
Stearoyl-CoA Desaturase (SCD) Induces Cardiac Dysfunction with Cardiac Lipid Overload and Angiotensin II AT1 Receptor Protein Up-Regulation

Abstract
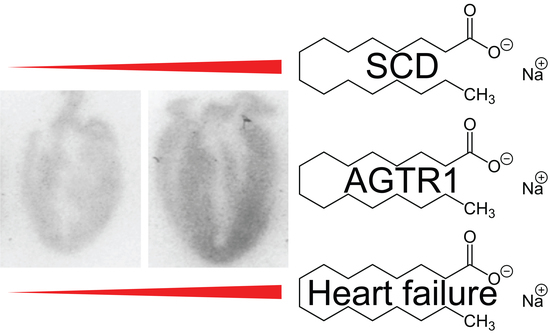
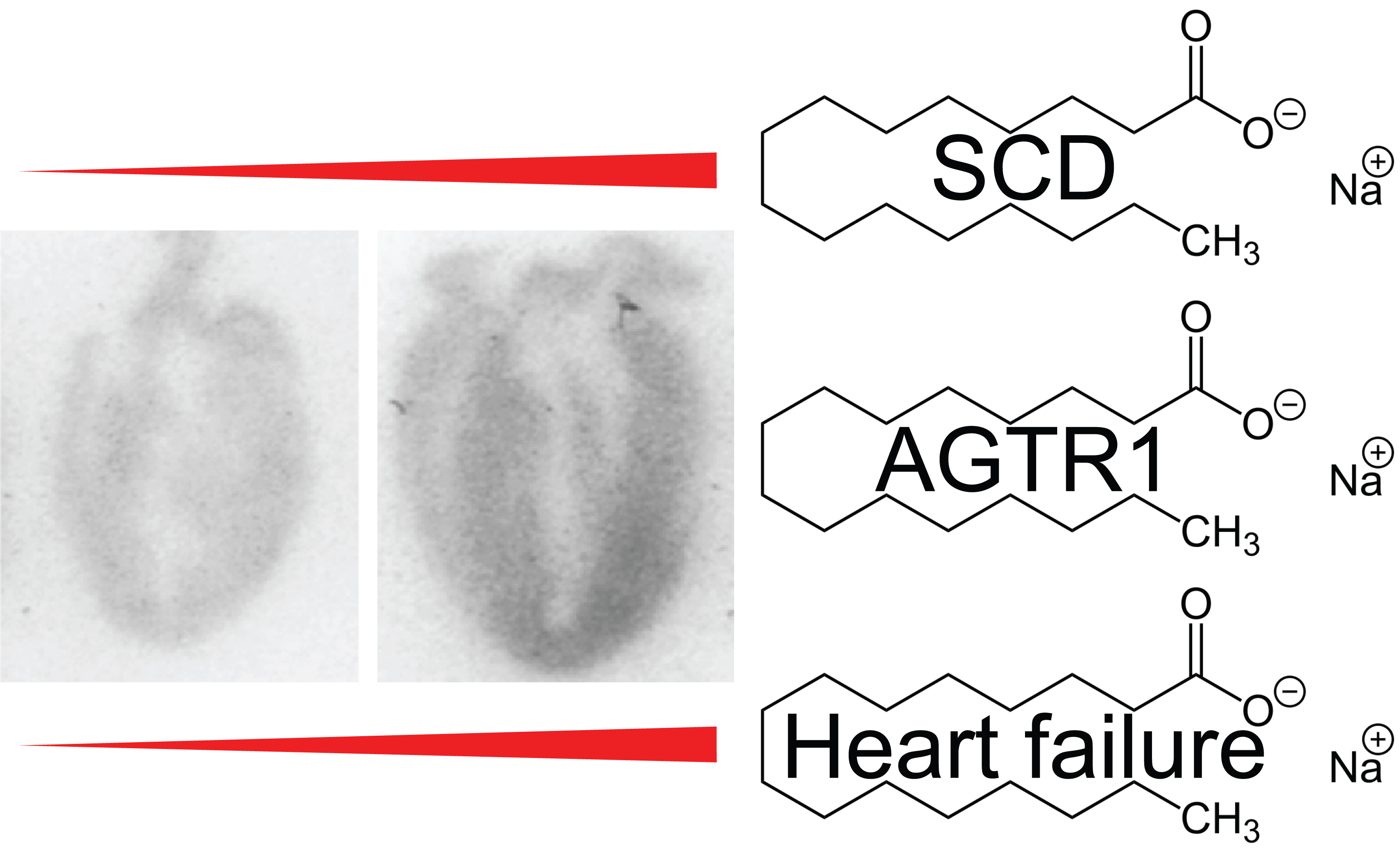
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
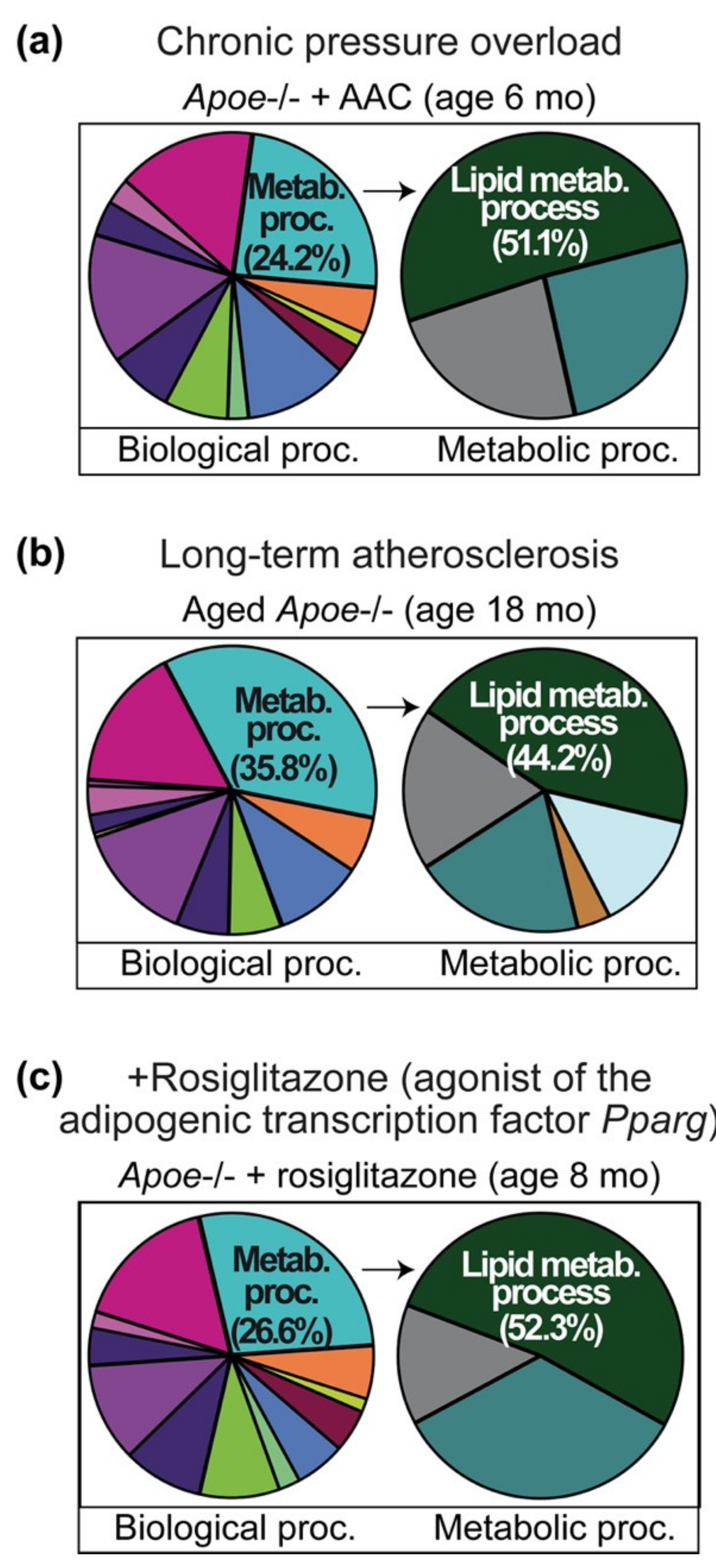
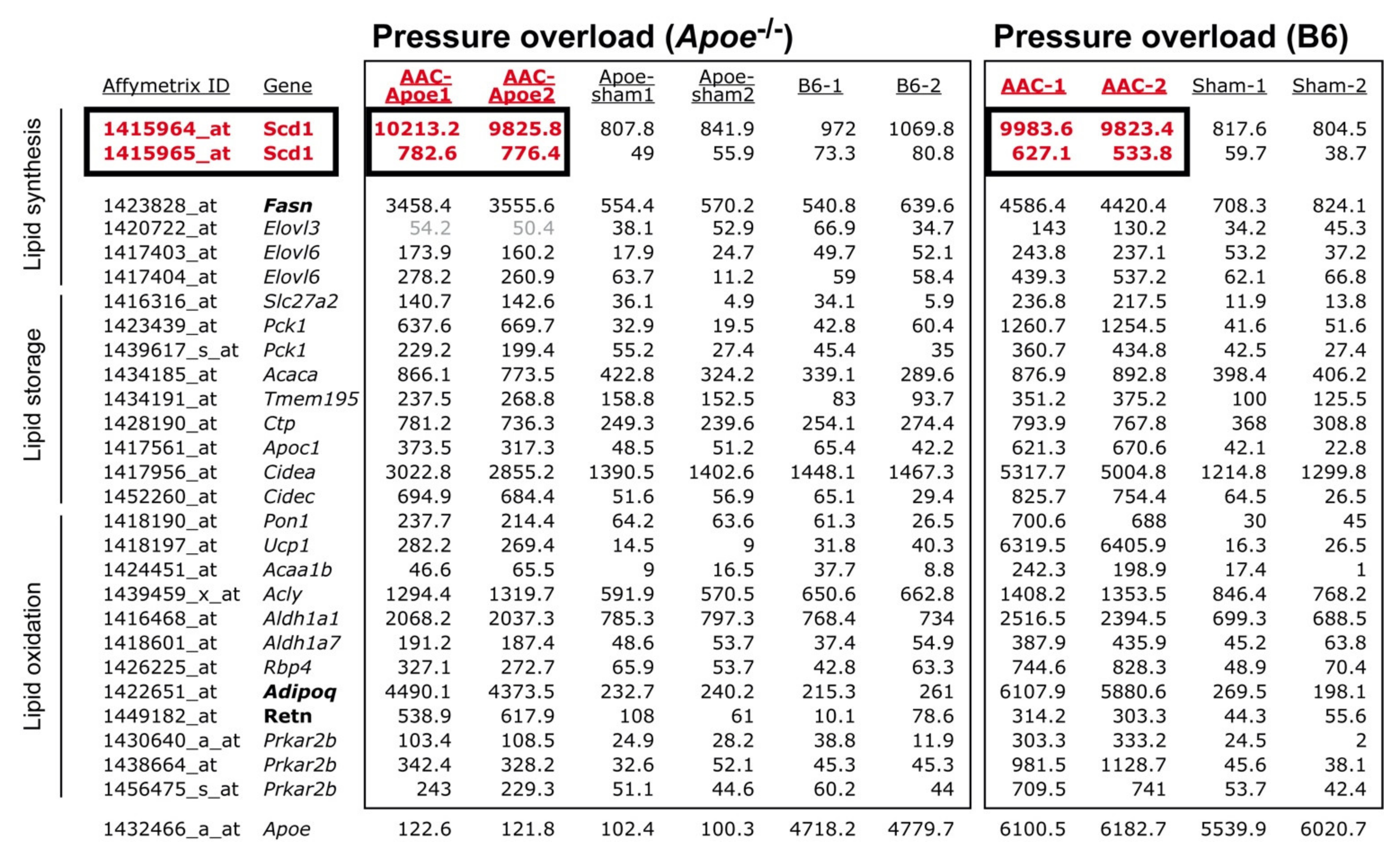
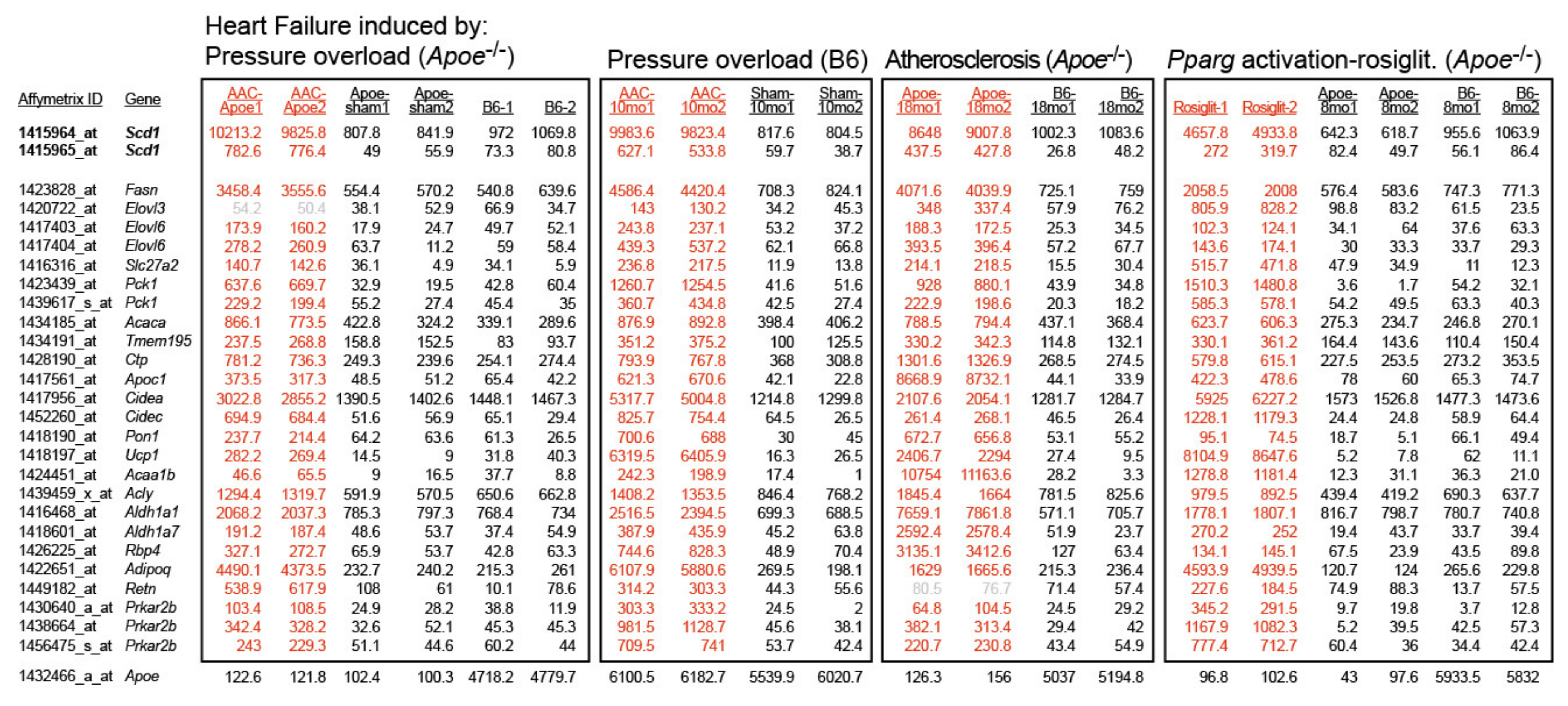
2.1. Cardiovascular Risk Factors Trigger Up-Regulation of the Cardiac Lipid Metabolic Process in Hypercholesterolemic Apoe-/- Mice
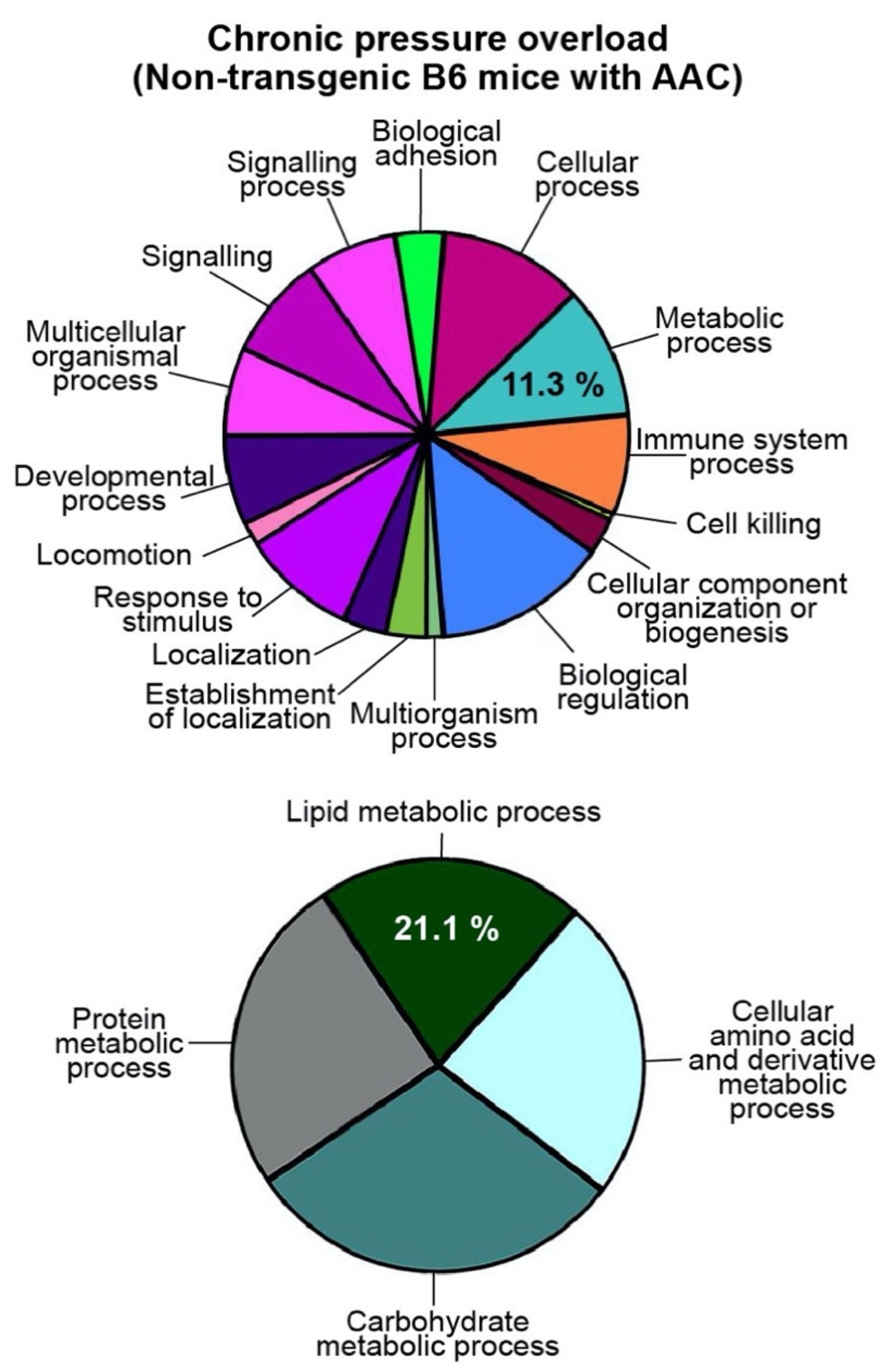
2.2. Up-Regulation of the Cardiac Lipid Metabolic Process of Non-Transgenic B6 Mice with Heart Failure Induced by Chronic Pressure Overload

2.3. Concordantly Up-Regulated Genes of the Cardiac Lipid Metabolic Process in Different Heart Failure Models
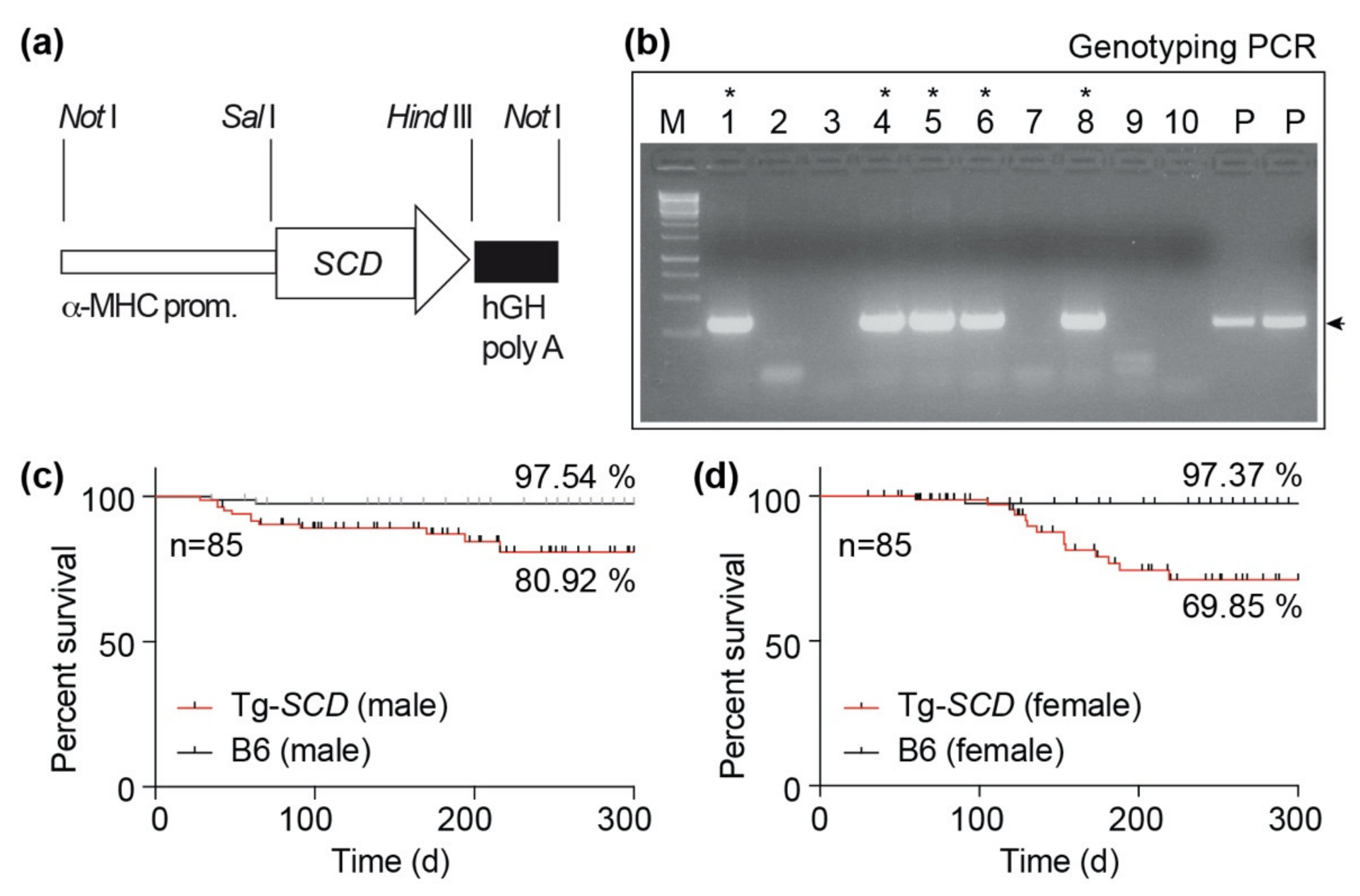
2.4. Generation of Tg-SCD Mice with Myocardium-Specific SCD Expression
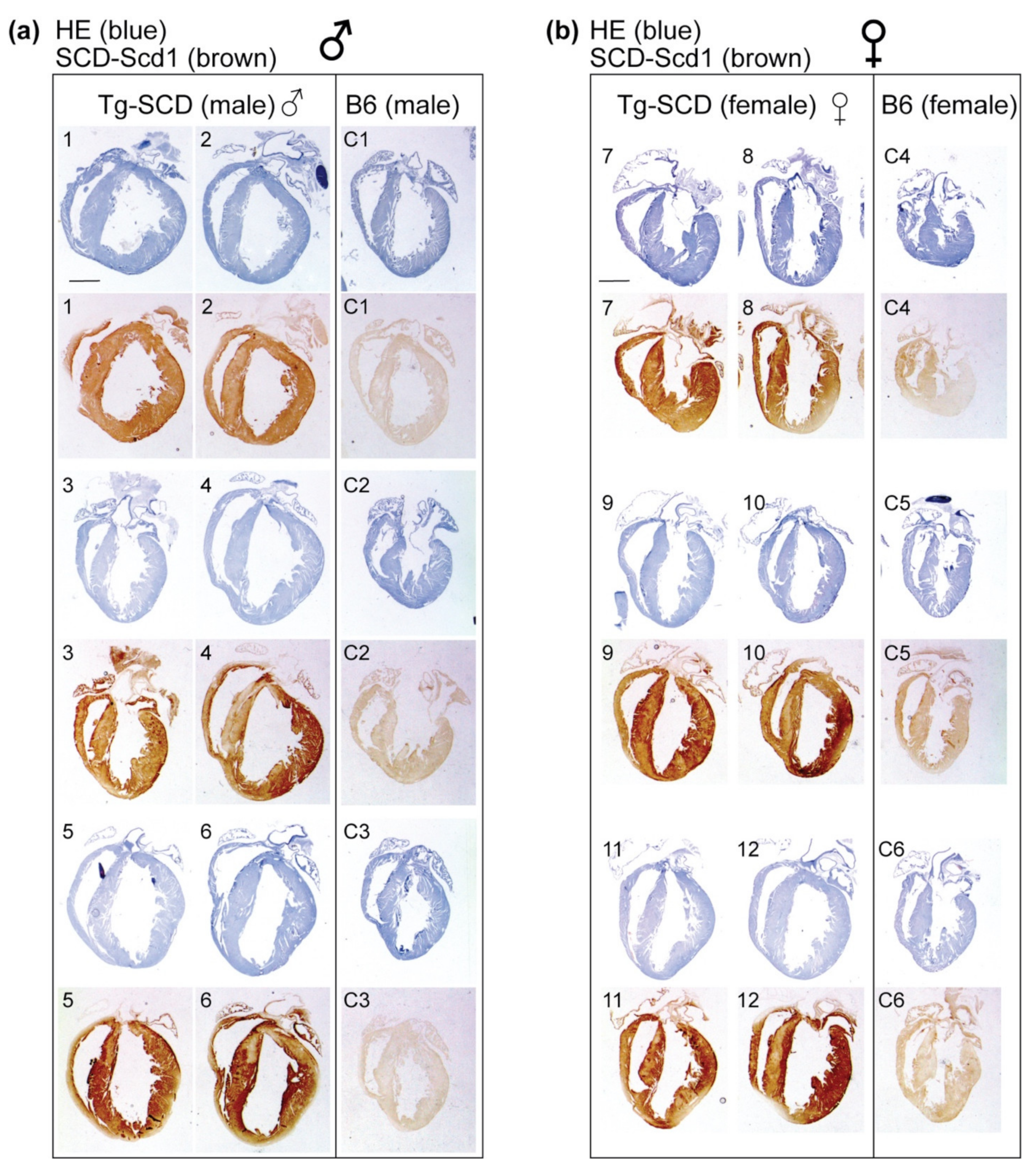
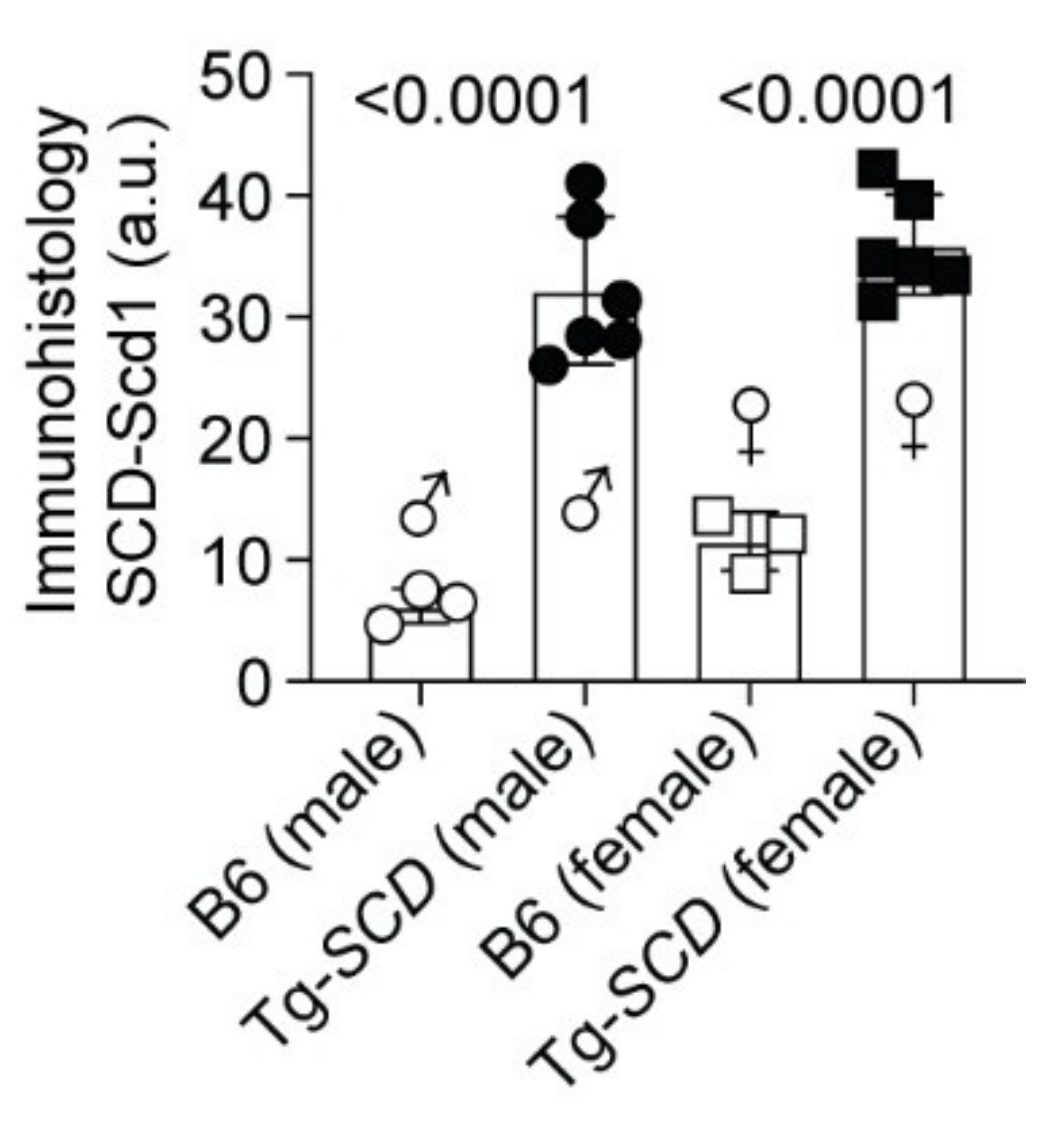
2.5. Immunohistological Analysis Shows Cardiac Enlargement of Tg-SCD Mice and Increased Cardiac SCD Protein Levels
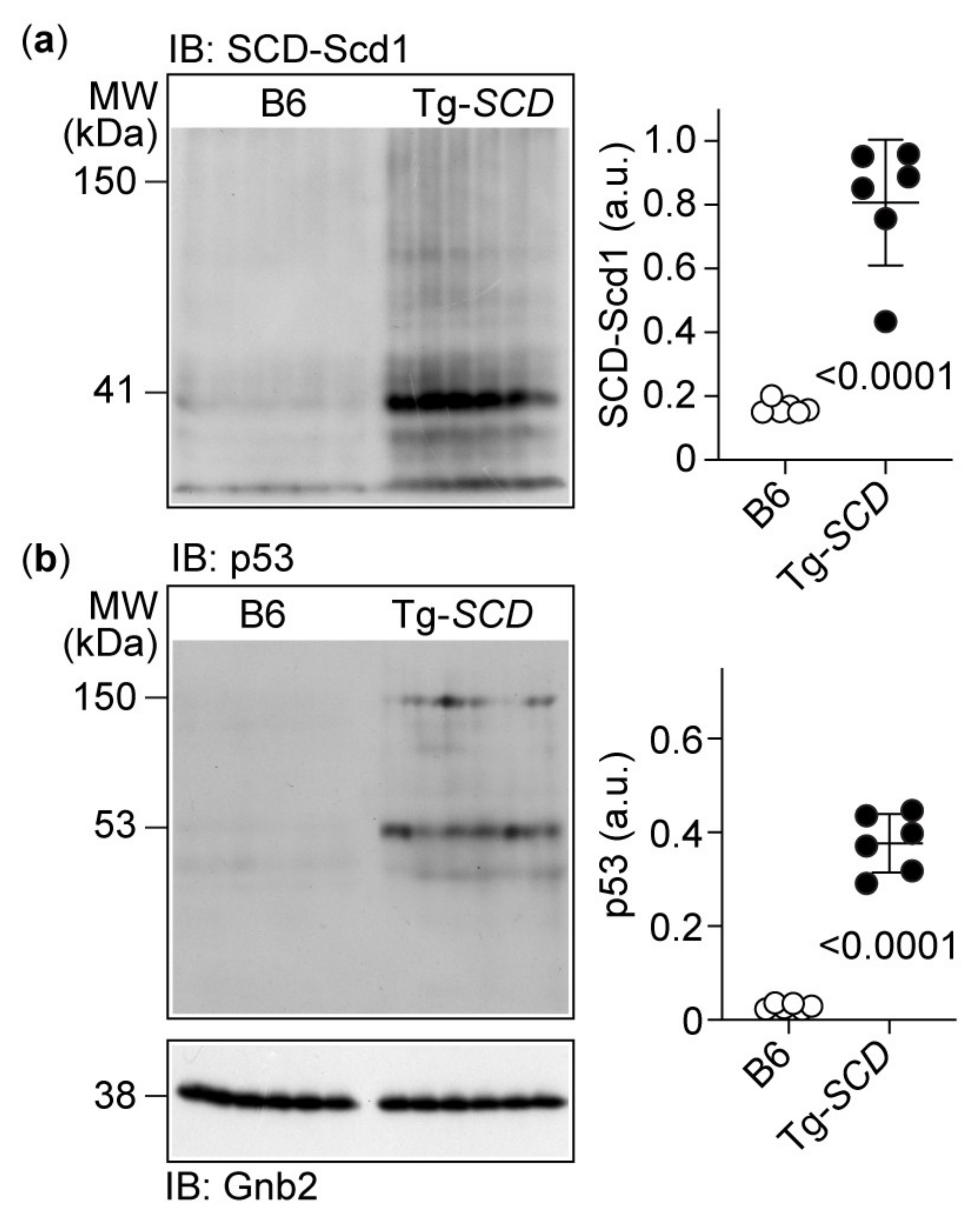
2.6. Immunoblot Analysis Shows Increased Cardiac SCD and Pro-Apoptotic p53 Protein Levels of Tg-SCD Mice
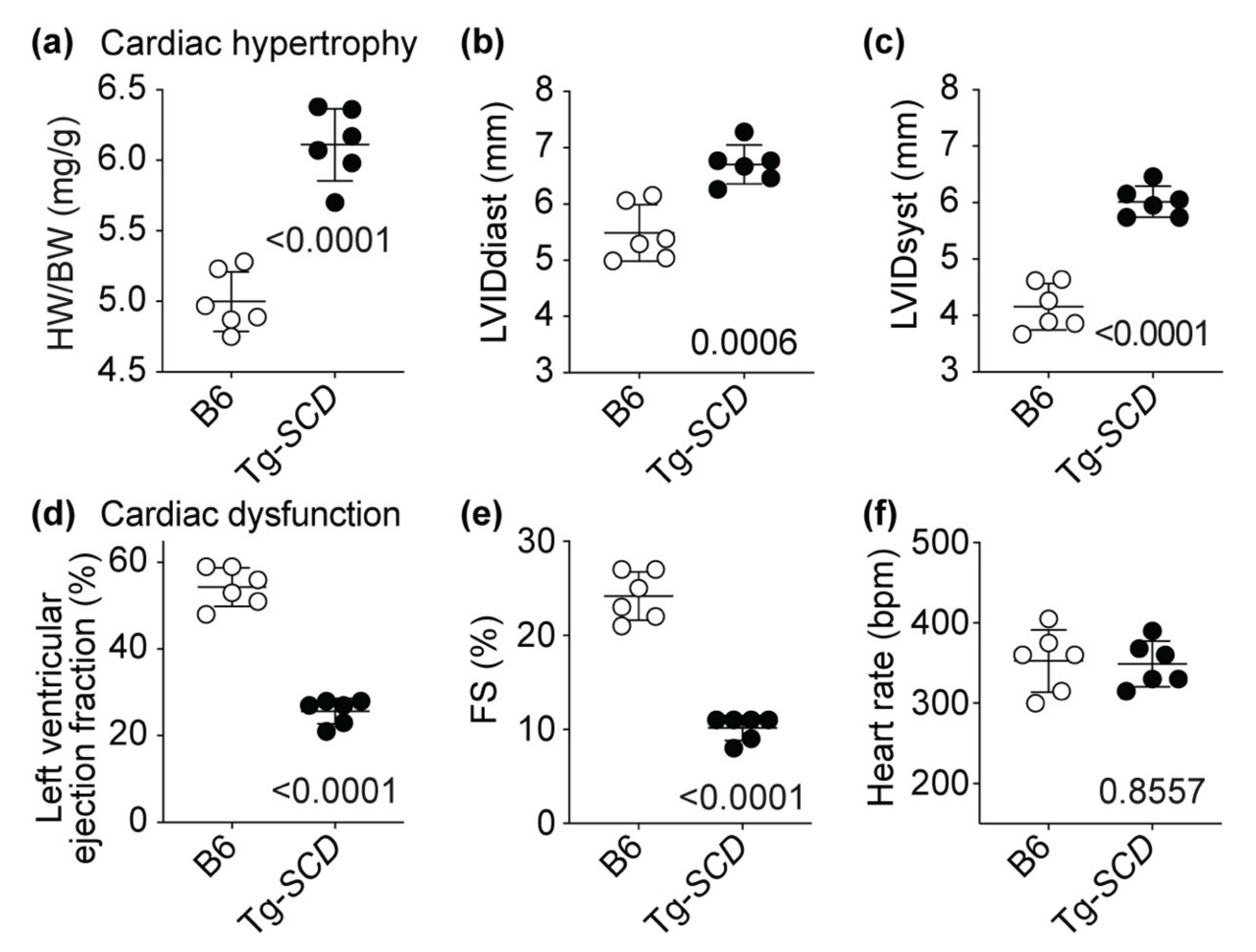
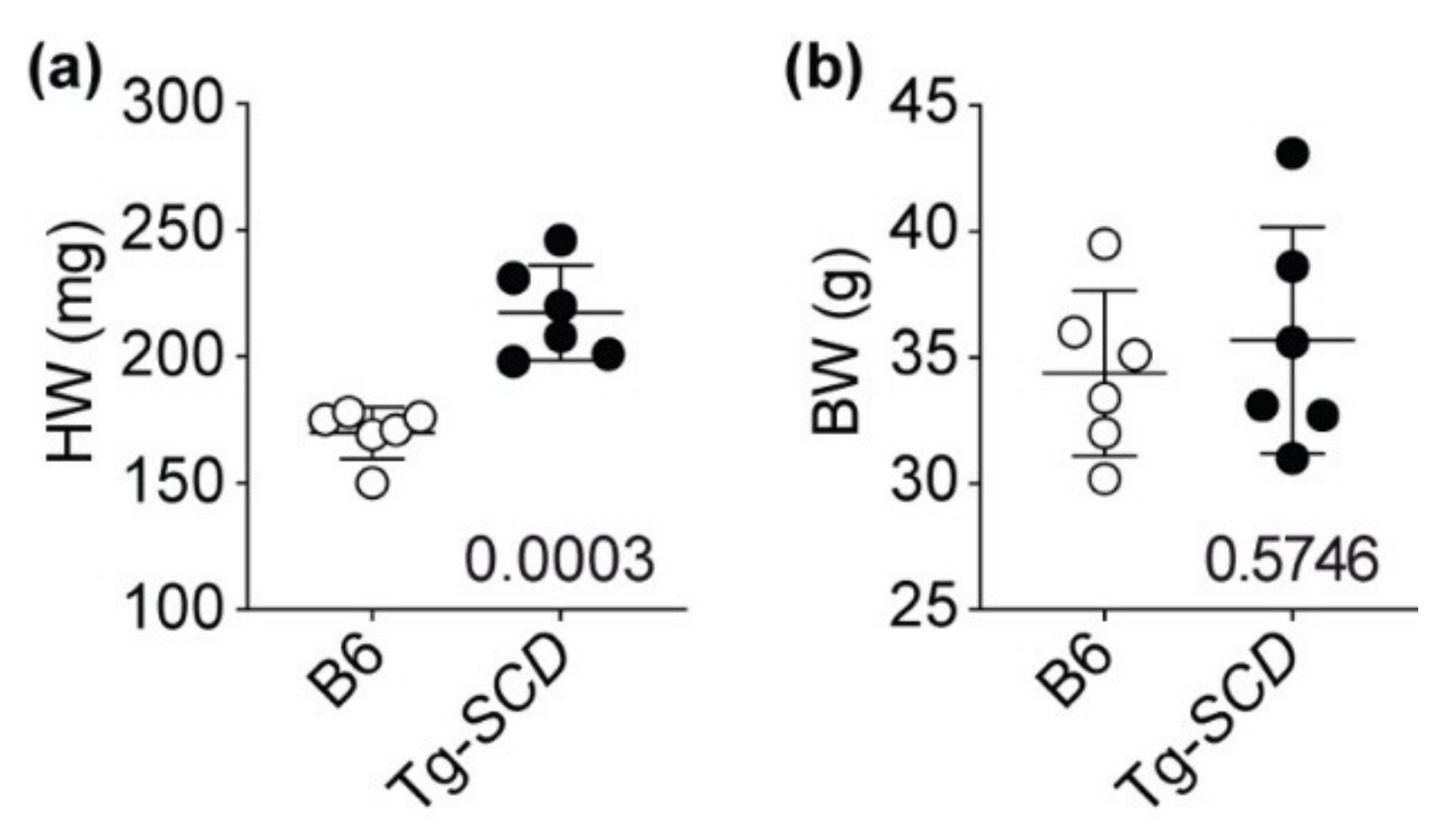
2.7. Tg-SCD Mice Have a Heart Failure Phenotype with Cardiac Hypertrophy and Cardiac Dysfunction
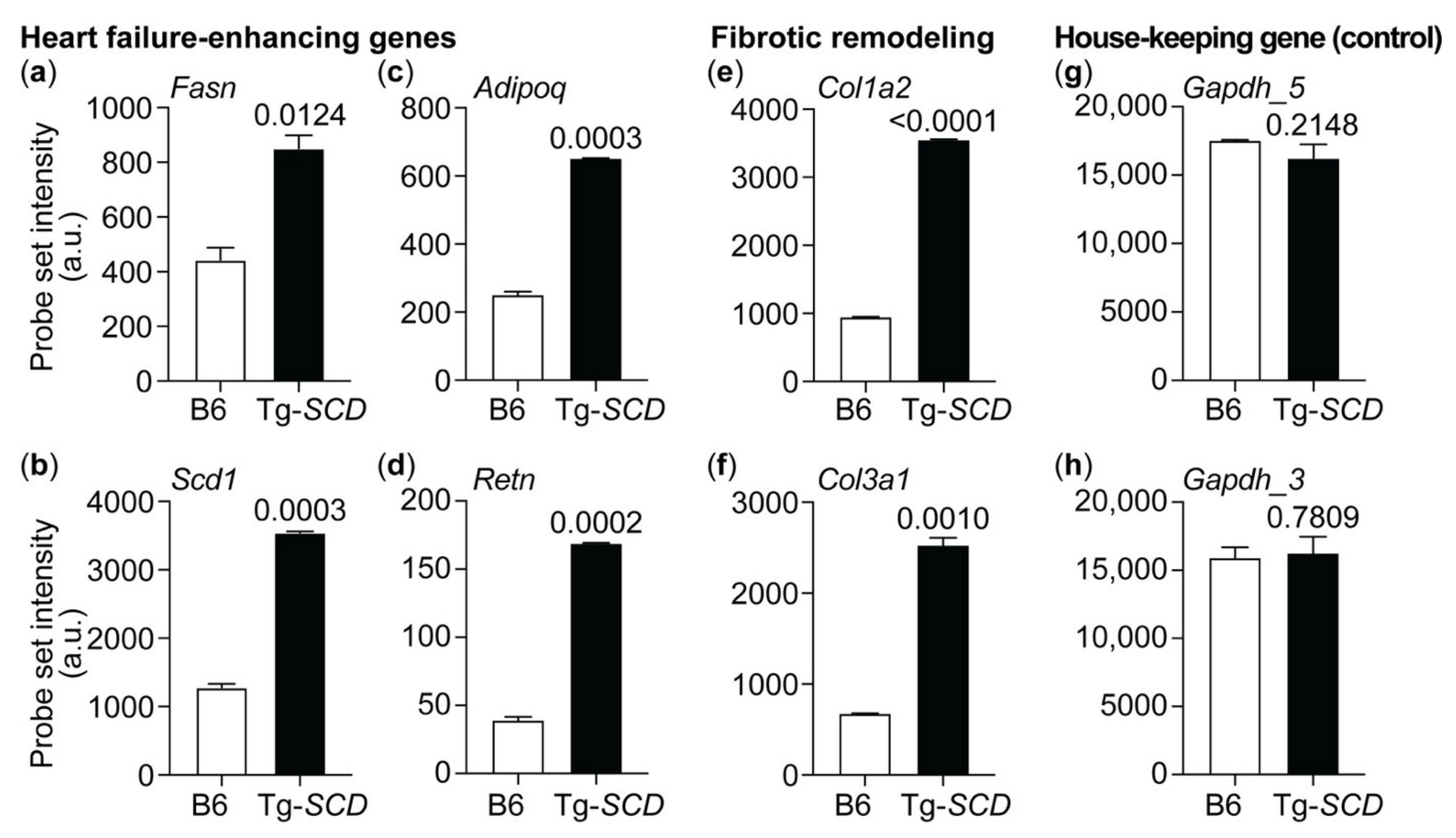
2.8. Gene Expression Profiling of Tg-SCD Mice Shows Up-Regulation of Heart Failure-Related Lipid Genes
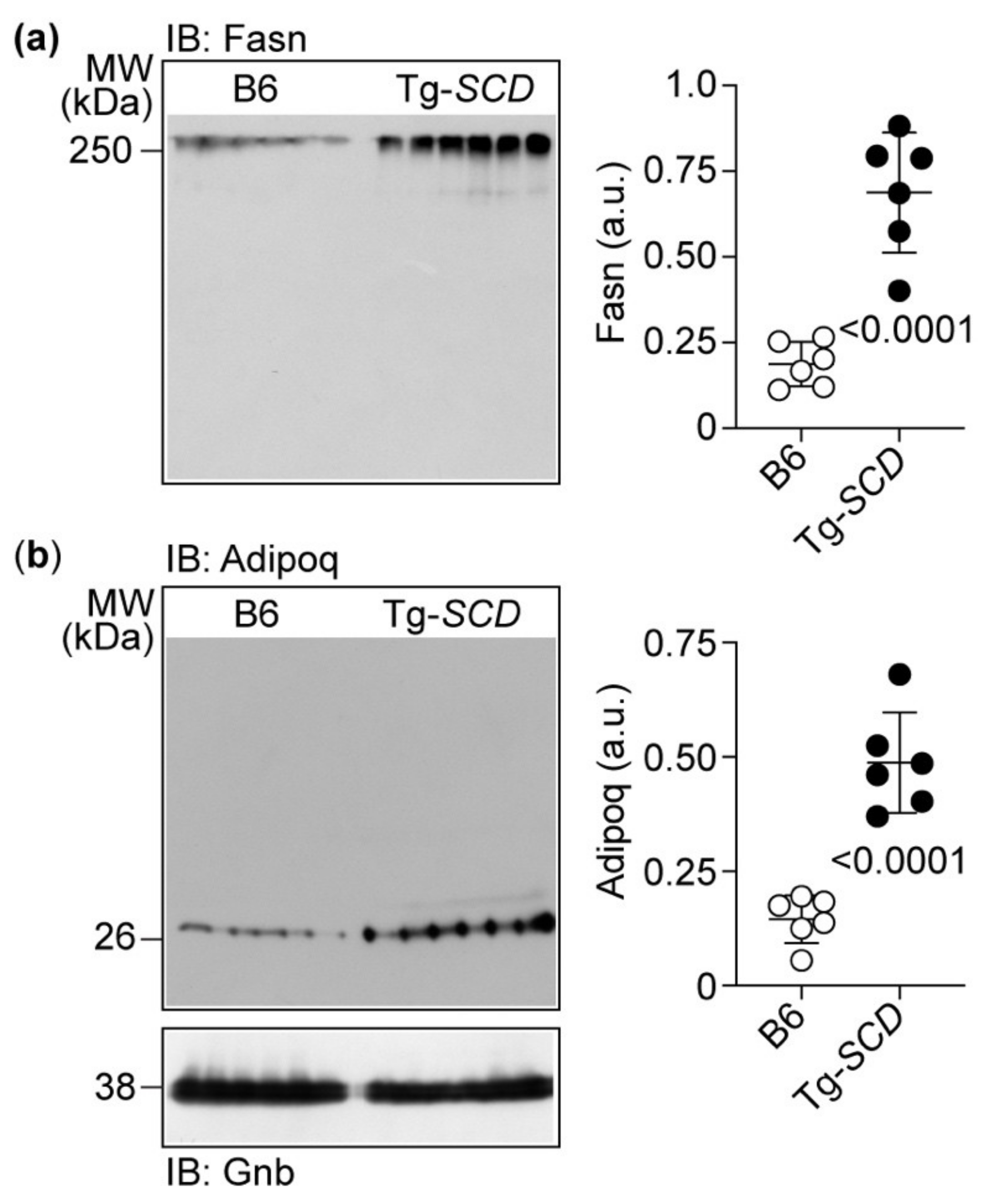
2.9. Immunoblot Detection Confirms Up-Regulation of Cardiac Fasn and Adipoq Proteins of Tg-SCD Mice
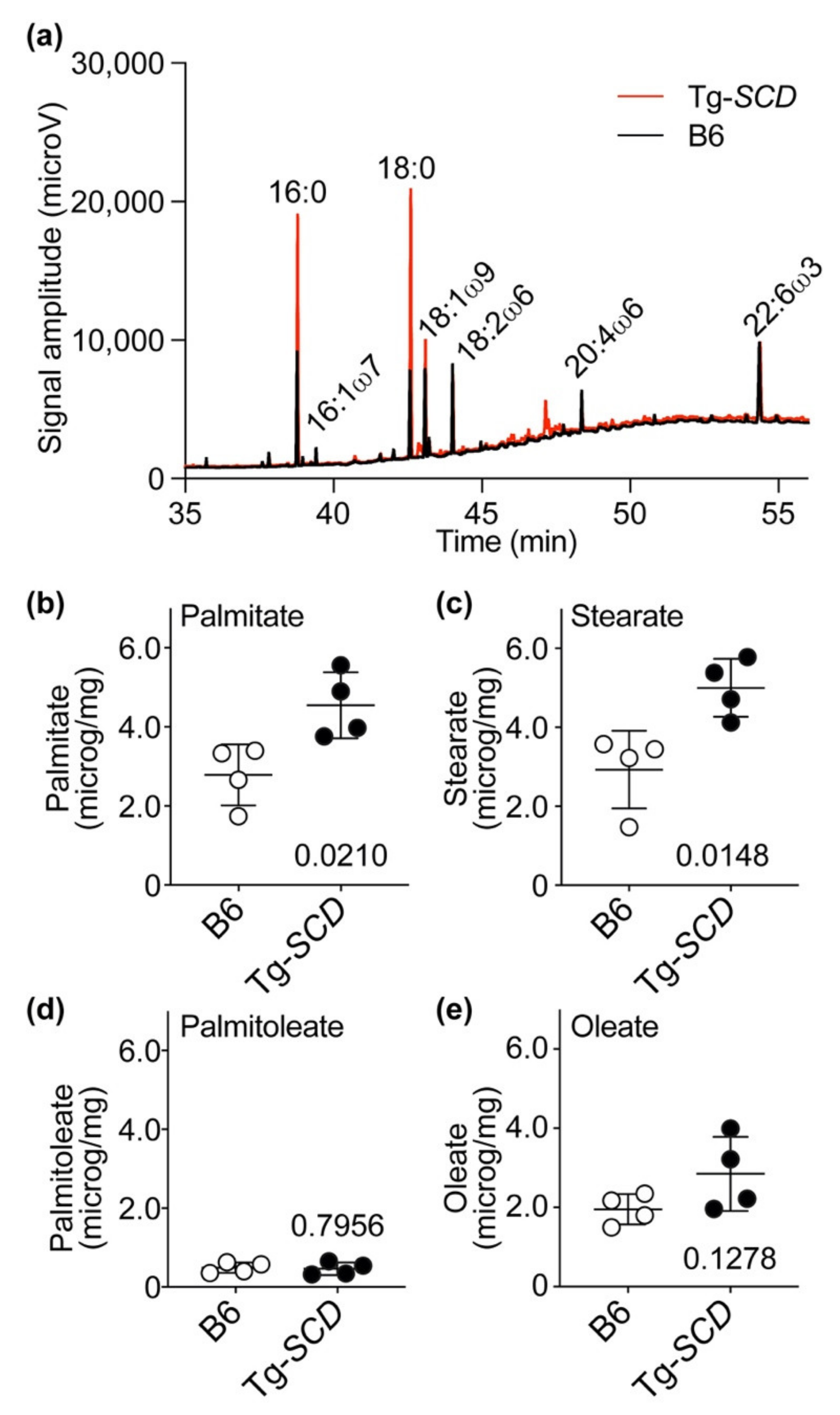
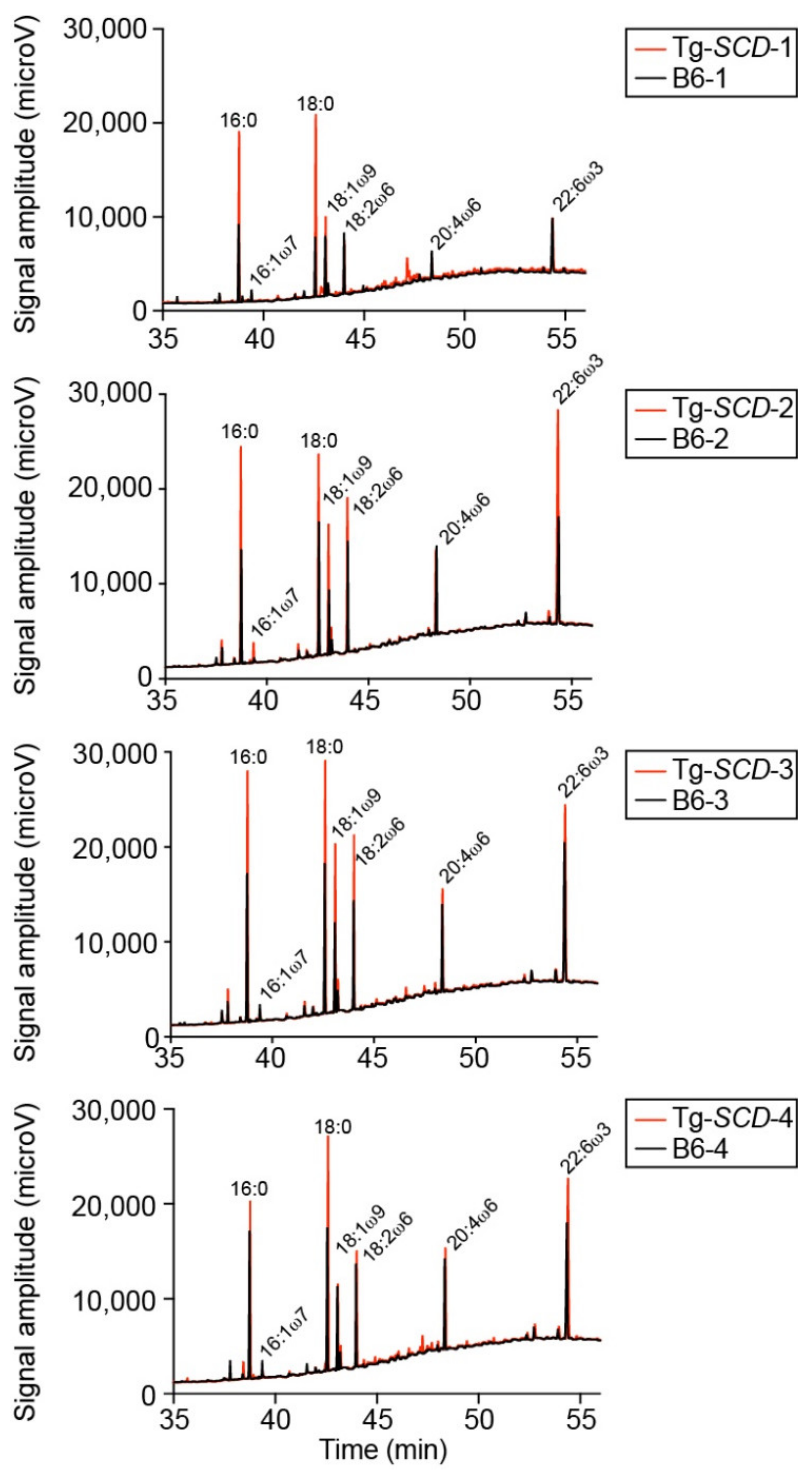
2.10. Accumulation of Saturated Lipids in Tg-SCD Hearts
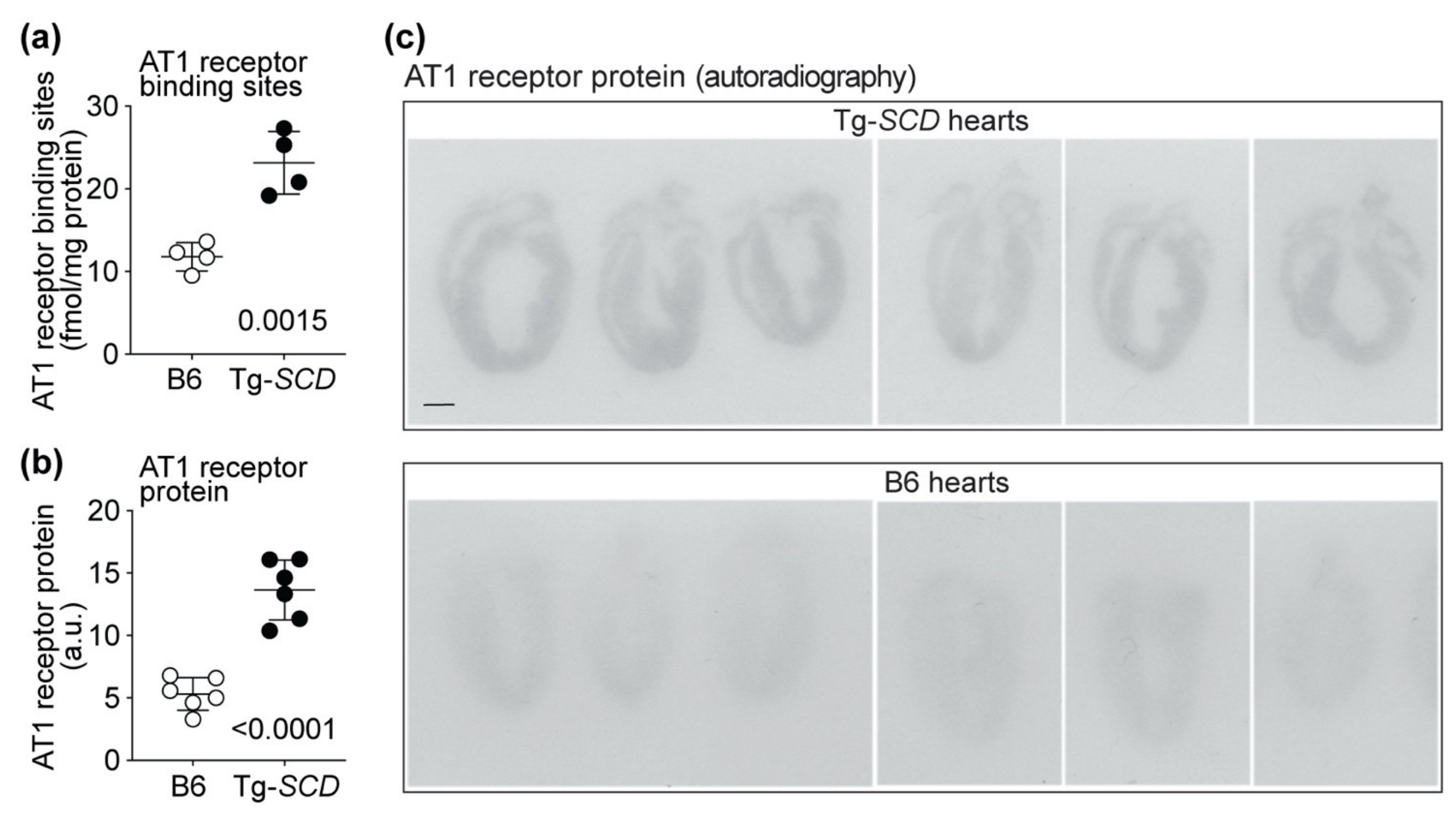
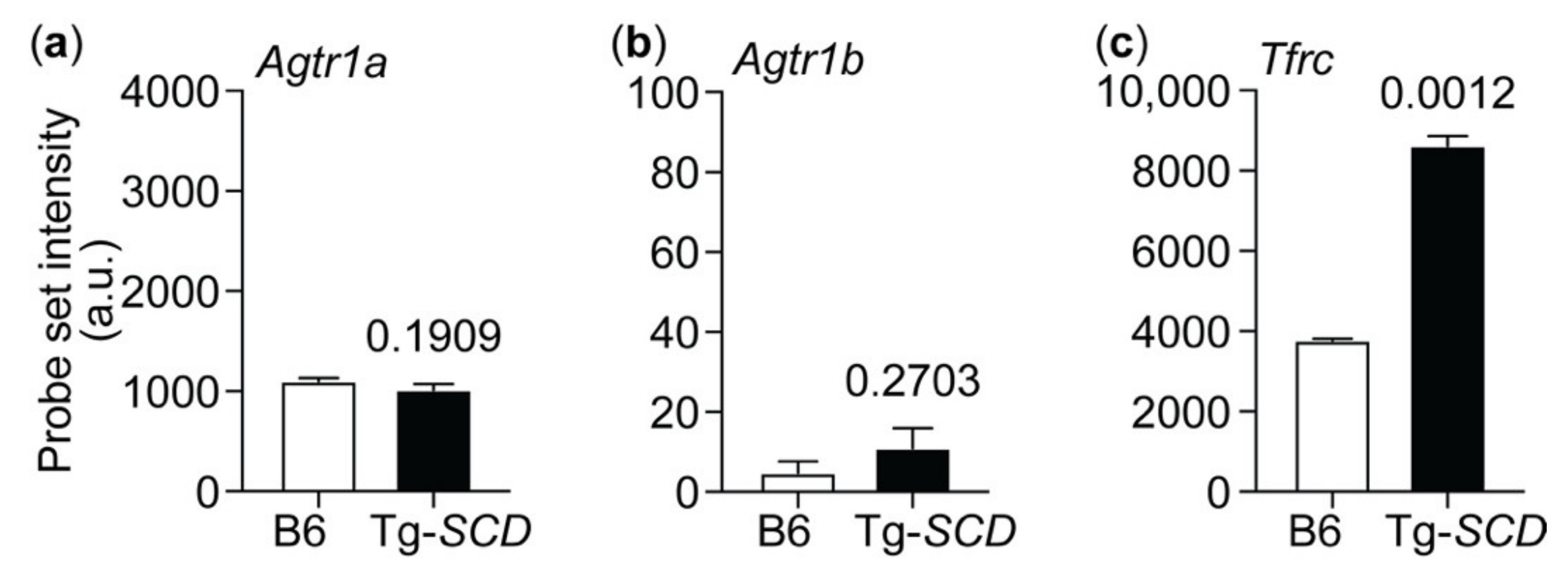
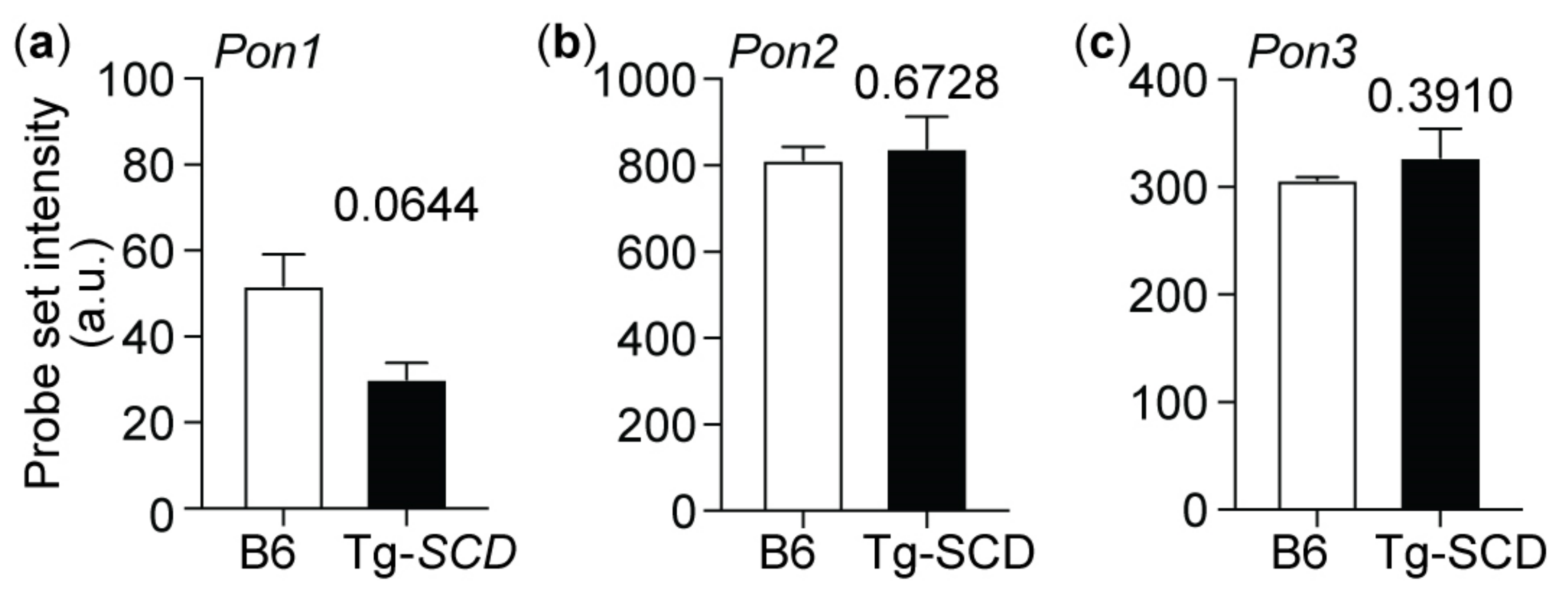
2.11. Up-Regulation of the Heart-Failure-Promoting AT1 Receptor in Tg-SCD Mice
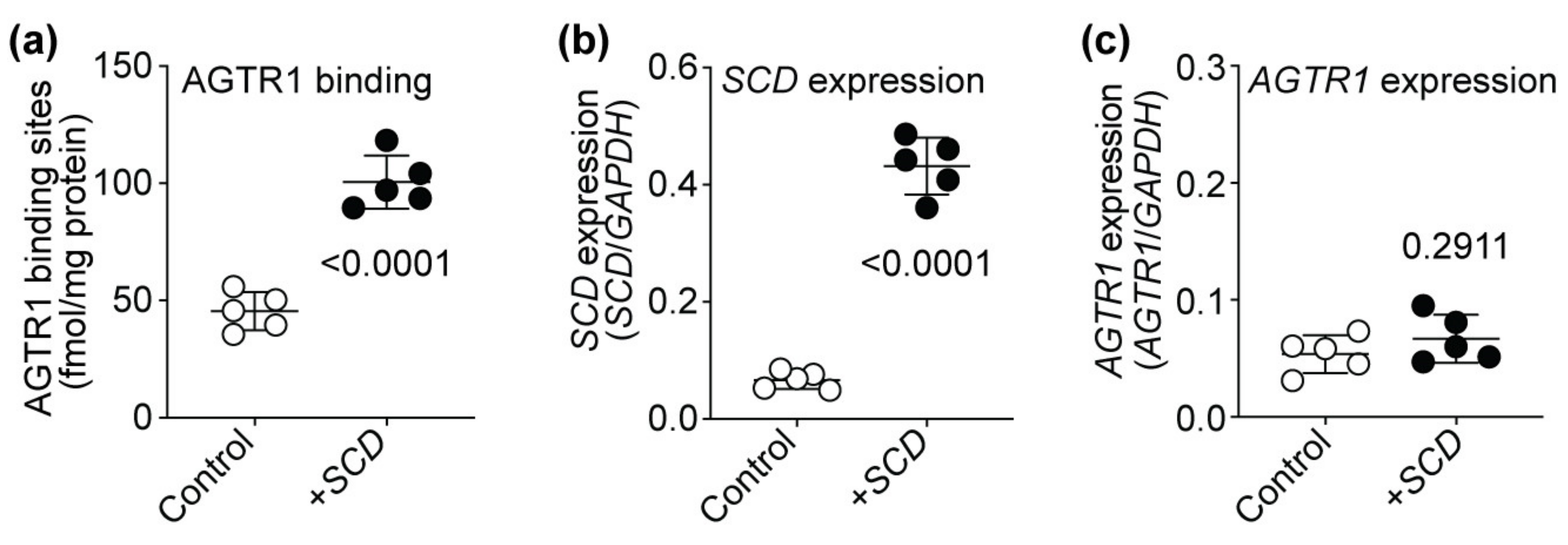
2.12. SCD Expression Enhances the Number of Cell-Surface AT1-Receptor-Binding Sites of HEK Cells
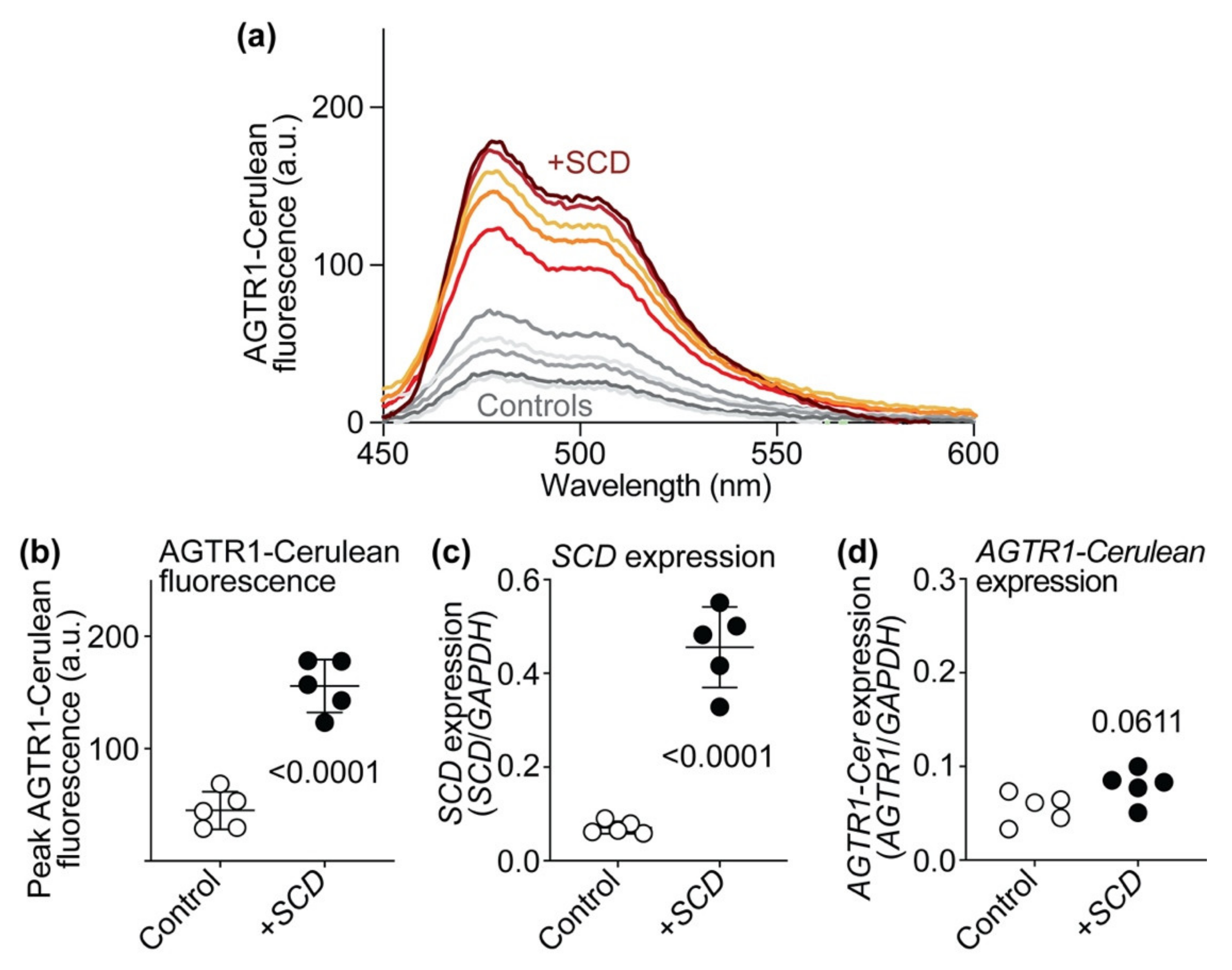
2.13. SCD Expression Increases AT1 Receptor-Cerulean Protein Levels of HEK Cells
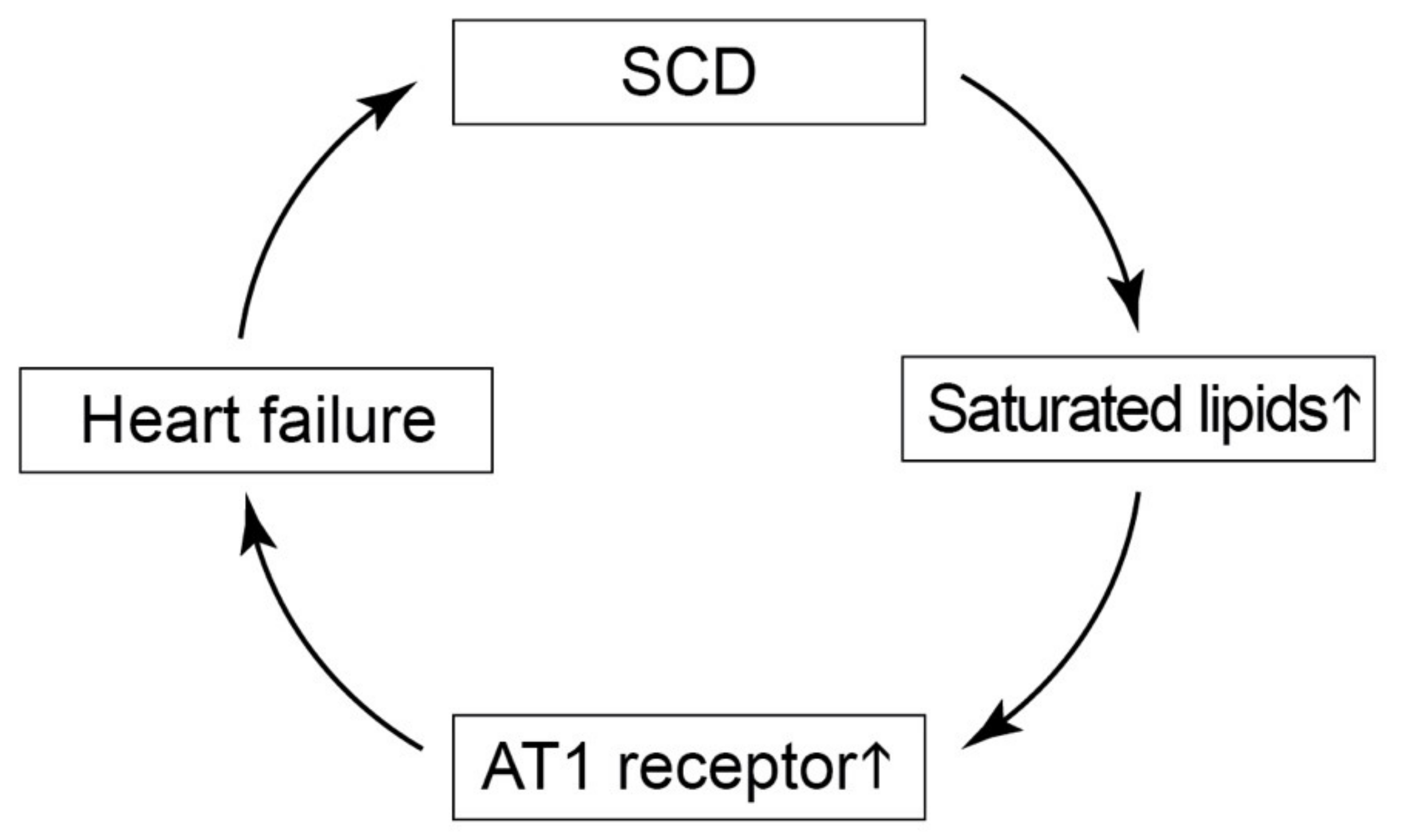
3. Discussion
4. Materials and Methods
