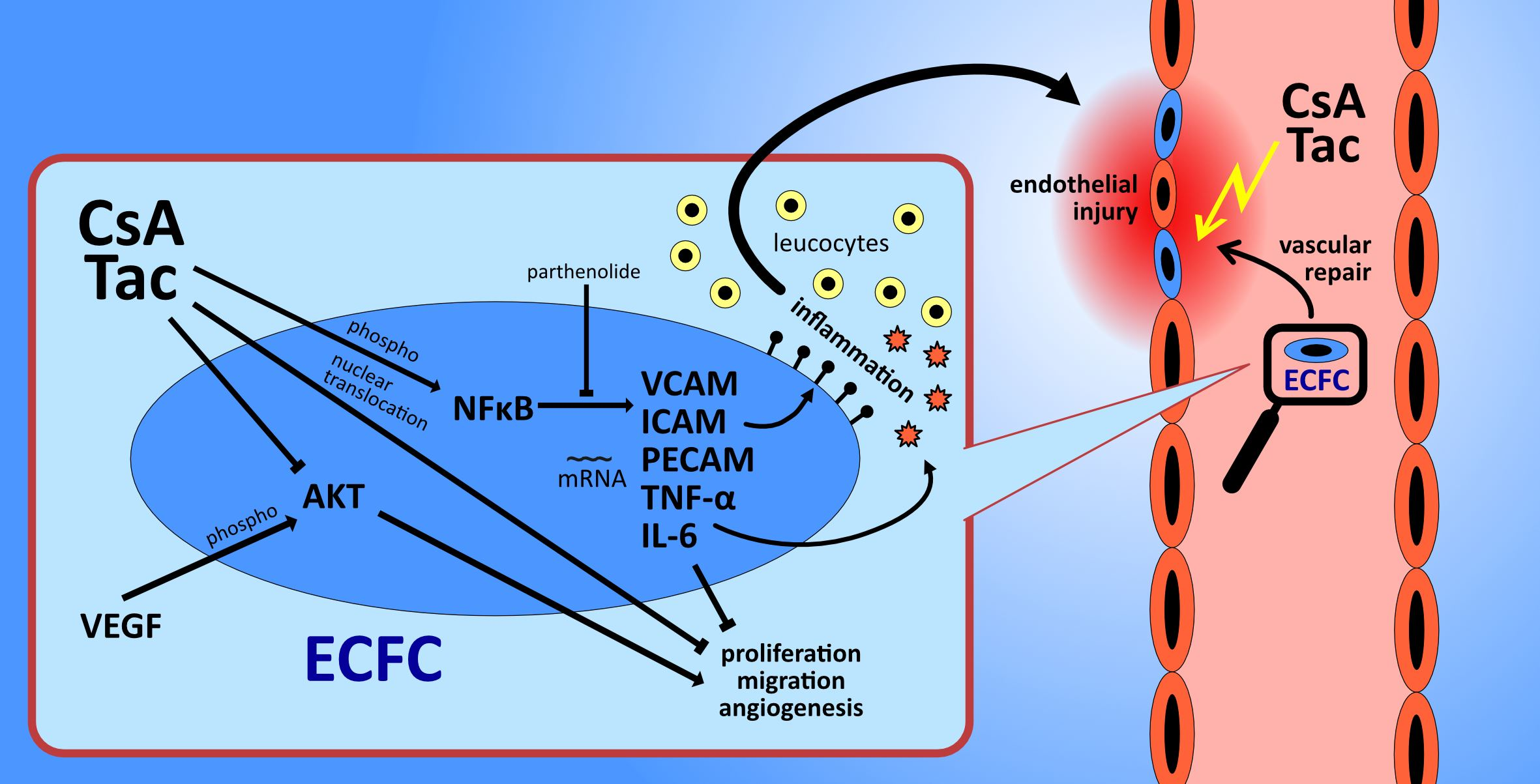
2.1. High-Dose Cyclosporine A and Tacrolimus Inhibit ECFC Proliferation
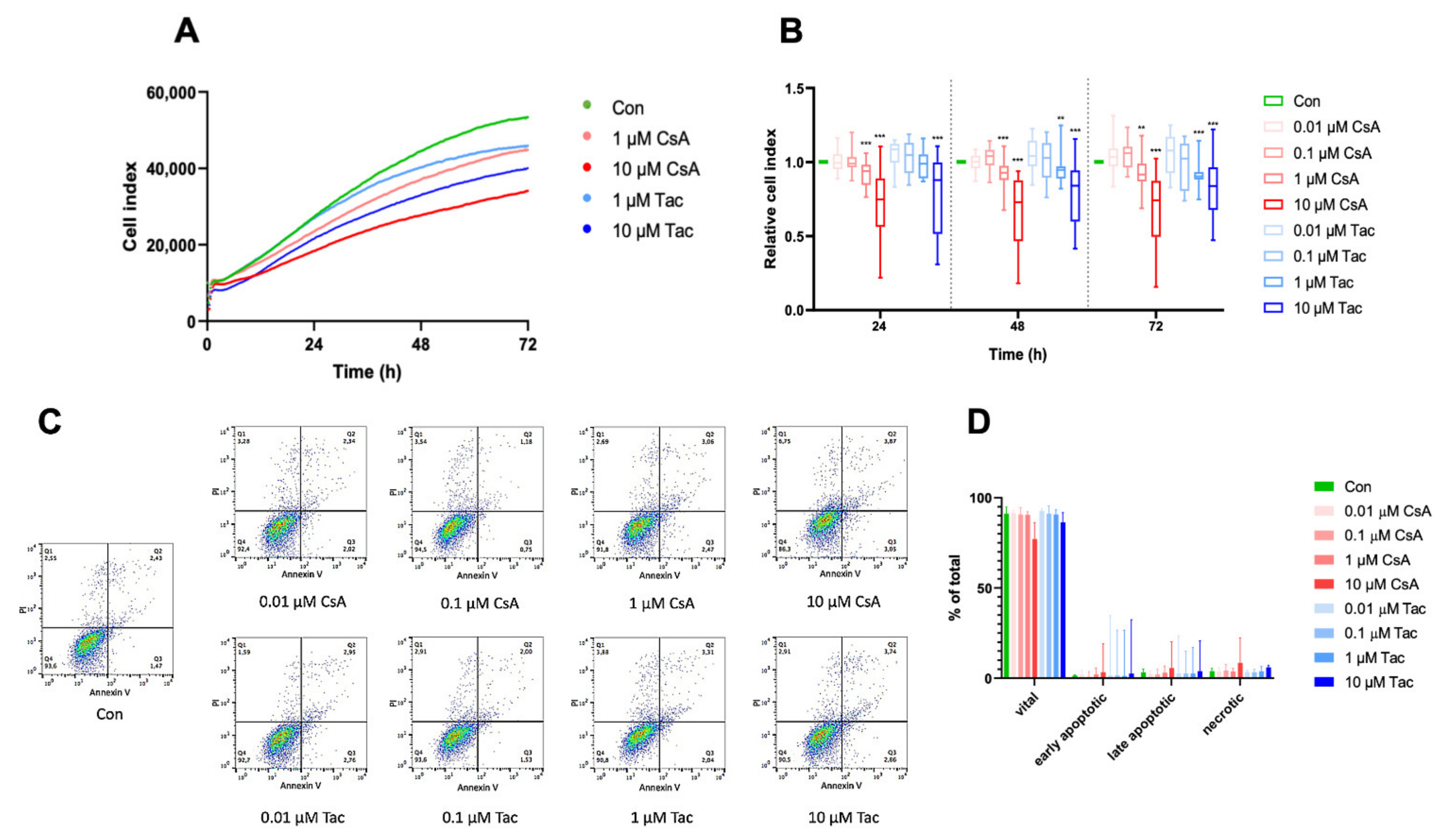
2.2. Cyclosporine A and Tacrolimus Impair ECFC Migration
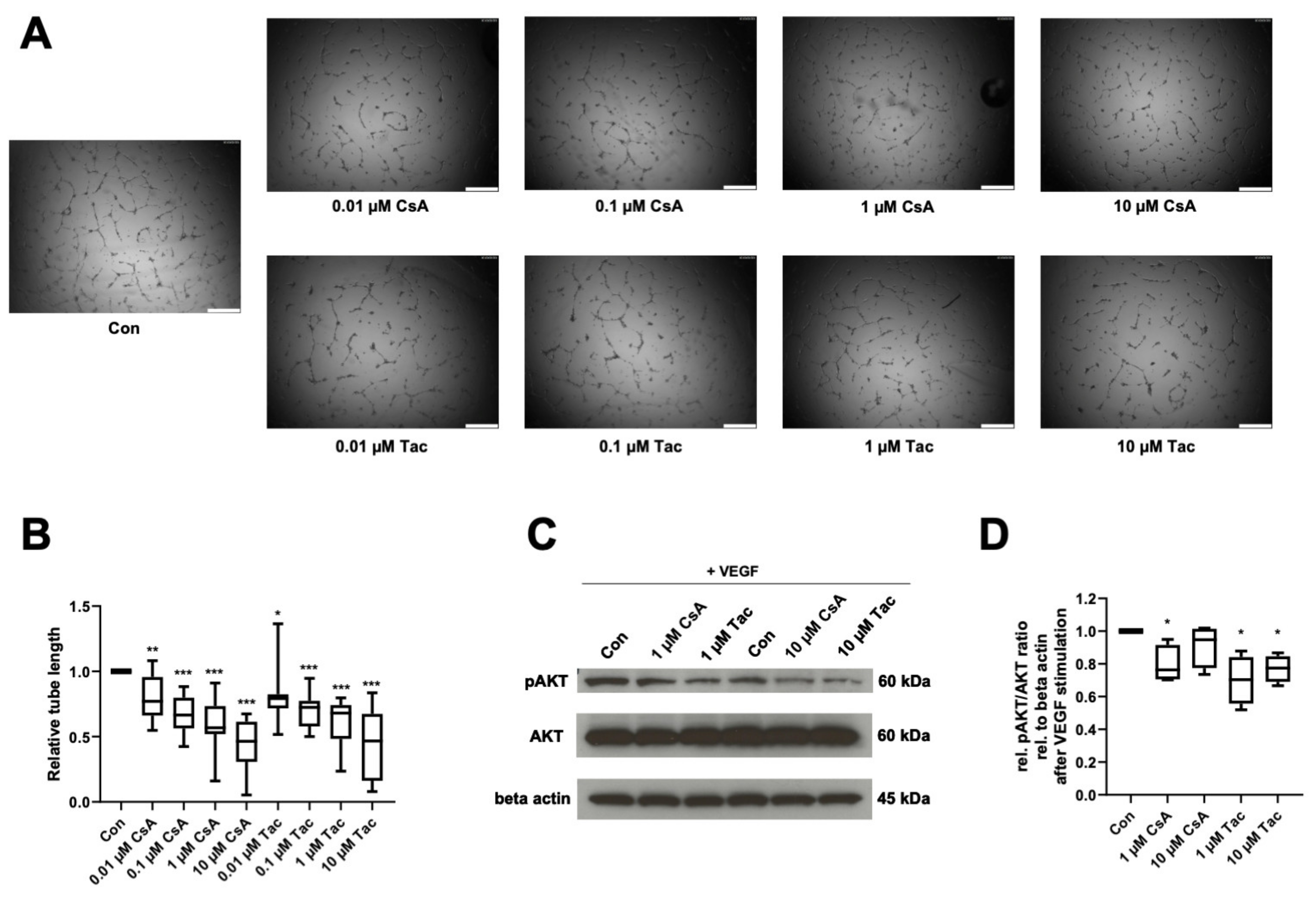
2.3. Cyclosporine A and Tacrolimus Decrease ECFC Angiogenic Capacity and AKT Phosphorylation
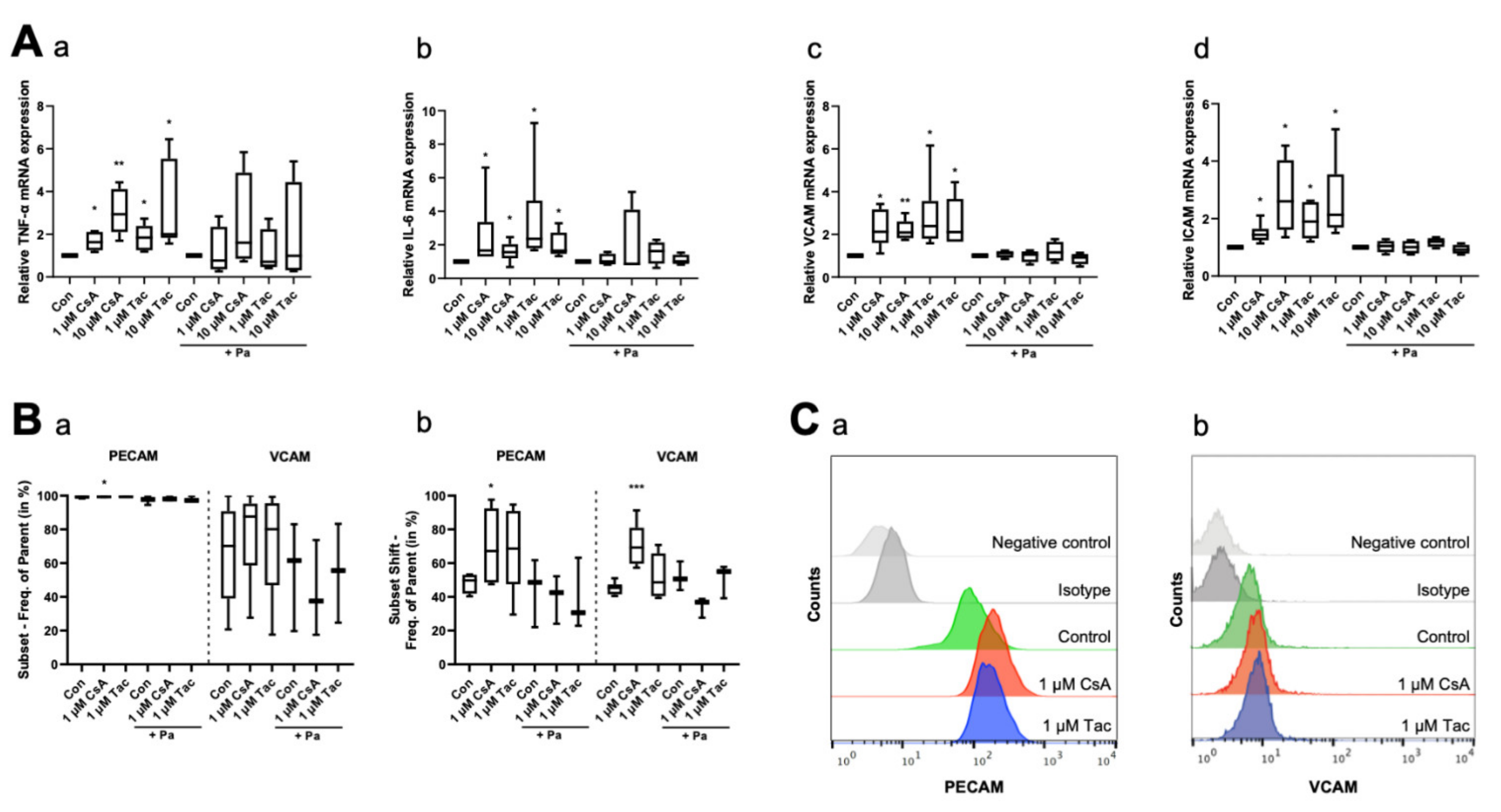
2.4. Cyclosporine A and Tacrolimus Affect Expression of Inflammatory Cytokines and Adhesion Molecules
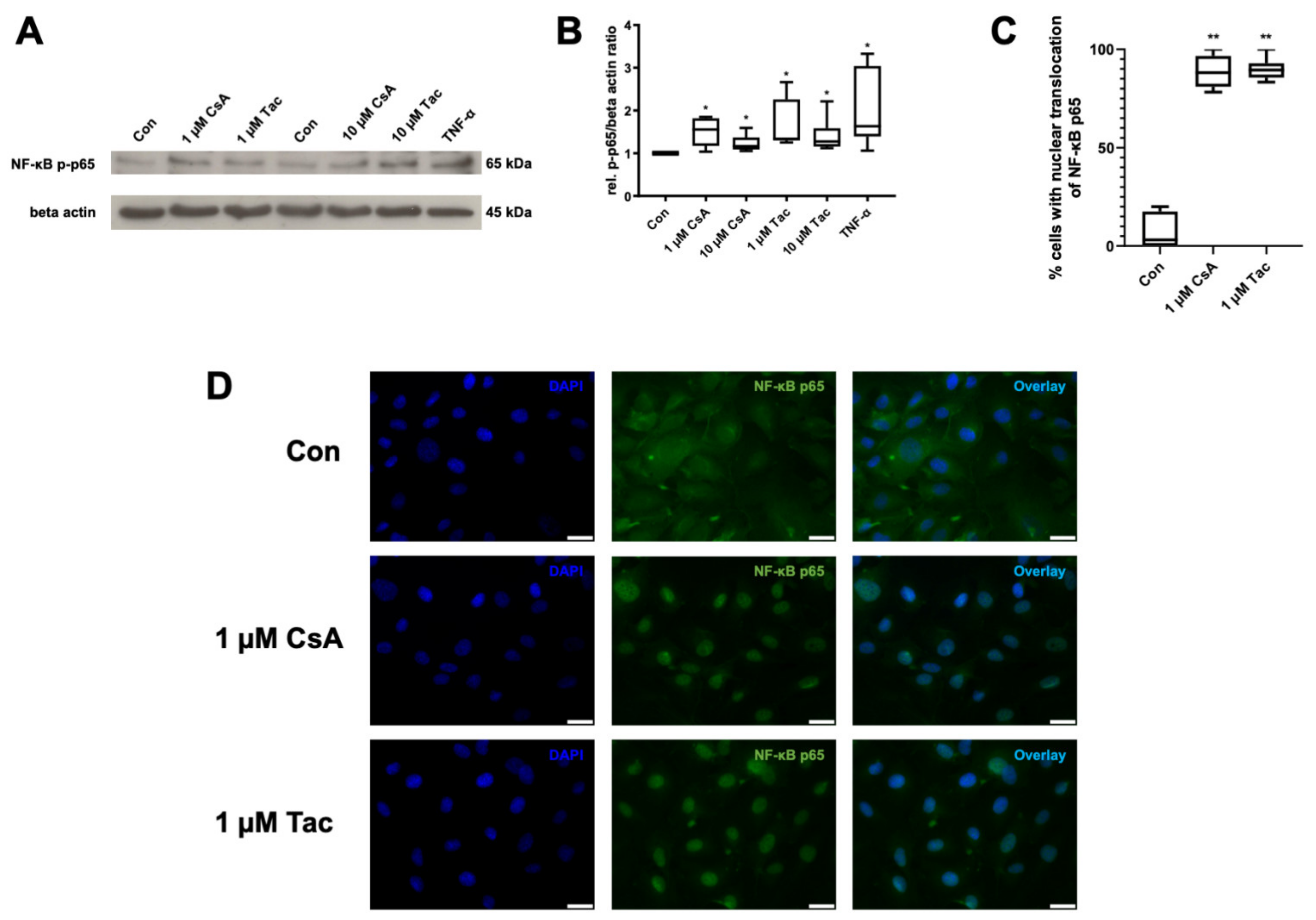
2.5. Cyclosporine A and Tacrolimus Induce Phosphorylation and Nuclear Translocation of NF-κB p65 Subunit
3. Discussion
4. Materials and Methods
4.1. ECFC Isolation, Culture, and Characterization
4.2. Cell Impedance Assay
4.3. Chemotaxis Assay
4.4. Migration Assay
4.5. In Vitro Angiogenesis Assay
4.6. Isolation of Proteins and Immunoblotting
4.7. Quantitative Real-Time PCR (qRT-PCR)
4.8. Flow Cytometry
4.9. Immunocytochemistry
4.10. Lactate Dehydrogenase Cytotoxicity Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waller, D.G.; Sampson, A.P. The immune response and immunosuppressant drugs. In Medical Pharmacology and Therapeutics; Elsevier: Amsterdam, The Netherlands, 2018; pp. 439–449. ISBN 9780702071676. [Google Scholar]
- Barbarino, J.M.; Staatz, C.E.; Venkataramanan, R.; Klein, T.E.; Altman, R.B. PharmGKB summary: Cyclosporine and tacrolimus pathways. Pharmacogenet. Genom. 2013, 23, 563–585. [Google Scholar] [CrossRef] [Green Version]
- Issa, N.; Braun, W.E. Immunosuppression for Renal Transplant Patients and Common Medical Problems in Renal Transplantation. Available online: http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/nephrology/immunosuppression-and-renal-transplant/ (accessed on 4 May 2018).
- Hardinger, K.; Magee, C.C. Pharmacology of Cyclosporine and Tacrolimus. Available online: https://www.uptodate.com/contents/pharmacology-of-cyclosporine-and-tacrolimus (accessed on 4 May 2018).
- Zoja, C.; Furci, L.; Ghilardi, F.; Zilio, P.; Benigni, A.; Remuzzi, G. Cyclosporin-induced endothelial cell injury. Lab. Invest. 1986, 55, 455–462. [Google Scholar] [PubMed]
- Trapp, A.; Weis, M. The impact of immunosuppression on endothelial function. J. Cardiovasc. Pharmacol. 2005, 45, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Michowitz, Y.; Goldstein, E.; Wexler, D.; Sheps, D.; Keren, G.; George, J. Circulating endothelial progenitor cells and clinical outcome in patients with congestive heart failure. Heart 2007, 93, 1046–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreou, I.; Tousoulis, D.; Tentolouris, C.; Antoniades, C.; Stefanadis, C. Potential role of endothelial progenitor cells in the pathophysiology of heart failure: Clinical implications and perspectives. Atherosclerosis 2006, 189, 247–254. [Google Scholar] [CrossRef]
- Luo, S.; Xia, W.; Chen, C.; Robinson, E.A.; Tao, J. Endothelial progenitor cells and hypertension: Current concepts and future implications. Clin. Sci. 2016, 130, 2029–2042. [Google Scholar] [CrossRef]
