
De Novo Transcriptome Assembly, Functional Annotation, and Transcriptome Dynamics Analyses Reveal Stress Tolerance Genes in Mangrove Tree (Bruguiera gymnorhiza)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Plant Material, Soil Salinity and Morphological Measurements
2.2. High Throughput Sequencing Output and De Novo Transcriptome Assembly
2.3. Quality Assessment of the Transcriptome Assembly
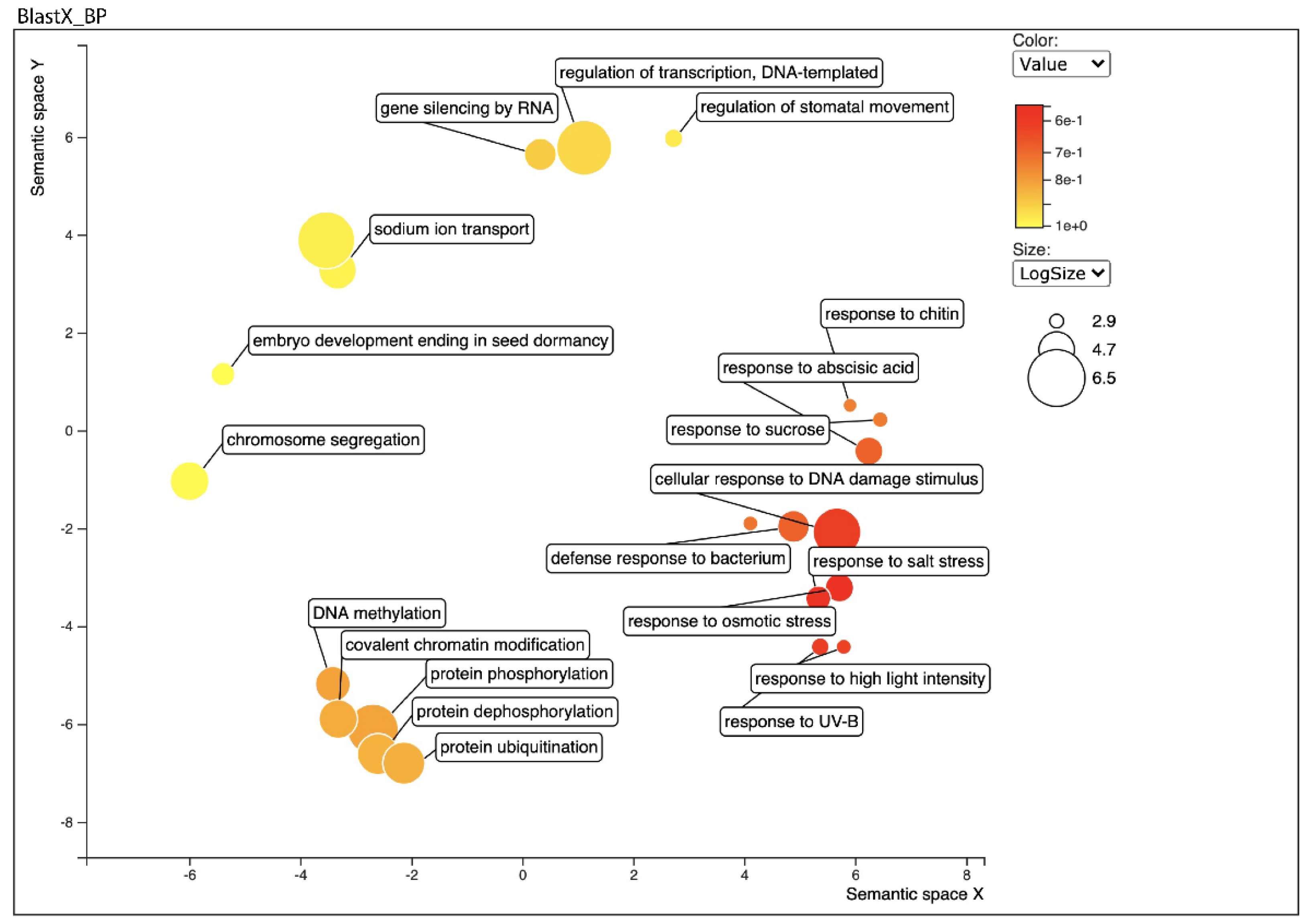
2.4. Functional Annotation of the Transcriptome by Homology and Gene Ontology (GO)
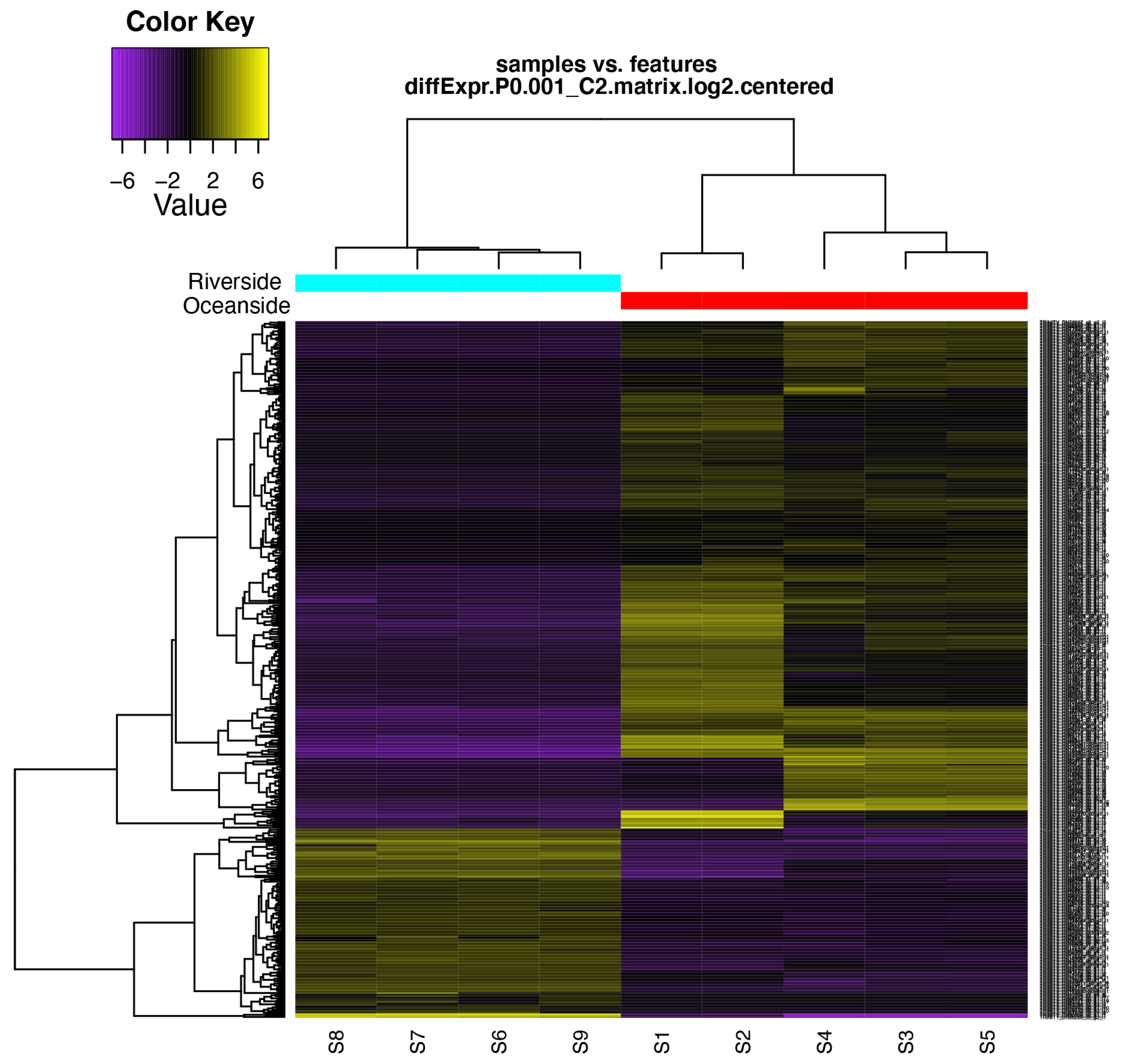
2.5. Differential Expression Analysis and Functional Classification
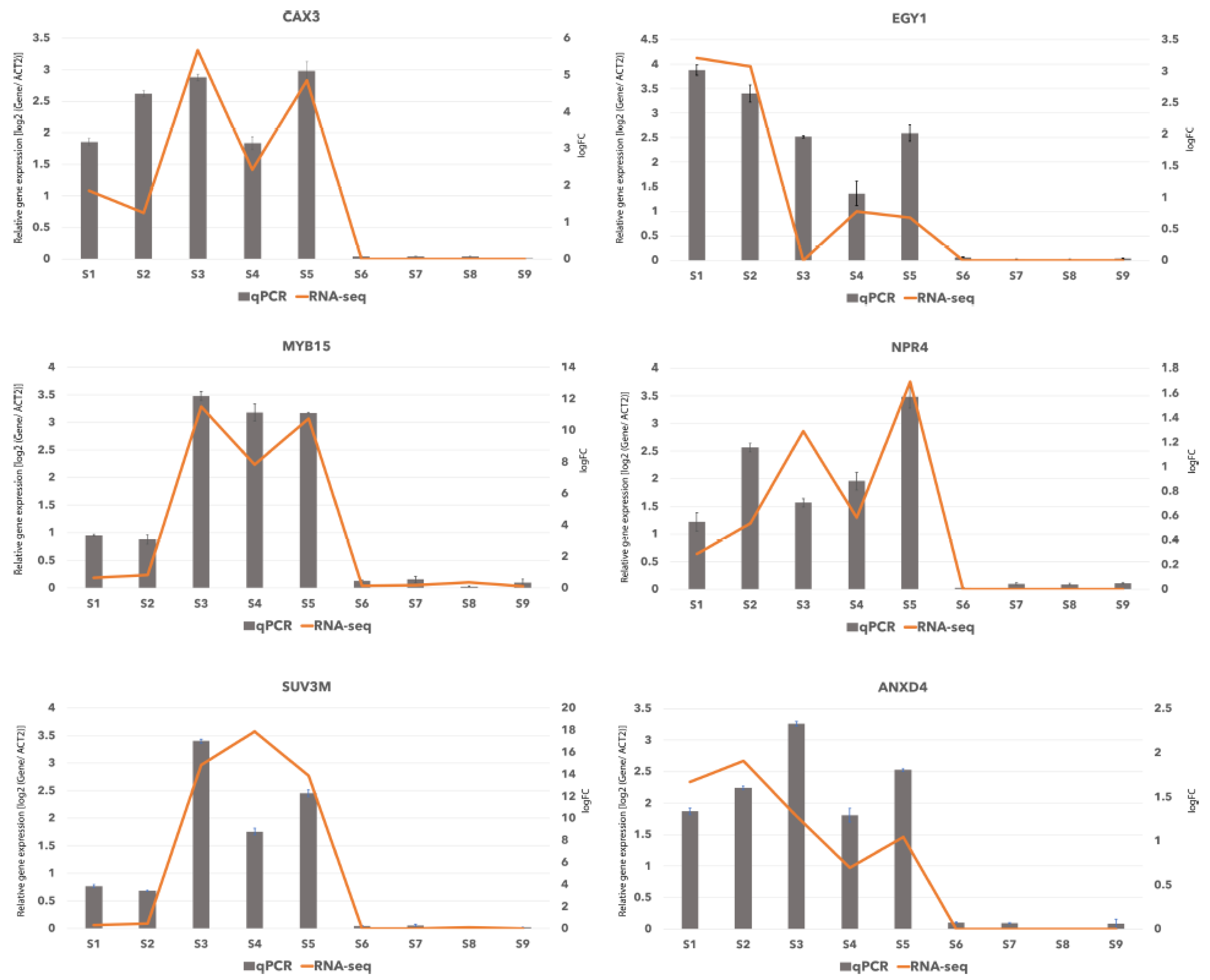
2.6. Verification of RNA-Seq Data by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Plant Material, Soil Salinity and Morphological Measurements
4.2. RNA-Seq and Transcription Analyses
4.2.1. RNA Extraction
4.2.2. Library Construction and Sequencing
4.2.3. De Novo Transcriptome Assembly
4.3. Functional Annotation of the Transcriptome
4.4. Transcript Abundance and Differential Expression Analysis
4.5. Validation of Differentially Expressed Genes by qRT-PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miryeganeh, M. Plants’ Epigenetic Mechanisms and Abiotic Stress. Genes 2021, 12, 1106. [Google Scholar] [CrossRef]
- Browne, L.; Mead, A.; Horn, C.; Chang, K.; Celikkol, Z.A.; Henriquez, C.L.; Ma, F.; Beraut, E.; Meyer, R.S.; Sork, V.L. Experimental DNA Demethylation Associates with Changes in Growth and Gene Expression of Oak Tree Seedlings. G3 Genes Genomes Genet. 2020, 10, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Balao, F.; Paun, O.; Alonso, C. Uncovering the contribution of epigenetics to plant phenotypic variation in Mediterranean ecosystems. Plant Biol. 2018, 20, 38–49. [Google Scholar] [CrossRef]
- Bräutigam, K.; Vining, K.J.; Lafon-Placette, C.; Fossdal, C.G.; Mirouze, M.; Marcos, J.G.; Fluch, S.; Fraga, M.F.; Guevara, M.Á.; Abarca, D.; et al. Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 2013, 3, 399–415. [Google Scholar] [CrossRef]