4.1. GO Analysis of Whole-Genome Microarray Gene Expression Data of Experimental Heart Failure Models
4.2. Generation of Tg-SCD Mice and Animal Experiments
4.3. Antibodies
4.4. Histology and Immunohistology
4.5. Immunoblot Detection of Proteins
4.6. RNA Isolation, Whole-Genome Microarray Gene Expression Profiling, and Real-Time qRT-PCR
4.7. GC Analysis of Cardiac Lipids
4.8. Radioligand Binding Studies
4.9. Imaging of Cardiac AT1 Receptors by Autoradiography
4.10. Culture of HEK Cells and Fluorescence Spectroscopy
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A






References
- Savarese, G.; Lund, L.H. Global public health burden of heart failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Murphy, S.P.; Ibrahim, N.E.; Januzzi, J.L. Heart failure with reduced ejection fraction: A review. JAMA 2020, 324, 488–504. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart failure with preserved ejection fraction in perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef]
- Writing Committee; Maddox, T.M.; Januzzi, J.L.; Allen, L.A.; Breathett, K.; Butler, J.; Davis, L.L.; Fonarow, G.C.; Ibrahim, N.E.; Lindenfeld, J.; et al. 2021 update to the 2017 ACC expert consensus decision pathway for optimization of heart failure treatment: Answers to 10 pivotal issues about heart failure with reduced ejection fraction: A report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2021, 77, 772–810. [Google Scholar] [CrossRef] [PubMed]
- Askoxylakis, V.; Thieke, C.; Pleger, S.T.; Most, P.; Tanner, J.; Lindel, K.; Katus, H.A.; Debus, J.; Bischof, M. Long-term survival of cancer patients compared to heart failure and stroke: A systematic review. BMC Cancer 2010, 10, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd Alla, J.; Graemer, M.; Fu, X.; Quitterer, U. Inhibition of G-protein-coupled receptor kinase 2 prevents the dysfunctional cardiac substrate metabolism in fatty acid synthase transgenic mice. J. Biol. Chem. 2016, 291, 2583–2600. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, S.; Fu, X.; Elzahwy, S.S.; Klaetschke, K.; Streichert, T.; Quitterer, U. Up-regulation of the cardiac lipid metabolism at the onset of heart failure. Cardiovasc. Hematol. Agents Med. Chem. 2011, 9, 190–206. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Xu, H.; DiSilvestre, D.; Halperin, V.L.; Tunin, R.; Tian, Y.; Yu, W.; Winslow, R.L.; Tomaselli, G.F. Transcriptome profiling of the canine tachycardia-induced heart failure model: Global comparison to human and murine heart failure. J. Mol. Cell. Cardiol. 