- Hill, J.M.; Zalos, G.; Halcox, J.P.J.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Sen, S.; McDonald, S.P.; Coates, P.T.H.; Bonder, C.S. Endothelial progenitor cells: Novel biomarker and promising cell therapy for cardiovascular disease. Clin. Sci. 2011, 120, 263–283. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Losordo, D.; Dimmeler, S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ. Res. 2012, 110, 624–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasa, M.; Fichtlscherer, S.; Aicher, A.; Adler, K.; Urbich, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 2001, 89, E1–E7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, D.A.; Mead, L.E.; Tanaka, H.; Meade, V.; Fenoglio, A.; Mortell, K.; Pollok, K.; Ferkowicz, M.J.; Gilley, D.; Yoder, M.C. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 2004, 104, 2752–2760. [Google Scholar] [CrossRef]
- Collett, J.A.; Mehrotra, P.; Crone, A.; Shelley, W.C.; Yoder, M.C.; Basile, D.P. Endothelial colony-forming cells ameliorate endothelial dysfunction via secreted factors following ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 2017, 312, F897–F907. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Zhou, J.; Gong, R.; Huang, X.; Pansuria, M.; Virtue, A.; Li, X.; Wang, H.; Yang, X.-F. Endothelial progenitor cells in atherosclerosis. Front. Biosci. 2012, 17, 2327–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smadja, D.M. Vasculogenic Stem and Progenitor Cells in Human: Future Cell Therapy Product or Liquid Biopsy for Vascular Disease. In Stem Cells: Therapeutic Applications; Ratajczak, M.Z., Ed.; Springer: Cham, Switzerland, 2020; pp. 215–237. ISBN 978-3-030-31206-0. [Google Scholar]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef]
- Dimmeler, S.; Aicher, A.; Vasa, M.; Mildner-Rihm, C.; Adler, K.; Tiemann, M.; Rütten, H.; Fichtlscherer, S.; Martin, H.; Zeiher, A.M. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J. Clin. Invest. 2001, 108, 391–397. [Google Scholar] [CrossRef]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willerson, J.T.; Ridker, P.M. Inflammation as a cardiovascular risk factor. Circulation 2004, 109, II-2–II-10. [Google Scholar] [CrossRef] [Green Version]
- Rusterholz, C.; Hahn, S.; Holzgreve, W. Role of placentally produced inflammatory and regulatory cytokines in pregnancy and the etiology of preeclampsia. Sem. Immunopathol. 2007, 29, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, F. Infection and inflammation in the cardiovascular system. Cardiovasc. Res. 2003, 60, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Van der Heiden, K.; Cuhlmann, S.; Le Luong, A.; Zakkar, M.; Evans, P.C. Role of nuclear factor kappaB in cardiovascular health and disease. Clin. Sci. 2010, 118, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; Trzyna, W.C.; McClintock, D.S.; Schumacker, P.T. Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J. Immunol. 2000, 165, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Brasier, A.R. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Maguire, O.; Collins, C.; O’Loughlin, K.; Miecznikowski, J.; Minderman, H. Quantifying nuclear p65 as a parameter for NF-κB activation: Correlation between ImageStream cytometry, microscopy, and Western blot. Cytometry A 2011, 79, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botella, L.M.; Puig-Kröger, A.; Almendro, N.; Sánchez-Elsner, T.; Muñoz, E.; Corbí, A.; Bernabéu, C. Identification of a functional NF-kappa B site in the platelet endothelial cell adhesion molecule-1 promoter. J. Immunol. 2000, 164, 1372–1378. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.-L.; Ju, J.H.; Kim, K.-W.; Moon, Y.-M.; Lee, S.-Y.; Min, S.-Y.; Cho, Y.-G.; Kim, H.-S.; Park, K.-S.; Yoon, C.-H.; et al. Cyclosporine A inhibits IL-15-induced IL-17 production in CD4+ T cells via down-regulation of PI3K/Akt and NF-kappaB. Immunol. Lett. 2007, 108, 88–96. [Google Scholar] [CrossRef]
- González-Guerrero, C.; Ocaña-Salceda, C.; Berzal, S.; Carrasco, S.; Fernández-Fernández, B.; Cannata-Ortiz, P.; Egido, J.; Ortiz, A.; Ramos, A.M. Calcineurin inhibitors recruit protein kinases JAK2 and JNK, TLR signaling and the UPR to activate NF-κB-mediated inflammatory responses in kidney tubular cells. Toxicol. Appl. Pharmacol. 2013, 272, 825–841. [Google Scholar] [CrossRef]
- Rodrigues-Diez, R.; González-Guerrero, C.; Ocaña-Salceda, C.; Rodrigues-Diez, R.R.; Egido, J.; Ortiz, A.; Ruiz-Ortega, M.; Ramos, A.M. Calcineurin inhibitors cyclosporine A and tacrolimus induce vascular inflammation and endothelial activation through TLR4 signaling. Sci. Rep. 2016, 6, 27915. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gaal, L.F.; Mertens, I.L.; de Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [CrossRef] [PubMed]
- Premer, C.; Kanelidis, A.J.; Hare, J.M.; Schulman, I.H. Rethinking Endothelial Dysfunction as a Crucial Target in Fighting Heart Failure. Mayo Clin. Proc. Innov. Qual. Outcomes 2019, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.; Barsalou, J.; Bradley, T.J.; Slorach, C.; Reynolds, J.A.; Hasni, S.; Thompson, B.; Ng, L.; Levy, D.; Silverman, E.; et al. Endothelial progenitor cell phenotype and function are impaired in childhood-onset systemic lupus erythematosus. Arthritis Rheumatol. 2015, 67, 2257–2262. [Google Scholar] [CrossRef]
- Földes, G.; Mioulane, M.; Kodagoda, T.; Lendvai, Z.; Iqbal, A.; Ali, N.N.; Schneider, M.D.; Harding, S.E. Immunosuppressive agents modulate function, growth, and survival of cardiomyocytes and endothelial cells derived from human embryonic stem cells. Stem Cells Dev. 2014, 23, 467–476. [Google Scholar] [CrossRef]
- Storogenko, M.; Pech-amsellem, M.-A.; Kerdine, S.; Rousselet, F.; Pallardy, M. Cyclosporin-A inhibits human endothelial cells proliferation through interleukin-6-dependent mechanisms. Life Sci. 1997, 60, 1487–1496. [Google Scholar] [CrossRef]
