
Arginine Methylation of hnRNPK Inhibits the DDX3-hnRNPK Interaction to Play an Anti-Apoptosis Role in Osteosarcoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids and Antibodies
2.2. Preparation of His6-Tagged Proteins
2.3. Preparation of GST-Tagged Fusion Proteins
2.4. Pull-Down Assay Using GST-Tagged Proteins
2.5. Cell Culture and Transfection
2.6. Immunoprecipitation and Western Blot Analysis
2.7. RNA Interference
- 5′-GAUUCACUGACCUUAGUGU[dT][dT]-3′;
- 5′-GCAAAUACUUGGUGUUAGA[dT][dT]-3′;
- 5′-GAAACAGUCGCUGGUGUGA[dT][dT]-3′;
- 5′-GAAAUACGAUGACAUUCCA[dT][dT]-3′.
2.8. TUNEL Assay
2.9. Statistical Analysis
3. Results
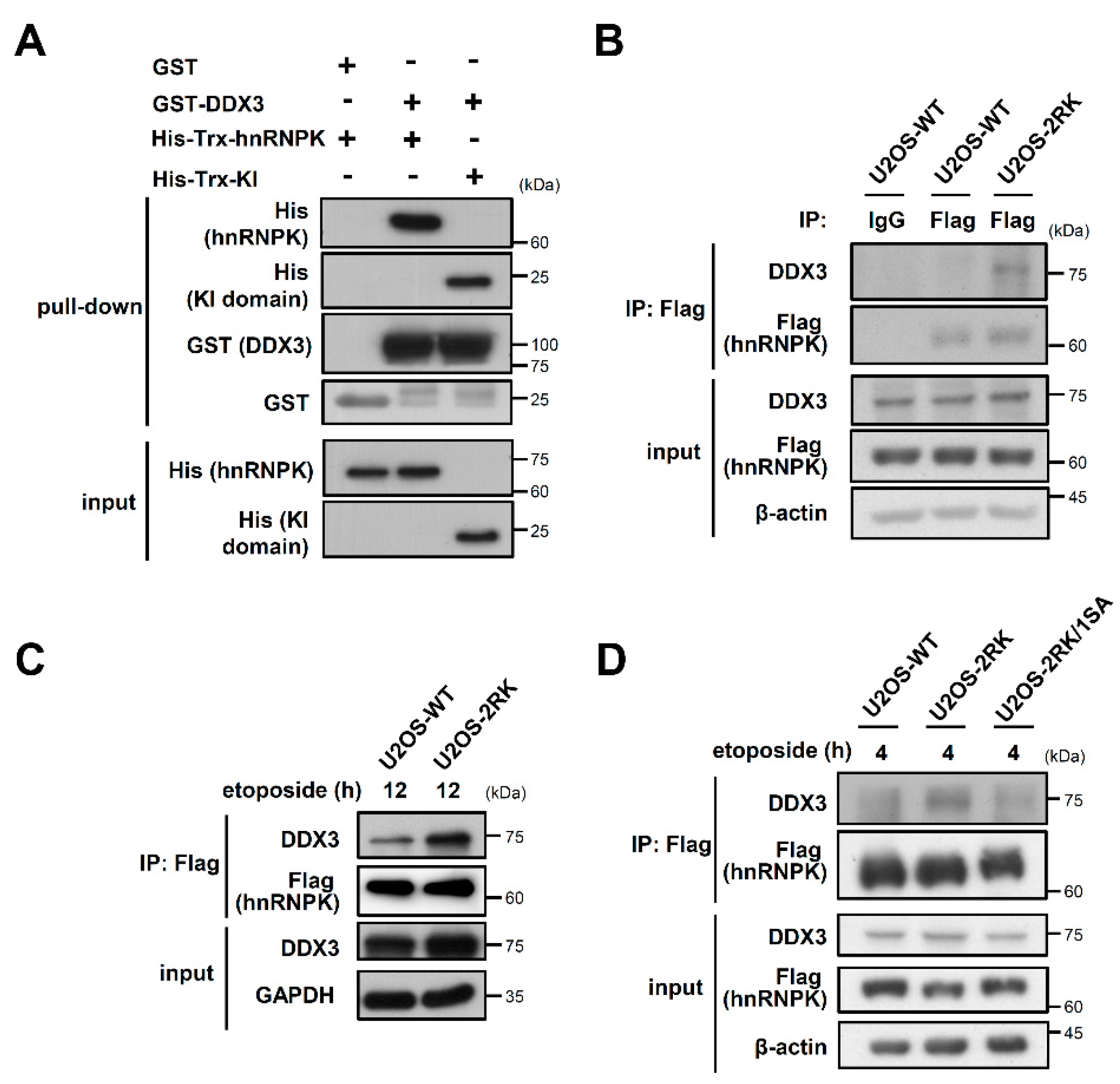
3.1. DDX3 Is Critical to the Apoptosis Enhancement Mediated by hnRNPKMD
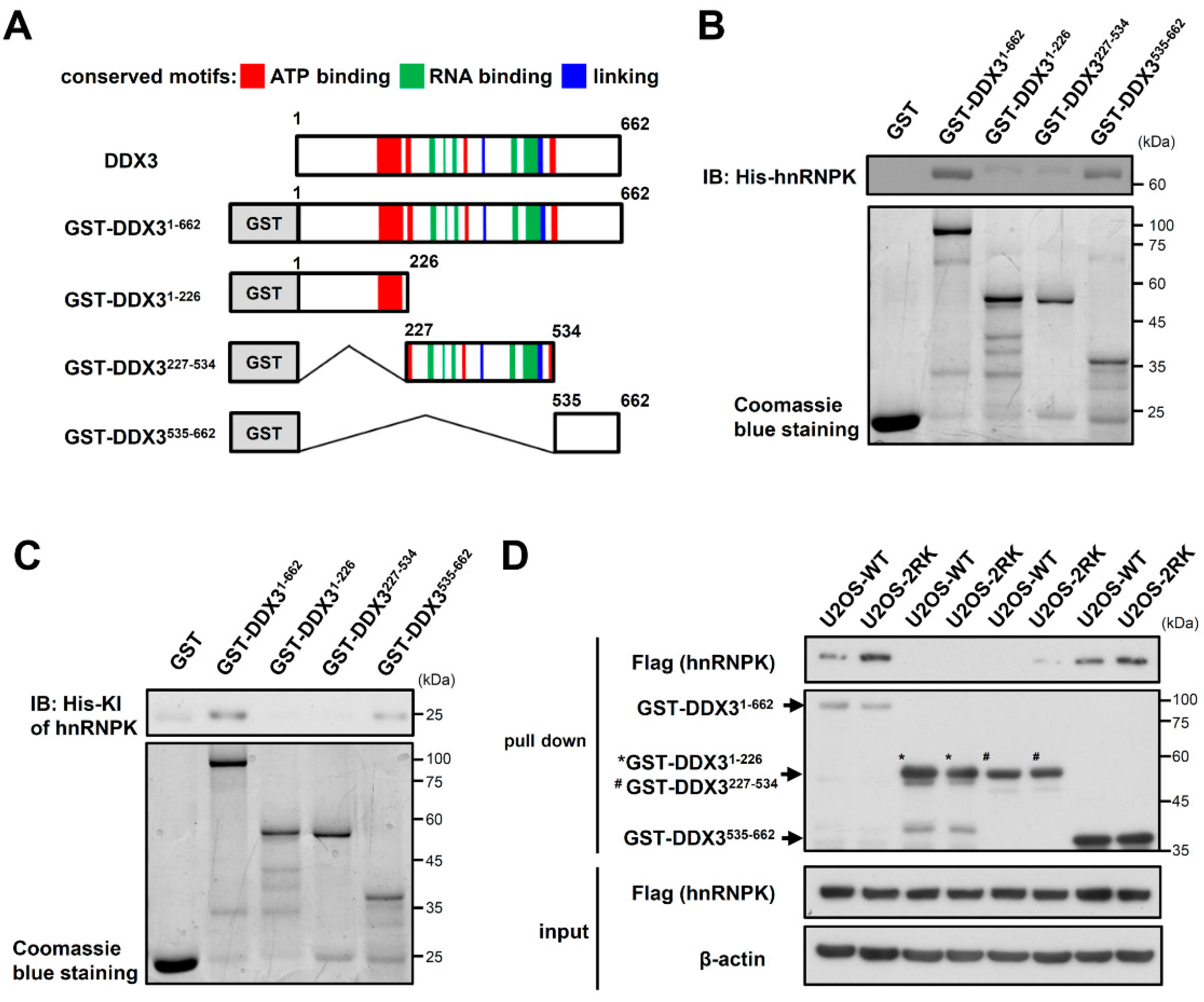
3.2. The C-Terminal Region of DDX3 Is Responsible for the hnRNPK–DDX3 Interaction
3.3. C-Terminus-Truncated DDX3 Could Not Support the Apoptosis Enhancement Mediated by hnRNPKMD upon DNA Damage
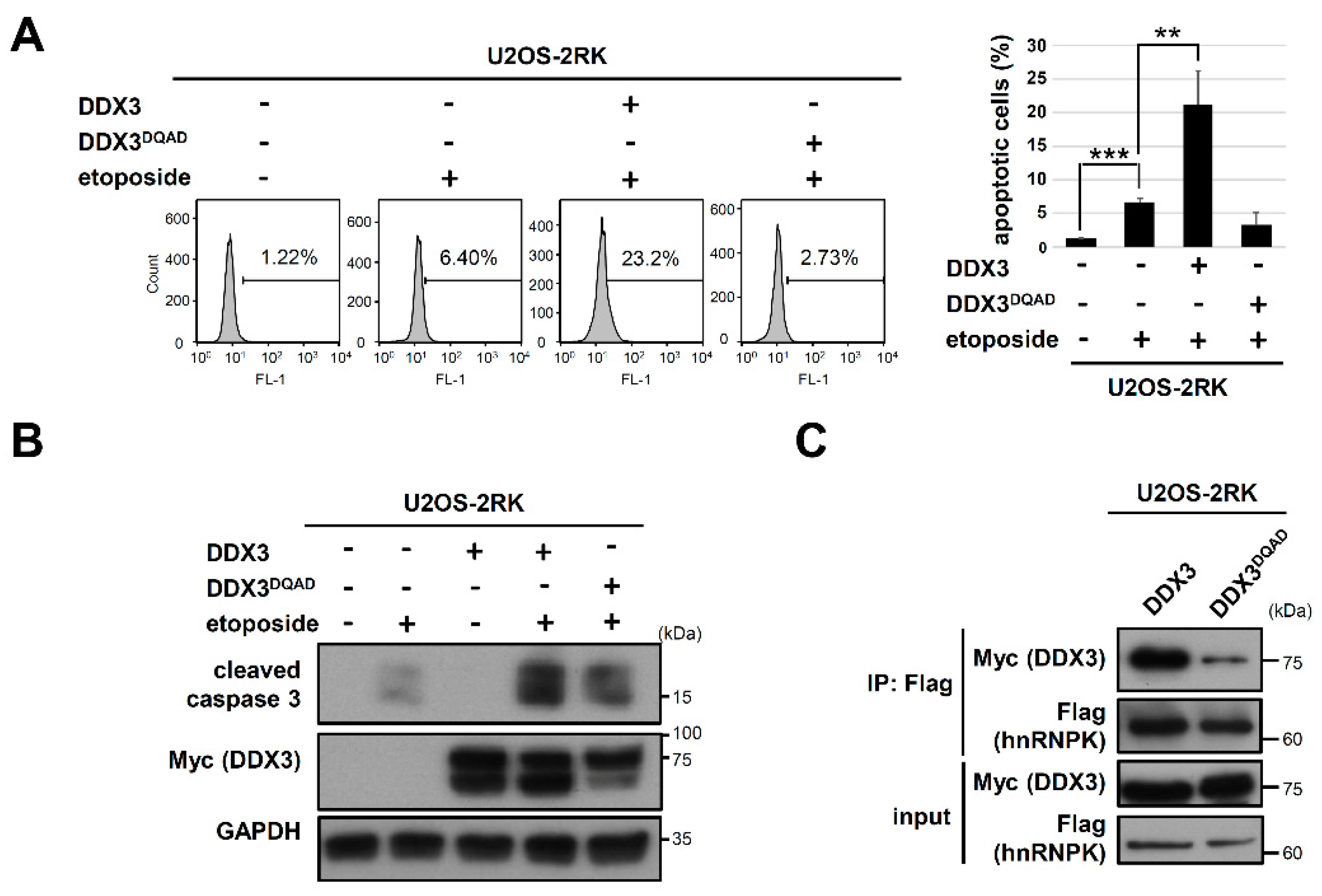
3.4. Mutation of the DEAD Motif in DDX3 Reduces Its hnRNPK Interaction and Impairs Its Apoptosis-Promoting Ability in U2OS Cells under DNA Damage
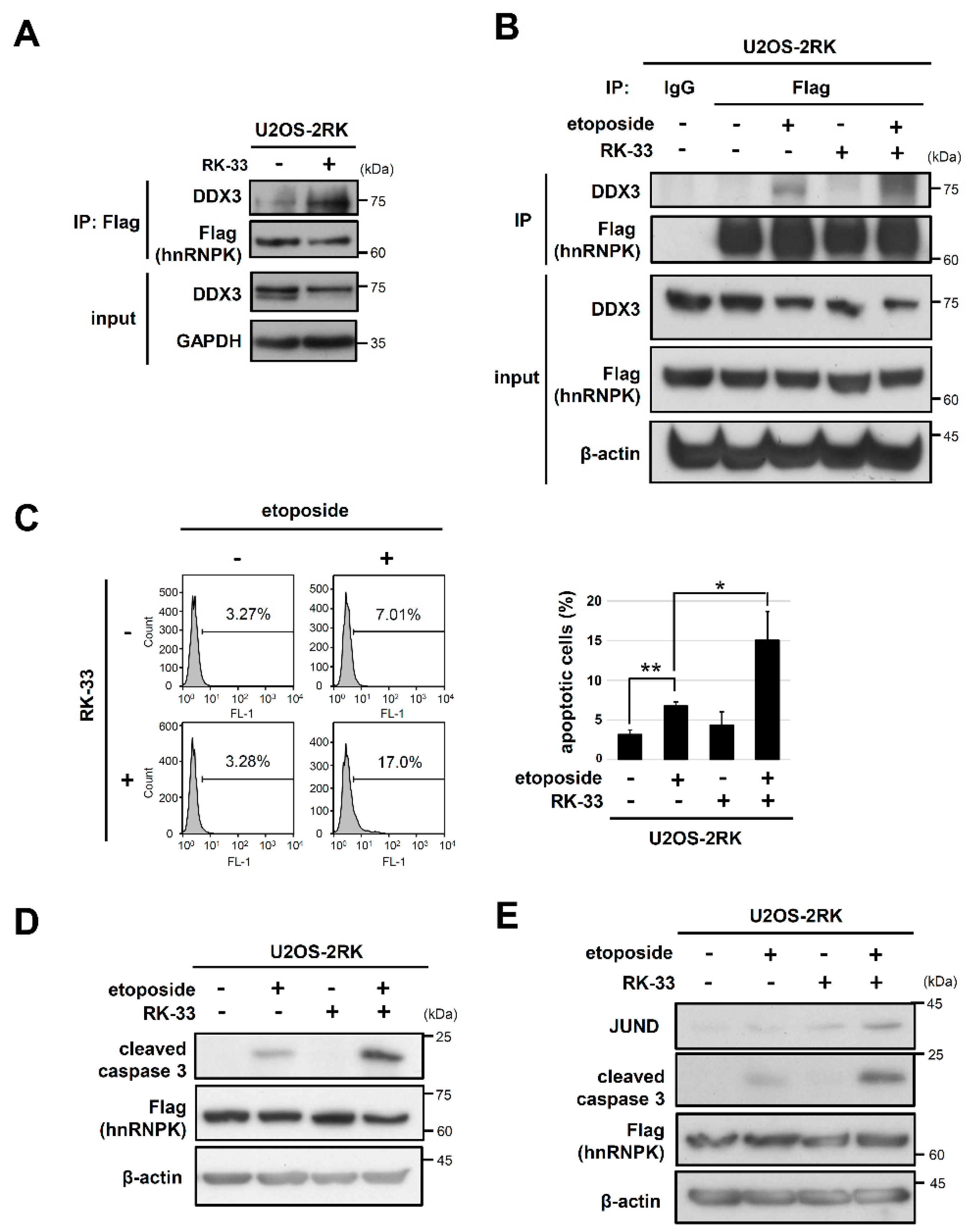
3.5. RK-33 Promotes the Apoptosis Enhancement Mediated by hnRNPKMD by Increasing the hnRNPK–DDX3 Interaction
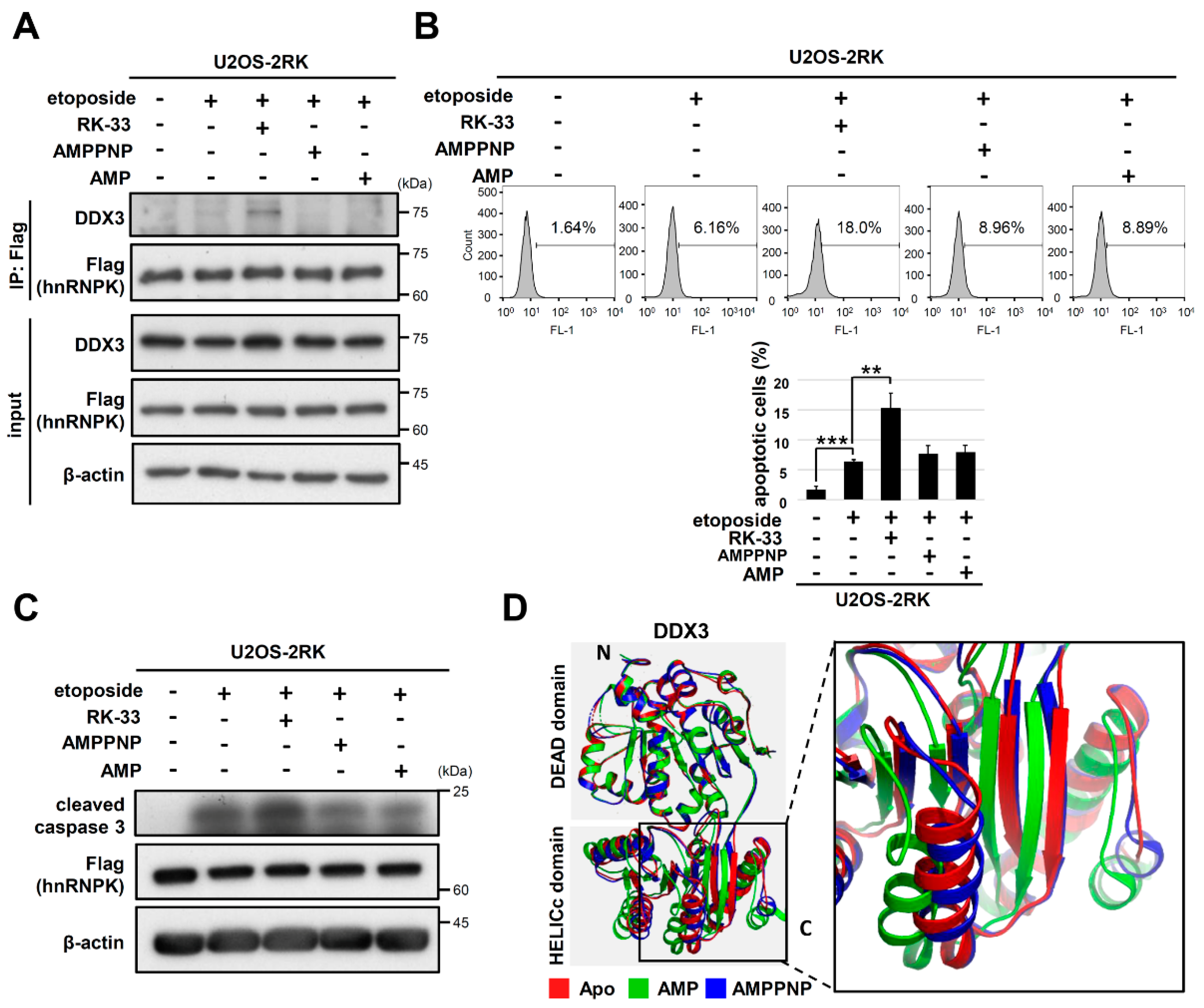
3.6. Other ATP Site Binding Ligands of DDX3 Did Not Promote hnRNPKMD-Mediated Apoptosis Enhancement
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Reyland, M.E.; Jones, D.N. Multifunctional roles of PKCδ: Opportunities for targeted therapy in human disease. Pharmacol. Ther. 2016, 165, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Liu, H.; Miki, Y. Protein kinase C delta regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J. Biol. Chem. 2006, 281, 5734–5740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Wang, H.G.; Miki, Y.; Kufe, D. Protein kinase Cdelta is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. EMBO J. 2003, 2003. 