- Stepien, P.; Johnson, G.N. Contrasting Responses of Photosynthesis to Salt Stress in the Glycophyte Arabidopsis and the Halophyte Thellungiella: Role of the Plastid Terminal Oxidase as an Alternative Electron Sink. Plant Physiol. 2009, 149, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- Khoon, G.W.; Tomlinson, P.B. The Botany of Mangroves; Cambridge University Press: Cambridge, UK, 1986; p. 413. ISBN 0-521-25567. [Google Scholar]
- Tomlinson, P.B. The Botany of Mangroves, 2nd ed.; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Donato, D.C.; Kauffman, J.B.; Murdiyarso, D.; Kurnianto, S.; Stidham, M.; Kanninen, M. Mangroves among the most carbon-rich forests in the tropics. Nat. Geosci. 2011, 4, 293–297. [Google Scholar] [CrossRef]
- Duke, N.C.; Meynecke, J.-O.; Dittmann, S.; Ellison, A.M.; Anger, K.; Berger, U.; Cannicci, S.; Diele, K.; Ewel, K.C.; Field, C.D.; et al. A World Without Mangroves? Science 2007, 317, 41–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, B.; Meng, Y.; Ma, X.; Lai, Y.; Si, E.; Yang, K.; Ren, P.; Shang, X.; Wang, H. Transcriptomic profiling of the salt-stress response in the halophyte Halogeton glomeratus. BMC Genom. 2015, 16, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelistella, C.; Valentini, A.; Ludovisi, R.; Firrincieli, A.; Fabbrini, F.; Scalabrin, S.; Cattonaro, F.; Morgante, M.; Mugnozza, G.S.; Keurentjes, J.; et al. De novo assembly, functional annotation, and analysis of the giant reed (Arundo donax L.) leaf transcriptome provide tools for the development of a biofuel feedstock. Biotechnol. Biofuels 2017, 10, 138. [Google Scholar] [CrossRef] [Green Version]
- Golldack, D.; Lüking, I.; Yang, O. Plant tolerance to drought and salinity: Stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Rep. 2011, 30, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Jin, H. Transcriptome sequencing and gene expression profiling of Populus wutunensis, a natural hybrid, during salinity stress. Trees 2020, 34, 1427–1438. [Google Scholar] [CrossRef]
- Razzaque, S.; Elias, S.M.; Haque, T.; Biswas, S.; Jewel, G.M.N.A.; Rahman, S.; Weng, X.; Ismail, A.M.; Walia, H.; Juenger, T.E.; et al. Gene Expression analysis associated with salt stress in a reciprocally crossed rice population. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Şahin-Çevik, M.; Çevik, B.; Coşkan, A. Identification and Expression Analysis of Salinity-Induced Genes in Rangpur Lime (Citrus limonia). Hortic. Plant J. 2020, 6, 267–276. [Google Scholar] [CrossRef]