2006, 40, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santolini, M.; Romay, M.C.; Yukhtman, C.L.; Rau, C.D.; Ren, S.; Saucerman, J.J.; Wang, J.J.; Weiss, J.N.; Wang, Y.; Lusis, A.J.; et al. A personalized, multiomics approach identifies genes involved in cardiac hypertrophy and heart failure. NPJ Syst. Biol. Appl. 2018, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Liu, H.; Ressom, H.W.; Tiwari, S.; Ecelbarger, C.M. Chronic rosiglitazone therapy normalizes expression of ACE1, SCD1 and other genes in the kidney of obese Zucker rats as determined by microarray analysis. Exp. Clin. Endocrinol. Diabetes 2008, 116, 315–325. [Google Scholar] [CrossRef]
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological functions of stearoyl-CoA desaturase. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E28–E37. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Yokoyama, T.; Sekiguchi, K.; Iijima, D.; Sunaga, H.; Maniwa, M.; Ueno, M.; Iso, T.; Arai, M.; Kurabayashi, M. Stearoyl-CoA desaturase-1 (SCD1) augments saturated fatty acid-induced lipid accumulation and inhibits apoptosis in cardiac myocytes. PLoS ONE 2012, 7, e33283. [Google Scholar] [CrossRef] [Green Version]
- Dalla Valle, A.; Vertongen, P.; Spruyt, D.; Lechanteur, J.; Suain, V.; Gaspard, N.; Brion, J.P.; Gangji, V.; Rasschaert, J. Induction of stearoyl-CoA 9-desaturase 1 protects human mesenchymal stromal cells against palmitic acid-induced lipotoxicity and inflammation. Front. Endocrinol. 2019, 10, 726. [Google Scholar] [CrossRef] [Green Version]
- Iwai, T.; Kume, S.; Chin-Kanasaki, M.; Kuwagata, S.; Araki, H.; Takeda, N.; Sugaya, T.; Uzu, T.; Maegawa, H.; Araki, S.I. Stearoyl-CoA desaturase-1 protects cells against lipotoxicity-mediated apoptosis in proximal tubular cells. Int. J. Mol. Sci. 2016, 17, 1868. [Google Scholar] [CrossRef] [Green Version]
- Dobrzyn, P.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 rescues cardiac function in obese leptin-deficient mice. J. Lipid Res. 2010, 51, 2202–2210. [Google Scholar] [CrossRef] [Green Version]