22, 1431–1441. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Pal, D. Two faces of protein kinase Cdelta: The contrasting roles of PKCdelta in cell survival and cell death. Sci. World J. 2010, 10, 2272–2284. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.H.; Wu, Y.L.; Zhao, M.; Liu, C.X.; Wang, L.S.; Chen, G.Q. Protein kinase C-delta mediates down-regulation of heterogeneous nuclear ribonucleoprotein K protein: Involvement in apoptosis induction. Exp. Cell Res. 2009, 315, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Barboro, P.; Ferrari, N.; Balbi, C. Emerging roles of heterogeneous nuclear ribonucleoprotein K (hnRNP K) in cancer progression. Cancer Lett. 2014, 352, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qiu, H.; He, J.; Liu, L.; Xue, W.; Fox, A.; Tickner, J.; Xu, J. The emerging roles of hnRNPK. J. Cell. Physiol. 2020, 235, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Yue, C.; Li, G.; He, B.; Cheng, W.; Wang, X.; Yan, M.; Long, Z.; Qiu, W.; Yuan, Z.; et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat. Commun. 2016, 7, 10180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziv-Av, A.; Giladi, N.D.; Lee, H.K.; Cazacu, S.; Finniss, S.; Xiang, C.; Pauker, M.H.; Barda-Saad, M.; Poisson, L.; Brodie, C. RTVP-1 regulates glioma cell migration and invasion via interaction with N-WASP and hnRNPK. Oncotarget 2015, 6, 19826–19840. [Google Scholar] [CrossRef]