- Esposito, C.; Fornoni, A.; Cornacchia, F.; Bellotti, N.; Fasoli, G.; Foschi, A.; Mazzucchelli, I.; Mazzullo, T.; Semeraro, L.; Dal Canton, A. Cyclosporine induces different responses in human epithelial, endothelial and fibroblast cell cultures. Kidney Int. 2000, 58, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, P.; Koppelstaetter, C.; Aydin, S.; Abberger, T.; Wolf, A.M.; Mayer, G.; Pfaller, W. Cyclosporine A induces senescence in renal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2007, 293, F831–F838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patschan, D.; Schwarze, K.; Tampe, B.; Zeisberg, M.; Patschan, S.; Müller, G.A. Endothelial Colony Forming Cells (ECFCs) in murine AKI—Implications for future cell-based therapies. BMC Nephrol. 2017, 18, 53. [Google Scholar] [CrossRef] [Green Version]
- Critser, P.J.; Yoder, M.C. Endothelial colony-forming cell role in neoangiogenesis and tissue repair. Curr. Opin. Organ Transplant. 2010, 15, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Au, P.; Daheron, L.M.; Duda, D.G.; Cohen, K.S.; Tyrrell, J.A.; Lanning, R.M.; Fukumura, D.; Scadden, D.T.; Jain, R.K. Differential in vivo potential of endothelial progenitor cells from human umbilical cord blood and adult peripheral blood to form functional long-lasting vessels. Blood 2008, 111, 1302–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keighron, C.; Lyons, C.J.; Creane, M.; O’Brien, T.; Liew, A. Recent Advances in Endothelial Progenitor Cells Toward Their Use in Clinical Translation. Front. Med. 2018, 5, 354. [Google Scholar] [CrossRef] [Green Version]
- Sim, S.-L.; Alexis, J.; Roy, E.; Shafiee, A.; Khosrotehrani, K.; Patel, J. Immunosuppression Agent Cyclosporine Reduces Self-Renewal and Vessel Regeneration Potentiation of Human Endothelial Colony Forming Cells. Stem Cells Transl. Med. 2019, 8, 162–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, R.; Kubo, S.; Ohta, T.; Kunimasa, K.; Okada, M.; Tamaki, H.; Kaji, K.; Wakabayashi, I.; Fujimori, Y.; Ogawa, H. FK506 induces endothelial dysfunction through attenuation of Akt and ERK1/2 independently of calcineurin inhibition and the caspase pathway. Cell. Signal. 2013, 25, 1731–1738. [Google Scholar] [CrossRef]
- Hernández, G.L.; Volpert, O.V.; Íñiguez, M.A.; Lorenzo, E.; Martínez-Martínez, S.; Grau, R.; Fresno, M.; Redondo, J.M. Selective Inhibition of Vascular Endothelial Growth Factor–Mediated Angiogenesis by Cyclosporin a: Roles of the Nuclear Factor of Activated T Cells and Cyclooxygenase 2. J. Exp. Med. 2001, 193, 607–620. [Google Scholar] [CrossRef]
- Banai, S.; Shweiki, D.; Pinson, A.; Chandra, M.; Lazarovici, G.; Keshet, E. Upregulation of vascular endothelial growth factor expression induced by myocardial ischaemia: Implications for coronary angiogenesis. Cardiovasc. Res. 1994, 28, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Wolff, T.; Valente, P.; Di Maggio, N.; Pellegrino, M.; Gürke, L.; Banfi, A.; Gianni-Barrera, R. Vascular endothelial growth factor biology for regenerative angiogenesis. Swiss Med. Wkly. 2019, 149, w20011. [Google Scholar] [CrossRef]
- Rafiee, P.; Heidemann, J.; Ogawa, H.; Johnson, N.A.; Fisher, P.J.; Li, M.S.; Otterson, M.F.; Johnson, C.P.; Binion, D.G. Cyclosporin A differentially inhibits multiple steps in VEGF induced angiogenesis in human microvascular endothelial cells through altered intracellular signaling. Cell Commun. Signal. 2004, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Chandakkar, P.; Zhao, H.; Papoin, J.; Chatterjee, P.K.; Christen, E.; Metz, C.N.; Blanc, L.; Campagne, F.; Marambaud, P. Tacrolimus rescues the signaling and gene expression signature of endothelial ALK1 loss-of-function and improves HHT vascular pathology. Hum. Mol. Genet. 2017, 26, 4786–4798. [Google Scholar] [CrossRef]