- Dobrzyn, P.; Bednarski, T.; Dobrzyn, A. Metabolic reprogramming of the heart through stearoyl-CoA desaturase. Prog. Lipid Res. 2015, 57, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Djousse, L.; Weir, N.L.; Hanson, N.Q.; Tsai, M.Y.; Gaziano, J.M. Plasma phospholipid concentration of cis-palmitoleic acid and risk of heart failure. Circ. Heart Fail. 2012, 5, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.J.; Folsom, A.R.; Ma, J.; Arnett, D.K.; McGovern, P.G.; Eckfeldt, J.H. Plasma fatty acid composition and 6-year incidence of hypertension in middle-aged adults: The Atherosclerosis Risk in Communities (ARIC) Study. Am. J. Epidemiol. 1999, 150, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Petersson, H.; Basu, S.; Cederholm, T.; Riserus, U. Serum fatty acid composition and indices of stearoyl-CoA desaturase activity are associated with systemic inflammation: Longitudinal analyses in middle-aged men. Br. J. Nutr. 2008, 99, 1186–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Folsom, A.R.; Zheng, Z.J.; Pankow, J.S.; Eckfeldt, J.H.; ARIC Study Investigators. Plasma fatty acid composition and incidence of diabetes in middle-aged adults: The Atherosclerosis Risk in Communities (ARIC) Study. Am. J. Clin. Nutr. 2003, 78, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Suresh, R.; Li, X.; Chiriac, A.; Goel, K.; Terzic, A.; Perez-Terzic, C.; Nelson, T.J. Transcriptome from circulating cells suggests dysregulated pathways associated with long-term recurrent events following first-time myocardial infarction. J. Mol. Cell. Cadiol. 2014, 74, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yu, F. SCD leads to the development and progression of acute myocardial infarction through the AMPK signaling pathway. BMC Cardiovasc. Disord. 2021, 21, 197. [Google Scholar] [CrossRef]
- Dobrzyn, P.; Sampath, H.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 inhibits fatty acid oxidation and increases glucose utilization in the heart. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E357–E364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednarski, T.; Olichwier, A.; Opasinska, A.; Pyrkowska, A.; Gan, A.M.; Ntambi, J.M.; Dobrzyn, P. Stearoyl-CoA desaturase 1 deficiency reduces lipid accumulation in the heart by activating lipolysis independently of peroxisome proliferator-activated receptor alpha. Biochim. Biophys. Acta 2016, 1861, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Yoshida, N.; Isobe, S.; Moriyama, H.; Goto, S.; et al. Sirt1 counteracts decrease in membrane phospholipid unsaturation and diastolic dysfunction during saturated fatty acid overload. J. Mol. Cell. Cardiol. 2019, 133, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.D.; Yee, M.; Porter, G.A.; Ritzer, E.; McDavid, A.N.; Brookes, P.S.; Pryhuber, G.S.; O’Reilly, M.A. Neonatal hyperoxia inhibits proliferation and survival of atrial cardiomyocytes by suppressing fatty acid synthesis. JCI Insight 2021, 6, e140785. [Google Scholar] [CrossRef]