- Gao, R.; Yu, Y.; Inoue, A.; Widodo, N.; Kaul, S.C.; Wadhwa, R. Heterogeneous nuclear ribonucleoprotein K (hnRNP-K) promotes tumor metastasis by induction of genes involved in extracellular matrix, cell movement and angiogenesis. J. Biol. Chem. 2013, 288, 15046–15056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-C.; Chung, I.-C.; Hsueh, C.; Tsang, N.-M.; Chi, L.-M.; Liang, Y.; Chen, C.-C.; Wang, L.-J.; Chang, Y.-S. The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. Cell Death Differ. 2010, 17, 1463–1473. [Google Scholar] [CrossRef] [Green Version]
- Barboro, P.; Salvi, S.; Rubagotti, A.; Boccardo, S.; Spina, B.; Truini, M.; Carmignani, G.; Introini, C.; Ferrari, N.; Boccardo, F.M.; et al. Prostate cancer: Prognostic significance of the association of heterogeneous nuclear ribonucleoprotein K and androgen receptor expression. Int. J. Oncol. 2014, 44, 1589–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Zeng, Y.; Xu, H.; Chen, Z.; Xiang, M.; Fu, Y.; Yin, Y.; Zhong, J.; Zeng, M.; Wang, P.; et al. Heterogeneous nuclear ribonucleoprotein K is overexpressed and associated with poor prognosis in gastric cancer. Oncol. Rep. 2016, 36, 929–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.W.; Lee, M.H.; Park, J.H.; Kang, S.H.; Yoo, H.M.; Ka, S.H.; Oh, Y.M.; Jeon, Y.J.; Chung, C.H. SUMOylation of hnRNP-K is required for p53-mediated cell-cycle arrest in response to DNA damage. EMBO J. 2012, 31, 4441–4452. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-H.; Chiou, Y.-Y.; Fu, S.-L.; Shih, I.-Y.; Weng, T.-H.; Lin, W.-J.; Lin, C.-H. Arginine methylation of hnRNPK negatively modulates apoptosis upon DNA damage through local regulation of phosphorylation. Nucleic Acids Res. 2014, 42, 9908–9924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poenisch, M.; Metz, P.; Blankenburg, H.; Ruggieri, A.; Lee, J.-Y.; Rupp, D.; Rebhan, I.; Diederich, K.; Kaderali, L.; Domingues, F.S.; et al. Identification of HNRNPK as Regulator of Hepatitis C Virus Particle Production. PLoS Pathog. 