- Charo, I.F.; Taubman, M.B. Chemokines in the pathogenesis of vascular disease. Circ. Res. 2004, 95, 858–866. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Theroux, P. Pathophysiology of coronary artery disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, R.L.; Tian, Y.; Caldwell, F.J.; Seeto, W.J.; Koehler, J.W.; Pascoe, D.A.; Fan, S.; Gaillard, P.; Lipke, E.A.; Wooldridge, A.A. Cell engraftment, vascularization, and inflammation after treatment of equine distal limb wounds with endothelial colony forming cells encapsulated within hydrogel microspheres. BMC Vet. Res. 2020, 16, 43. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Invest. 2013, 123, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, A.; Montezano, A.C.; Touyz, R.M. Vascular biology of ageing-Implications in hypertension. J. Mol. Cell. Cardiol. 2015, 83, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z. Aging, arterial stiffness, and hypertension. Hypertension 2015, 65, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Popa, C.; Netea, M.G.; van Riel, P.L.C.M.; van der Meer, J.W.M.; Stalenhoef, A.F.H. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J. Lipid Res. 2007, 48, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Mena, H.A.; Lokajczyk, A.; Dizier, B.; Strier, S.E.; Voto, L.S.; Boisson-Vidal, C.; Schattner, M.; Negrotto, S. Acidic preconditioning improves the proangiogenic responses of endothelial colony forming cells. Angiogenesis 2014, 17, 867–879. [Google Scholar] [CrossRef] [Green Version]
- Schröder-Heurich, B.; von Hardenberg, S.; Brodowski, L.; Kipke, B.; Meyer, N.; Borns, K.; von Kaisenberg, C.S.; Brinkmann, H.; Claus, P.; von Versen-Höynck, F. Vitamin D improves endothelial barrier integrity and counteracts inflammatory effects on endothelial progenitor cells. FASEB J. 2019, 33, 9142–9153. [Google Scholar] [CrossRef]
- Deng, X.L.; Li, X.X.; Liu, X.Y.; Sun, L.; Liu, R. Comparative study on circulating endothelial progenitor cells in systemic lupus erythematosus patients at active stage. Rheumatol. Int. 2010, 30, 1429–1436. [Google Scholar] [CrossRef]
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bautista, L.E.; Vera, L.M.; Arenas, I.A.; Gamarra, G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J. Hum. Hypertens. 2005, 19, 149–154. [Google Scholar] [CrossRef]
- Smykiewicz, P.; Segiet, A.; Keag, M.; Żera, T. Proinflammatory cytokines and ageing of the cardiovascular-renal system. Mech. Ageing Dev. 2018, 175, 35–45. [Google Scholar] [CrossRef]