- George, J.; Patal, S.; Wexler, D.; Sharabi, Y.; Peleg, E.; Kamari, Y.; Grossman, E.; Sheps, D.; Keren, G.; Roth, A. Circulating adiponectin concentrations in patients with congestive heart failure. Heart 2006, 92, 1420–1424. [Google Scholar] [CrossRef] [Green Version]
- Frankel, D.S.; Vasan, R.S.; D’Agostino, R.B.; Benjamin, E.J.; Levy, D.; Wang, T.J.; Meigs, J.B. Resistin, adiponectin, and risk of heart failure the Framingham offspring study. J. Am. Coll. Cardiol. 2009, 53, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Conte, J.V., Jr.; Foster, A.H.; McLaughlin, J.S.; Wei, C. Increased p53 protein expression in human failing myocardium. J. Heart Lung Transplant. 1999, 18, 744–749. [Google Scholar] [CrossRef]
- Querejeta, R.; Lopez, B.; Gonzalez, A.; Sanchez, E.; Larman, M.; Martinez Ubago, J.L.; Diez, J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: Relation to myocardial fibrosis. Circulation 2004, 110, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Bashey, R.I.; Martinez-Hernandez, A.; Jimenez, S.A. Isolation, characterization, and localization of cardiac collagen type VI. Associations with other extracellular matrix components. Circ. Res. 1992, 70, 1006–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, J.E.; Vork, M.M.; Roemen, T.H.; de Jong, Y.F.; Cleutjens, J.P.; van der Vusse, G.J.; van Bilsen, M. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J. Lipid Res. 1997, 38, 1384–1394. [Google Scholar] [CrossRef]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Yoshida, N.; Isobe, S.; Moriyama, H.; Goto, S.; et al. Decrease in membrane phospholipids unsaturation correlates with myocardial diastolic dysfunction. PLoS ONE 2018, 13, e0208396. [Google Scholar] [CrossRef]
- Jacquemyn, J.; Cascalho, A.; Goodchild, R.E. The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 2017, 18, 1905–1921. [Google Scholar] [CrossRef]
- Larkin, J.E.; Frank, B.C.; Gaspard, R.M.; Duka, I.; Gavras, H.; Quackenbush, J. Cardiac transcriptional response to acute and chronic angiotensin II treatments. Physiol. Genom. 2004, 18, 152–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakishita, M.; Nakamura, K.; Asanuma, M.; Morita, H.; Saito, H.; Kusano, K.; Nakamura, Y.; Emori, T.; Matsubara, H.; Sugaya, T.; et al. Direct evidence for increased hydroxyl radicals in angiotensin II-induced cardiac hypertrophy through angiotensin II type 1a receptor. J. Cardiovasc. Pharmacol. 