2015, 11, e1004573. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Xiong, H.; Wei, J.; Gao, X.; Feng, Y.; Liu, X.; Zhang, G.; He, Q.Y.; Xu, J.; Liu, L. Cytoplasmic hnRNPK interacts with GSK3beta and is essential for the osteoclast differentiation. Sci. Rep. 2015, 5, 17732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostareck-Lederer, A.; Ostareck, D.H.; Rucknagel, K.P.; Schierhorn, A.; Moritz, B.; Huttelmaier, S.; Flach, N.; Handoko, L.; Wahle, E. Asymmetric arginine dimethylation of heterogeneous nuclear ribonucleoprotein K by protein-arginine methyltransferase 1 inhibits its interaction with c-Src. J. Biol. Chem. 2006, 281, 11115–11125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikula, M.; Dzwonek, A.; Karczmarski, J.; Rubel, T.; Dadlez, M.; Wyrwicz, L.S.; Bomsztyk, K.; Ostrowski, J. Landscape of the hnRNP K protein–protein interactome. Proteomics 2006, 6, 2395–2406. [Google Scholar] [CrossRef]
- Mikula, M.; Rubel, T.; Karczmarski, J.; Statkiewicz, M.; Bomsztyk, K.; Ostrowski, J. Beads-free protein immunoprecipitation for a mass spectrometry-based interactome and posttranslational modifications analysis. Proteome Sci. 2015, 13, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Mao, Y.; Zhou, J.; Zhao, Y.; Cao, Y.; Chen, X. Multifunctional DDX3: Dual roles in various cancer development and its related signaling pathways. Am. J. Cancer Res. 2016, 6, 387–402. [Google Scholar]
- Ariumi, Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front. Genet. 2014, 5, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.-Y.; Lin, T.-C.; Lin, Y.-F.; Chen, M.-H.; Lee, C.-H.; Wang, H.-Y.; Lee, Y.-C.; Liu, Y.-P.; Chen, C.