- Brodowski, L.; Schröder-Heurich, B.; Kipke, B.; Schmidt, C.; von Kaisenberg, C.S.; von Versen-Höynck, F. Low Ethanol Concentrations Promote Endothelial Progenitor Cell Capacity and Reparative Function. Cardiovasc. Ther. 2020, 2020, 4018478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ingram, D.A.; Murphy, M.P.; Saadatzadeh, M.R.; Mead, L.E.; Prater, D.N.; Rehman, J. Release of proinflammatory mediators and expression of proinflammatory adhesion molecules by endothelial progenitor cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1675–H1682. [Google Scholar] [CrossRef] [Green Version]
- D’audigier, C.; Cochain, C.; Rossi, E.; Guérin, C.L.; Bièche, I.; Blandinières, A.; Marsac, B.; Silvestre, J.-S.; Gaussem, P.; Smadja, D.M. Thrombin receptor PAR-1 activation on endothelial progenitor cells enhances chemotaxis-associated genes expression and leukocyte recruitment by a COX-2-dependent mechanism. Angiogenesis 2015, 18, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Lehle, K.; Schreml, S.; Kunz-Schughart, L.A.; Rupprecht, L.; Birnbaum, D.E.; Schmid, C.; Preuner, J.G. mTOR inhibitors and calcineurin inhibitors do not affect adhesion molecule expression of human macro- and microvascular endothelial cells. J. Vasc. Res. 2008, 45, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Sasakawa, T.; Sasakawa, Y.; Masunaga, T.; Fujitsu, T.; Hirayama, Y.; Ohkubo, Y.; Mutoh, S. FK506 suppresses E-selectin, ICAM-1 and VCAM-1 expression on vascular endothelial cells by inhibiting tumor necrosis factor alpha secretion from peripheral blood mononuclear cells. Cytokine 2005, 29, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Badiwala, M.V.; Guha, D.; Tumiati, L.; Joseph, J.; Ghashghai, A.; Ross, H.J.; Delgado, D.H.; Rao, V. Epidermal growth factor-like domain 7 is a novel inhibitor of neutrophil adhesion to coronary artery endothelial cells injured by calcineurin inhibition. Circulation 2011, 124, S197–S203. [Google Scholar] [CrossRef] [Green Version]
- Kidokoro, K.; Satoh, M.; Nagasu, H.; Sakuta, T.; Kuwabara, A.; Yorimitsu, D.; Nishi, Y.; Tomita, N.; Sasaki, T.; Kashihara, N. Tacrolimus induces glomerular injury via endothelial dysfunction caused by reactive oxygen species and inflammatory change. Kidney Blood Press. Res. 2012, 35, 549–557. [Google Scholar] [CrossRef]
- Riesinger, L.; Saemisch, M.; Nickmann, M.; Methe, H. CD34+ circulating cells display signs of immune activation in patients with acute coronary syndrome. Heart Vessels 2018, 33, 1559–1569. [Google Scholar] [CrossRef]
- Csiszar, A.; Wang, M.; Lakatta, E.G.; Ungvari, Z. Inflammation and endothelial dysfunction during aging: Role of NF-kappaB. J. Appl. Physiol. 2008, 105, 1333–1341. [Google Scholar] [CrossRef] [Green Version]
- Savoia, C.; Schiffrin, E.L. Inflammation in hypertension. Curr. Opin. Nephrol. Hypertens. 2006, 15, 152–158. [Google Scholar] [CrossRef]
- Savoia, C.; Schiffrin, E.L. Vascular inflammation in hypertension and diabetes: Molecular mechanisms and therapeutic interventions. Clin. Sci. 2007, 112, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.-S.; Schulman, I.H.; Raij, L. Vascular inflammation, insulin resistance, and endothelial dysfunction in salt-sensitive hypertension: Role of nuclear factor kappa B activation. J. Hypertens. 2010, 28, 527–535. [Google Scholar] [CrossRef] [PubMed]