2003, 42 (Suppl. S1), S67–S70. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Orekhov, A.N.; Bobryshev, Y.V. Paraoxonase and atherosclerosis-related cardiovascular diseases. Biochimie 2017, 132, 19–27. [Google Scholar] [CrossRef]
- Pei, J.F.; Yan, Y.F.; Tang, X.; Zhang, Y.; Cui, S.S.; Zhang, Z.Q.; Chen, H.Z.; Liu, D.P. Human paraoxonase gene cluster overexpression alleviates angiotensin II-induced cardiac hypertrophy in mice. Sci. China Life Sci. 2016, 59, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Sorescu, D.; Griendling, K.K. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest. Heart Fail. 2002, 8, 132–140. [Google Scholar] [CrossRef]
- Leri, A.; Claudio, P.P.; Li, Q.; Wang, X.; Reiss, K.; Wang, S.; Malhotra, A.; Kajstura, J.; Anversa, P. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J. Clin. Investig. 1998, 101, 1326–1342. [Google Scholar] [CrossRef] [Green Version]
- Williams, B. Angiotensin II and the pathophysiology of cardiovascular remodeling. Am. J. Cardiol. 2001, 87, 10C–17C. [Google Scholar] [CrossRef]
- Wei, Y.; Clark, S.E.; Morris, E.M.; Thyfault, J.P.; Uptergrove, G.M.; Whaley-Connell, A.T.; Ferrario, C.M.; Sowers, J.R.; Ibdah, J.A. Angiotensin II-induced non-alcoholic fatty liver disease is mediated by oxidative stress in transgenic TG(mRen2)27(Ren2) rats. J. Hepatol. 2008, 49, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.H.; Standridge, M.K.; Moustaid, N. Angiotensin increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology 1997, 138, 1512–1519. [Google Scholar] [CrossRef]
- Goossens, G.H.; Blaak, E.E.; Arner, P.; Saris, W.H.; van Baak, M.A. Angiotensin II: A hormone that affects lipid metabolism in adipose tissue. Int. J. Obes. 2007, 31, 382–384. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Cuenca, S.; Whyte, L.; Hagen, R.; Vidal-Puig, A.; Fuller, M. Stearoyl-CoA desaturase 1 is a key determinant of membrane lipid composition in 3T3-L1 adipocytes. PLoS ONE 2016, 11, e0162047. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.C. Progress in understanding the role of lipids in membrane protein folding. Biochim. Biophys. Acta 2012, 1818, 951–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntambi, J.M. Dietary regulation of stearoyl-CoA desaturase 1 gene expression in mouse liver. J. Biol. Chem. 1992, 267, 10925–10930. [Google Scholar] [CrossRef]