-L.; Hsiao, M. DDX3 as a strongest prognosis marker and its downregulation promotes metastasis in colorectal cancer. Oncotarget 2015, 6, 18602–18612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Zhou, T.; Jonasch, E.; Jope, R.S. DDX3 regulates DNA damage-induced apoptosis and p53 stabilization. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1833, 1489–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, A.; Haemmerle, M.W.; Oguh, A.U.; Doliba, N.M.; Stoffers, D.A. Metabolic stress activates an ERK/hnRNPK/DDX3X pathway in pancreatic β cells. Mol. Metab. 2019, 26, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Chiou, Y.-Y.; Lin, W.-J.; Fu, S.-L.; Lin, C.-H. Direct mass-spectrometric identification of Arg296 and Arg299 as the methylation sites of hnRNP K protein for methyltransferase PRMT. Protein J. 2007, 26, 87–93. [Google Scholar] [CrossRef]
- Bol, G.M.; Xie, M.; Raman, V. DDX3, a potential target for cancer treatment. Mol. Cancer 2015, 14, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pause, A.; Sonenberg, N. Mutational analysis of a DEAD box RNA helicase: The mammalian translation initiation factor elF-4A. EMBO J. 1992, 11, 2643–2654. [Google Scholar] [CrossRef]
- de Bisschop, G.; Ameur, M.; Ulryck, N.; Benattia, F.; Ponchon, L.; Sargueil, B.; Chamond, N. HIV-1 gRNA, a biological substrate, uncovers the potency of DDX3X biochemical activity. Biochimie 2019, 164, 83–94. [Google Scholar] [CrossRef]
- Bol, G.; Vesuna, F.; Xie, M.; Zeng, J.; Aziz, K.; Gandhi, N.; Levine, A.; Irving, A.; Korz, D.; Tantravedi, S.; et al. Targeting DDX 3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol. Med. 2015, 7, 648–669. [Google Scholar] [CrossRef] [PubMed]