- Suwa, R.; Analuddin, K.; Khan, N.I.; Hagihara, A. Structure and productivity along a tree height gradient in a Kandelia obovata mangrove forest in the Manko Wetland, Okinawa Island, Japan. Wetl. Ecol. Manag. 2007, 16, 331–343. [Google Scholar] [CrossRef]
- Lira-Medeiros, C.F.; Parisod, C.; Fernandes, R.A.; Mata, C.S.; Cardoso, M.A.; Ferreira, P.C.G. Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS ONE 2010, 5, e10326. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer-Novelli, Y.; Cintrón-Molero, G.; Adaime, R.R.; De Camargo, T.M. Variability of Mangrove Ecosystems along the Brazilian Coast. Estuaries 1990, 13, 204–218. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Wu, H.; Zhang, Z.; Yang, C.; Hu, L.; Shi, X.; Jian, S.; Shi, S.; Huang, Y. Comparative Analysis of Transcriptomes in Rhizophoraceae Provides Insights into the Origin and Adaptive Evolution of Mangrove Plants in Intertidal Environments. Front. Plant Sci. 2017, 8, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Smuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-Y.; Li, Y.-C.; Zhang, T.-H.; Qin, L.-L.; An, Y.-D.; Pang, Y.-K.; Jiang, G.-F. Characterization of the complete chloroplast genome of mangrove Bruguiera gymnorrhiza (L.) Lam. ex Savigny. Mitochondrial DNA Part B Resour. 2021, 6, 2076–2078. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Basyuni, M.; Baba, S.; Inafuku, M.; Iwasaki, H.; Kinjo, K.; Oku, H. Expression of terpenoid synthase mRNA and terpenoid content in salt stressed mangrove. J. Plant Physiol. 2009, 166, 1786–1800. [Google Scholar] [CrossRef]
- Larionov, A.; Krause, A.; Miller, W. A standard curve based method for relative real time PCR data processing. BMC Bioinform. 2005, 6, 62. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Morgante, M.; De Paoli, E.; Radovic, S. Transposable elements and the plant pan-genomes. Curr. Opin. Plant Biol. 2007, 10, 149–155. [Google Scholar] [CrossRef]
- Li, H.-Z.; Gao, X.; Li, X.-Y.; Chen, Q.-J.; Dong, J.; Zhao, W.-C. Evaluation of Assembly Strategies Using RNA-Seq Data Associated with Grain Development of Wheat (Triticum aestivum L.). PLoS ONE 2013, 8, e83530. [Google Scholar] [CrossRef] [Green Version]