- Tabaczar, S.; Wolosiewicz, M.; Filip, A.; Olichwier, A.; Dobrzyn, P. The role of stearoyl-CoA desaturase in the regulation of cardiac metabolism. Postepy Biochem. 2018, 64, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Steffen, B.T.; Duprez, D.; Szklo, M.; Guan, W.; Tsai, M.Y. Circulating oleic acid levels are related to greater risks of cardiovascular events and all-cause mortality: The Multi-Ethnic Study of Atherosclerosis. J. Clin. Lipidol. 2018, 12, 1404–1412. [Google Scholar] [CrossRef]
- Yokozawa, J.; Sasaki, T.; Ohwada, K.; Sasaki, Y.; Ito, J.I.; Saito, T.; Kawata, S. Down-regulation of hepatic stearoyl-CoA desaturase 1 expression by angiotensin II receptor blocker in the obese fa/fa Zucker rat: Possible role in amelioration of insulin resistance and hepatic steatosis. J. Gastroenterol. 2009, 44, 583–591. [Google Scholar] [CrossRef]
- Reuter, H.; Adam, C.; Grönke, S.; Zobel, C.; Frank, K.F.; Müller-Ehmsen, J.; Brabender, J.; Schwinger, R.H. The increased angiotensin II (type 1) receptor density in myocardium of type 2 diabetic patients is prevented by blockade of the renin-angiotensin system. Diabetologia 2006, 49, 3067–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd Alla, J.; Langer, A.; Elzahwy, S.S.; Arman-Kalcek, G.; Streichert, T.; Quitterer, U. Angiotensin-converting enzyme inhibition down-regulates the pro-atherogenic chemokine receptor 9 (CCR9)-chemokine ligand 25 (CCL25) axis. J. Biol. Chem. 2010, 285, 23496–23505. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Rudel, L.L. Stearoyl-coenzyme A desaturase 1 inhibition and the metabolic syndrome: Considerations for future drug discovery. Curr. Opin. Lipidol. 2010, 21, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Chung, S.; Sawyer, J.K.; Degirolamo, C.; Alger, H.M.; Nguyen, T.M.; Zhu, X.; Duong, M.N.; Brown, A.L.; Lord, C.; et al. Combined therapy of dietary fish oil and stearoyl-CoA desaturase 1 inhibition prevents the metabolic syndrome and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Kurikawa, N.; Takagi, T.; Wakimoto, S.; Uto, Y.; Terashima, H.; Kono, K.; Ogata, T.; Oshumi, J. A novel inhibitor of stearoyl-CoA desaturase-1 attenuates hepatic lipid accumulation, liver injury and inflammation in model of nonalcoholic steatohepatitis. Biol. Pharm. Bull. 2013, 36, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oballa, R.M.; Belair, L.; Black, W.C.; Bleasby, K.; Chan, C.C.; Desroches, C.; Du, X.; Gordon, R.; Guay, J.; Guiral, S.; et al. Development of a liver-targeted stearoyl-CoA desaturase (SCD) inhibitor (MK-8245) to establish a therapeutic window for the treatment of diabetes and dyslipidemia. J. Med. Chem. 2011, 54, 5082–5096. [Google Scholar] [CrossRef]