- Kondaskar, A.; Kondaskar, S.; Fishbein, J.C.; Carter-Cooper, B.A.; Lapidus, R.G.; Sadowska, M.; Edelman, M.; Hosmane, R.S. Structure-based drug design and potent anti-cancer activity of tricyclic 5:7:5-fused diimidazo[4,5-d:4′,5′-f][1,3]diazepines. Bioorg. Med. Chem. 2013, 21, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, A.; Cannon, C.E.; Haemmerle, M.W.; Yang, J.; Stanescu, D.E.; Doliba, N.M.; Birnbaum, M.J.; Stoffers, D.A. JUND regulates pancreatic β cell survival during metabolic stress. Mol. Metab. 2019, 25, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Papoudou-Bai, A.; Hatzimichael, E.; Barbouti, A.; Kanavaros, P. Expression patterns of the activator protein-1 (AP-1) family members in lymphoid neoplasms. Clin. Exp. Med. 2017, 17, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Yang, P.; Li, Z.; Liu, W.; Amin, S.; Li, Z. Avenanthramide A triggers potent ROS-mediated anti-tumor effects in colorectal cancer by directly targeting DDX. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Floor, S.N.; Condon, K.J.; Sharma, D.; Jankowsky, E.; Doudna, J.A. Autoinhibitory interdomain interactions and subfamily-specific extensions redefine the catalytic core of the human DEAD-box protein DDX. J. Biol. Chem. 2016, 291, 2412–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, P.; Jankowsky, E. From unwinding to clamping—the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Jankowsky, E. The ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.-W.; Tsai, T.-Y.; Chao, C.-H.; Lee, Y.-H.W. Candidate tumor suppressor DDX3 RNA helicase specifically represses cap-dependent translation by acting as an eIF4E inhibitory protein. Oncogene 2007, 27, 700–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, J.-W.; Wang, W.-T.; Tsai, T.-Y.; Kuo, C.-Y.; Li, H.-K.; Lee, Y.-H.W. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 2011, 441, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Ji, X. The mechanism of RNA duplex recognition and unwinding by DEAD-box helicase DDX3X. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Gao, F.-H. Role and molecular mechanism of heterogeneous nuclear ribonucleoprotein K in tumor development and progression. Biomed. Rep. 2016, 4, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo, M.; Malaney, P.; Aitken, M.; Zhang, X.; Link, T.M.; Shah, V.; Alybayev, S.; Wu, M.-H.; Pageon, L.R.; Ma, H.; et al. Uncovering the role of RNA-binding protein hnRNP K in B-cell lymphomas. J. Natl. Cancer Inst. 2020, 112, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.R.; Messenger, Z.J.; Tam, H.W.; Phillips, S.L.; Recio, L.; Smart, R.C. Long noncoding RNA lincRNA-p21 is the major mediator of UVB-induced and p53-dependent apoptosis in keratinocytes. Cell Death Dis. 2015, 6, e1700. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-H.; Yu, H.-I.; Cho, W.-C.; Tarn, W.-Y. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene 2014, 34, 2790–2800. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schütz, P.; Karlberg, T.; Berg, S.V.D.; Collins, R.; Lehtiö, L.; Högbom, M.; Holmberg-Schiavone, L.; Tempel, W.; Park, H.-W.; Hammarström, M.; et al. Comparative structural analysis of human DEAD-Box RNA helicases. PLoS ONE 2010, 5, e12791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilbert, M.; Karow, A.R.; Klostermeier, D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol. Chem. 2009, 390, 1237–1250. [Google Scholar] [CrossRef] [Green Version]
- Radi, M.; Falchi, F.; Garbelli, A.; Samuele, A.; Bernardo, V.; Paolucci, S.; Baldanti, F.; Schenone, S.; Manetti, F.; Maga, G.; et al. Discovery of the first small molecule inhibitor of human DDX3 specifically designed to target the RNA binding site: Towards the next generation HIV-1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2094–2098. [Google Scholar] [CrossRef] [PubMed]