- De Winther, M.P.J.; Kanters, E.; Kraal, G.; Hofker, M.H. Nuclear factor kappaB signaling in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Franco, O.; Hernández-Vargas, P.; Ortiz-Muñoz, G.; Sanjuán, G.; Suzuki, Y.; Ortega, L.; Blanco, J.; Egido, J.; Gómez-Guerrero, C. Parthenolide modulates the NF-kappaB-mediated inflammatory responses in experimental atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1864–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.-W.; Tian, N.; Shparago, M.; Tan, W.; Bailey, A.P.; Manning, R.D. Renal NF-kappaB activation and TNF-alpha upregulation correlate with salt-sensitive hypertension in Dahl salt-sensitive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1817–R1824. [Google Scholar] [CrossRef]
- Cui, R.; Tieu, B.; Recinos, A.; Tilton, R.G.; Brasier, A.R. RhoA mediates angiotensin II-induced phospho-Ser536 nuclear factor kappaB/RelA subunit exchange on the interleukin-6 promoter in VSMCs. Circ. Res. 2006, 99, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Iturbe, B.; Ferrebuz, A.; Vanegas, V.; Quiroz, Y.; Mezzano, S.; Vaziri, N.D. Early and sustained inhibition of nuclear factor-kappaB prevents hypertension in spontaneously hypertensive rats. J. Pharmacol. Exp. Ther. 2005, 315, 51–57. [Google Scholar] [CrossRef]
- Elks, C.M.; Mariappan, N.; Haque, M.; Guggilam, A.; Majid, D.S.A.; Francis, J. Chronic NF-{kappa}B blockade reduces cytosolic and mitochondrial oxidative stress and attenuates renal injury and hypertension in SHR. Am. J. Physiol. Renal Physiol. 2009, 296, F298–F305. [Google Scholar] [CrossRef] [Green Version]
- Taylor, F.; Huffman, M.D.; Macedo, A.F.; Moore, T.H.M.; Burke, M.; Smith, G.D.; Ward, K.; Ebrahim, S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013, CD004816. [Google Scholar] [CrossRef]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar]
- Kwak, B.; Mulhaupt, F.; Myit, S.; Mach, F. Statins as a newly recognized type of immunomodulator. Nat. Med. 2000, 6, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Tangney, C.C. Antiatherothrombotic properties of statins: Implications for cardiovascular event reduction. JAMA 1998, 279, 1643–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hölschermann, H.; Schuster, D.; Parviz, B.; Haberbosch, W.; Tillmanns, H.; Muth, H. Statins prevent NF-kappaB transactivation independently of the IKK-pathway in human endothelial cells. Atherosclerosis 2006, 185, 240–245. [Google Scholar] [CrossRef]
- Lin, C.-P.; Huang, P.-H.; Lai, C.F.; Chen, J.-W.; Lin, S.-J.; Chen, J.-S. Simvastatin Attenuates Oxidative Stress, NF-κB Activation, and Artery Calcification in LDLR−/− Mice Fed with High Fat Diet via Down-regulation of Tumor Necrosis Factor-α and TNF Receptor 1. PLoS ONE 2015, 10, e0143686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, N.; Brodowski, L.; Richter, K.; von Kaisenberg, C.S.; Schröder-Heurich, B.; von Versen-Höynck, F. Pravastatin Promotes Endothelial Colony-Forming Cell Function, Angiogenic Signaling and Protein Expression In Vitro. J. Clin. Med. 2021, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Haidar, M.; Placzko, S.; Niendorf, R.; Darashchonak, N.; Hubel, C.A.; von Versen-Höynck, F. Vitamin D improves the angiogenic properties of endothelial progenitor cells. Am. J. Physiol. Cell Physiol. 2012, 303, C954–C962. [Google Scholar] [CrossRef]