- Krasileva, K.; Buffalo, V.; Bailey, P.; Pearce, S.; Ayling, S.; Tabbita, F.; Soria, M.; Wang, S.; Akhunov, E.; et al.; IWGS Consortium Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol. 2013, 14, R66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mevy, J.-P.; Loriod, B.; Liu, X.; Corre, E.; Torres, M.; Büttner, M.; Haguenauer, A.; Reiter, I.M.; Fernandez, C.; Gauquelin, T. Response of Downy Oak (Quercus pubescens Willd.) to Climate Change: Transcriptome Assembly, Differential Gene Analysis and Targeted Metabolomics. Plants 2020, 9, 1149. [Google Scholar] [CrossRef] [PubMed]
- Schwarzbach, A.E.; Ricklefs, R.E. Systematic affinities of Rhizophoraceae and Anisophylleaceae, and intergeneric relationships within Rhizophoraceae, based on chloroplast DNA, nuclear ribosomal DNA, and morphology. Am. J. Bot. 2000, 87, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.J.; Flagel, L.E.; Paterson, A.H.; Rapp, R.A.; Soltis, D.E.; Soltis, P.S.; Wendel, J.F. Evolutionary Genetics of Genome Merger and Doubling in Plants. Annu. Rev. Genet. 2008, 42, 443–461. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Kim, B.G.; Cheong, Y.H.P.; Girdhar, K.P.; Sheng, L. A Ca2+ signaling pathway regulates a K+ channel for low-K response in Arabidopsis. Proc. Natl. Acad. Sci. USA 2006, 103, 12625. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Shao, H.; Shao, C.; Chen, P.; Zhao, S.; Brestic, M.; Chen, X. Physiological adaptive mechanisms of plants grown in saline soil and implications for sustainable saline agriculture in coastal zone. Acta Physiol. Plant. 2013, 35, 2867–2878. [Google Scholar] [CrossRef]
- Li, F.; Zhang, H.; Zhao, H.; Gao, T.; Song, A.; Jiang, J.; Chen, F.; Chen, S. Chrysanthemum CmHSFA4 gene positively regulates salt stress tolerance in transgenic chrysanthemum. Plant Biotechnol. J. 2018, 16, 1311–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Du, H.; Li, T.; Wuyun, T.-N. De novo transcriptome sequencing and identification of genes related to salt stress in Eucommia ulmoides Oliver. Trees 2017, 32, 151–163. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Hong, Y.; Yao, J.; Ren, Z.; Shi, H.; Zhu, J.-K. The Flowering Repressor SVP Confers Drought Resistance in Arabidopsis by Regulating Abscisic Acid Catabolism. Mol. Plant 2018, 11, 1184–1197. [Google Scholar] [CrossRef] [Green Version]