- Wilky, B.A.; Kim, C.; Mccarty, G.; Montgomery, E.A.; Kammers, K.; Devine, L.; Cole, R.N.; Raman, V.; Loeb, D.M. RNA helicase DDX3: A novel therapeutic target in Ewing sarcoma. Oncogene 2016, 35, 2574–2583. [Google Scholar] [CrossRef]
- Xie, M.; Vesuna, F.; Tantravedi, S.; Bol, G.M.; van Voss, M.H.; Nugent, K.; Malek, R.; Gabrielson, K.; Van Diest, P.J.; Tran, P.T.; et al. RK-33 radiosensitizes prostate cancer cells by blocking the RNA helicase DDX. Cancer Res. 2016, 76, 6340–6350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerma van Voss, M.R.; Kammers, K.; Vesuna, F.; Brilliant, J.; Bergman, Y.; Tantravedi, S.; Wu, X.; Cole, R.N.; Holland, A.; van Diest, P.J.; et al. Global effects of DDX3 inhibition on cell cycle regulation identified by a combined phosphoproteomics and single cell tracking approach. Transl. Oncol. 2018, 11, 755–763. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-C.; Yang, J.-H.; Fu, S.-L.; Lin, W.-J.; Lin, C.-H. Arginine Methylation of hnRNPK Inhibits the DDX3-hnRNPK Interaction to Play an Anti-Apoptosis Role in Osteosarcoma Cells. Int. J. Mol. Sci. 2021, 22, 9764. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189764
Chen C-C, Yang J-H, Fu S-L, Lin W-J, Lin C-H. Arginine Methylation of hnRNPK Inhibits the DDX3-hnRNPK Interaction to Play an Anti-Apoptosis Role in Osteosarcoma Cells. International Journal of Molecular Sciences. 2021; 22(18):9764. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189764
Chicago/Turabian StyleChen, Chiao-Che, Jen-Hao Yang, Shu-Ling Fu, Wey-Jinq Lin, and Chao-Hsiung Lin. 2021. "Arginine Methylation of hnRNPK Inhibits the DDX3-hnRNPK Interaction to Play an Anti-Apoptosis Role in Osteosarcoma Cells" International Journal of Molecular Sciences 22, no. 18: 9764. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189764