- Tiefenthaler, M.; Hofer, S.; Ebner, S.; Ivarsson, L.; Neyer, S.; Herold, M.; Mayer, G.; Fritsch, P.; Heufler, C. In vitro treatment of dendritic cells with tacrolimus: Impaired T-cell activation and IP-10 expression. Nephrol. Dial. Transplant 2004, 19, 553–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusting, G.J.; Akita, K.; Hickey, H.; Smith, M.; Gurevich, V. Cyclosporin A and tacrolimus (FK506) suppress expression of inducible nitric oxide synthase in vitro by different mechanisms. Br. J. Pharmacol. 1999, 128, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, J.; Rohrbach, A.; Borns, K.; Hillemanns, P.; Feng, L.; Hubel, C.A.; von Versen-Höynck, F. Vitamin D rescues dysfunction of fetal endothelial colony forming cells from individuals with gestational diabetes. Placenta 2015, 36, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Brodowski, L.; Burlakov, J.; Myerski, A.C.; von Kaisenberg, C.S.; Grundmann, M.; Hubel, C.A.; von Versen-Höynck, F. Vitamin D prevents endothelial progenitor cell dysfunction induced by sera from women with preeclampsia or conditioned media from hypoxic placenta. PLoS ONE 2014, 9, e98527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Versen-Höynck, F.; Brodowski, L.; Dechend, R.; Myerski, A.C.; Hubel, C.A. Vitamin D antagonizes negative effects of preeclampsia on fetal endothelial colony forming cell number and function. PLoS ONE 2014, 9, e98990. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense | Antisense |
|---|---|---|
| TNF-α | CCCAGGCAGTCAGATCATCTT | TCAGCTTGAGGGTTTGCTACA |
| IL-6 | GGTACATCCTCGACGGCATCT | GTGCCTCTTTGCTGCTTTCAC |
| VCAM | ATGGTCGTGATCCTTGGAGC | AGATTCTGGGGTGGTCTCGA |
| ICAM | GAACCAGAGCCAGGAGACAC | CTTCACTGTCACCTCGGTCC |
| RNA18S1 | ACATCCAAGGAAGGCAGCAG | TTTTCGTCACTACCTCCCCG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, N.; Brodowski, L.; von Kaisenberg, C.; Schröder-Heurich, B.; von Versen-Höynck, F. Cyclosporine A and Tacrolimus Induce Functional Impairment and Inflammatory Reactions in Endothelial Progenitor Cells. Int. J. Mol. Sci. 2021, 22, 9696. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189696
Meyer N, Brodowski L, von Kaisenberg C, Schröder-Heurich B, von Versen-Höynck F. Cyclosporine A and Tacrolimus Induce Functional Impairment and Inflammatory Reactions in Endothelial Progenitor Cells. International Journal of Molecular Sciences. 2021; 22(18):9696. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189696
Chicago/Turabian StyleMeyer, Nadia, Lars Brodowski, Constantin von Kaisenberg, Bianca Schröder-Heurich, and Frauke von Versen-Höynck. 2021. "Cyclosporine A and Tacrolimus Induce Functional Impairment and Inflammatory Reactions in Endothelial Progenitor Cells" International Journal of Molecular Sciences 22, no. 18: 9696. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189696