- Cheng, N.-H.; Pittman, J.; Shigaki, T.; Lachmansingh, J.; LeClere, S.; Lahner, B.; Salt, D.E.; Hirschi, K.D. Functional Association of Arabidopsis CAX1 and CAX3 Is Required for Normal Growth and Ion Homeostasis. Plant Physiol. 2005, 138, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Bi, Y.R.; Li, N. EGY1 encodes a membrane-associated and ATP-independent metalloprotease that is required for chloroplast development. Plant J. 2004, 41, 364–375. [Google Scholar] [CrossRef]
- Ding, Z.; Li, S.; An, X.; Liu, X.; Qin, H.; Wang, D. Transgenic expression of MYB15 confers enhanced sensitivity to abscisic acid and improved drought tolerance in Arabidopsis thaliana. J. Genet. Genom. 2009, 36, 17–29. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhong, S.; Li, Q.; Zhu, Z.; Lou, Y.; Wang, L.; Wang, J.; Wang, M.; Li, Q.; Yang, D.; et al. Functional analysis of rice NPR1-like genes reveals that OsNPR1/NH1 is the rice orthologue conferring disease resistance with enhanced herbivore susceptibility. Plant Biotechnol. J. 2007, 5, 313–324. [Google Scholar] [CrossRef]
- Tuteja, N.; Sahoo, R.K.; Garg, B.; Tuteja, R. OsSUV3 dual helicase functions in salinity stress tolerance by maintaining photosynthesis and antioxidant machinery in rice (Oryza sativa L. cv. IR64). Plant J. 2013, 76, 115–127. [Google Scholar] [CrossRef]
- Liu, L.-L.; Ren, H.-M.; Chen, L.-Q.; Wang, Y.; Wu, W.-H. A Protein Kinase, Calcineurin B-Like Protein-Interacting Protein Kinase9, Interacts with Calcium Sensor Calcineurin B-Like Protein3 and Regulates Potassium Homeostasis under Low-Potassium Stress in Arabidopsis. Plant Physiol. 2012, 161, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, E.J.; Yang, E.J.; Lee, J.E.; Park, A.R.; Song, W.H.; Park, O.K. Proteomic Identification of Annexins, Calcium-Dependent Membrane Binding Proteins That Mediate Osmotic Stress and Abscisic Acid Signal Transduction in Arabidopsis. Plant Cell 2004, 16, 1378–1391. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Wang, H.; Yu, H.; Zhong, C.; Zhang, X.; Peng, J.; Wang, X. GASA14 regulates leaf expansion and abiotic stress resistance by modulating reactive oxygen species accumulation. J. Exp. Bot. 2013, 64, 1637–1647. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Briefings Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhssassi, N.; Doblas, V.G.; Rosado, A.; del Valle, A.E.; Posé, D.; Jimenez, A.J.; Castillo, A.G.; Valpuesta, V.; Borsani, O.; Botella, M.A. The Arabidopsis Tetratricopeptide Thioredoxin-Like Gene Family Is Required for Osmotic Stress Tolerance and Male Sporogenesis. Plant Physiol. 2012, 158, 1252–1266. [Google Scholar] [CrossRef] [Green Version]
- Julkowska, M.; Testerink, C. Tuning plant signaling and growth to survive salt. Trends Plant Sci. 2015, 20, 586–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Wu, Y.; Di, L.; Wang, G.; Shen, Y. Retracted Article: The AtCCX1 transporter mediates salinity tolerance in both Arabidopsis and yeast. Plant Cell Tissue Organ Cult. (PCTOC) 2011, 109, 91–99. [Google Scholar] [CrossRef]
- Clough, S.J.; Fengler, K.A.; Yu, I.-C.; Lippok, B.; Smith, R.K.; Bent, A. The Arabidopsis dnd1 "defense, no death" gene encodes a mutated cyclic nucleotide-gated ion channel. Proc. Natl. Acad. Sci. USA 2000, 97, 9323–9328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-Q.; Feechan, A.; Yun, B.-W.; Shafiei, R.; Hofmann, A.; Taylor, P.; Xue, P.; Yang, F.-Q.; Xie, Z.-S.; Pallas, J.; et al. S-Nitrosylation of AtSABP3 Antagonizes the Expression of Plant Immunity. J. Biol. Chem. 2009, 284, 2131–2137. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Boisson-Dernier, A.; Israelsson-Nordström, M.; Böhmer, M.; Xue, S.; Ries, A.; Godoski, J.; Kuhn, J.M.; Schroeder, J.I. Carbonic anhydrases are upstream regulators of CO2-controlled stomatal movements in guard cells. Nat. Cell Biol. 2009, 12, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Saze, H.; Kitayama, J.; Takashima, K.; Miura, S.; Harukawa, Y.; Ito, T.; Kakutani, T. Mechanism for full-length RNA processing of Arabidopsis genes containing intragenic heterochromatin. Nat. Commun. 2013, 4, 2301. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Duan, C.G.; Tang, K.; Wang, B.; Zhang, H.; Lei, M.; Lu, K.; Mangrauthia, S.K.; Wang, P.; Zhu, G.; et al. RNA-binding protein regulates plant DNA methylation by controlling mRNA processing at the intronic heterochromatin-containing gene IBM1. Proc. Natl. Acad. Sci. USA 2013, 110, 15467–15472. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Koussevitzky, S.; Mittler, R.; Miller, G. ROS and redox signalling in the response of plants to abiotic stress. Plant Cell Environ. 2011, 35, 259–270. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Cassagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Schopfer, C.R.; Kochs, G.; Lottspeich, F.; Ebel, J. Molecular characterization and functional expression of dihydroxypterocarpan 6a-hydroxylase, an enzyme specific for pterocarpanoid phytoalexin biosynthesis in soybean (Glycine max L.). FEBS Lett. 1998, 432, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Tognolli, M.; Penel, C.; Greppin, H.; Simon, P. Analysis and expression of the class III peroxidase large gene family in Arabidopsis thaliana. Gene 2002, 288, 129–138. [Google Scholar] [CrossRef]
- Kim, S.-J.; Chandrasekar, B.; Rea, A.C.; Danhof, L.; Zemelis-Durfee, S.; Thrower, N.; Shepard, Z.S.; Pauly, M.; Brandizzi, F.; Keegstra, K. The synthesis of xyloglucan, an abundant plant cell wall polysaccharide, requires CSLC function. Proc. Natl. Acad. Sci. USA 2020, 117, 20316–20324. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Ito, Y.; Yamaguchi-Shinozaki, K. Transcriptional Regulatory Networks in Response to Abiotic Stresses in Arabidopsis and Grasses. Plant Physiol. 2009, 149, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.N.; Zuo, Y.Q.; Liang, X.Q.; Yin, B.; Wang, G.D.; Meng, Q.W. The multiple stress-responsive transcription factor SlNAC1 improves the chilling tolerance of tomato. Physiol. Plant 2013, 149, 474–486. [Google Scholar] [CrossRef]
- Yu, C.; Xu, S.; Yin, Y. Transcriptome analysis of the Taxodium ‘Zhongshanshan 405’ roots in response to salinity stress. Plant Physiol. Biochem. 2016, 100, 156–165. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukrong, S.; Yun, K.-Y.; Stadler, P.; Kumar, C.; Facciuolo, T.; Moffatt, B.A.; Falcone, D.L. Improved Growth and Stress Tolerance in the Arabidopsis oxt1 Mutant Triggered by Altered Adenine Metabolism. Mol. Plant 2012, 5, 1310–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, H.; Ishiga, Y.; Watanabe, S.; Konishi, T.; Egusa, M.; Akiyoshi, N.; Matsuura, T.; Mori, I.C.; Hirayama, T.; Kaminaka, H.; et al. Allantoin, a stress-related purine metabolite, can activate jasmonate signaling in a MYC2-regulated and abscisic acid-dependent manner. J. Exp. Bot. 2016, 67, 2519–2532. [Google Scholar] [CrossRef]