- Miyazaki, M.; Man, W.C.; Ntambi, J.M. Targeted disruption of stearoyl-CoA desaturase1 gene in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid. J. Nutr. 2001, 131, 2260–2268. [Google Scholar] [CrossRef]
- Gulick, J.; Subramaniam, A.; Neumann, J.; Robbins, J. Isolation and characterization of the mouse cardiac myosin heavy chain genes. J. Biol. Chem. 1991, 266, 9180–9185. [Google Scholar] [CrossRef]
- Quitterer, U.; Fu, X.; Pohl, A.; Bayoumy, K.M.; Langer, A.; AbdAlla, S. Beta-arrestin1 prevents preeclampsia by downregulation of mechanosensitive AT1-B2 receptor heteromers. Cell 2019, 176, 318–333.e19. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.; Abd Alla, J.; Quitterer, U. Sensitization of the angiotensin II AT1 receptor contributes to RKIP-induced symptoms of heart failure. Front. Med. 2019, 5, 359. [Google Scholar] [CrossRef] [Green Version]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Quitterer, U.; Pohl, A.; Langer, A.; Koller, S.; Abdalla, S. A cleavable signal peptide enhances cell surface delivery and heterodimerization of Cerulean-tagged angiotensin II AT1 and bradykinin B2 receptor. Biochem. Biophys. Res. Commun. 2011, 409, 544–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd Alla, J.; Jamous, Y.F.; Quitterer, U. Stearoyl-CoA Desaturase (SCD) Induces Cardiac Dysfunction with Cardiac Lipid Overload and Angiotensin II AT1 Receptor Protein Up-Regulation. Int. J. Mol. Sci. 2021, 22, 9883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189883
Abd Alla J, Jamous YF, Quitterer U. Stearoyl-CoA Desaturase (SCD) Induces Cardiac Dysfunction with Cardiac Lipid Overload and Angiotensin II AT1 Receptor Protein Up-Regulation. International Journal of Molecular Sciences. 2021; 22(18):9883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189883
Chicago/Turabian StyleAbd Alla, Joshua, Yahya F. Jamous, and Ursula Quitterer. 2021. "Stearoyl-CoA Desaturase (SCD) Induces Cardiac Dysfunction with Cardiac Lipid Overload and Angiotensin II AT1 Receptor Protein Up-Regulation" International Journal of Molecular Sciences 22, no. 18: 9883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189883