- Wang, D.; Lu, X.; Chen, X.; Wang, S.; Wang, J.; Guo, L.; Yin, Z.; Chen, Q.; Ye, W. Temporal salt stress-induced transcriptome alterations and regulatory mechanisms revealed by PacBio long-reads RNA sequencing in Gossypium hirsutum. BMC Genom. 2020, 21, 1–15. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, Y.; Li, X.; Zhang, L.; Fan, S. Transcriptomic analysis identifies novel genes and pathways for salt stress responses in Suaeda salsa leaves. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Sangha, J.S.; Gu, K.; Kaur, J.; Yin, Z. An improved method for RNA isolation and cDNA library construction from immature seeds of Jatropha curcas L. BMC Res. Notes 2010, 3, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39 (Suppl. 2), W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Punta, M.; Coggill, P.C.; Eberhardt, R.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, S.K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2011, 40, D290–D301. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. Signal P 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, A.; Domingues, F.S.; Rahnenführer, J.; Lengauer, T. A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinform. 2006, 7, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miryeganeh, M.; Saze, H. De Novo Transcriptome Assembly, Functional Annotation, and Transcriptome Dynamics Analyses Reveal Stress Tolerance Genes in Mangrove Tree (Bruguiera gymnorhiza). Int. J. Mol. Sci. 2021, 22, 9874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189874
Miryeganeh M, Saze H. De Novo Transcriptome Assembly, Functional Annotation, and Transcriptome Dynamics Analyses Reveal Stress Tolerance Genes in Mangrove Tree (Bruguiera gymnorhiza). International Journal of Molecular Sciences. 2021; 22(18):9874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189874
Chicago/Turabian StyleMiryeganeh, Matin, and Hidetoshi Saze. 2021. "De Novo Transcriptome Assembly, Functional Annotation, and Transcriptome Dynamics Analyses Reveal Stress Tolerance Genes in Mangrove Tree (Bruguiera gymnorhiza)" International Journal of Molecular Sciences 22, no. 18: 9874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189874