
Probabilistic Critical Controllability Analysis of Protein Interaction Networks Integrating Normal Brain Ageing Gene Expression Profiles



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- (1)
- We developed a controllability algorithm based on MDS that efficiently identified critical nodes in large-scale probabilistic protein networks. Because of the weighted edges, it was a challenging problem to efficiently compute critical nodes among the multiple possible solutions. We bypassed this problem by introducing three novel mathematical propositions that significantly reduced the computational complexity and time, and extended the computable network size of the networks. This algorithm is one of the main theoretical contributions of this work.
- (2)
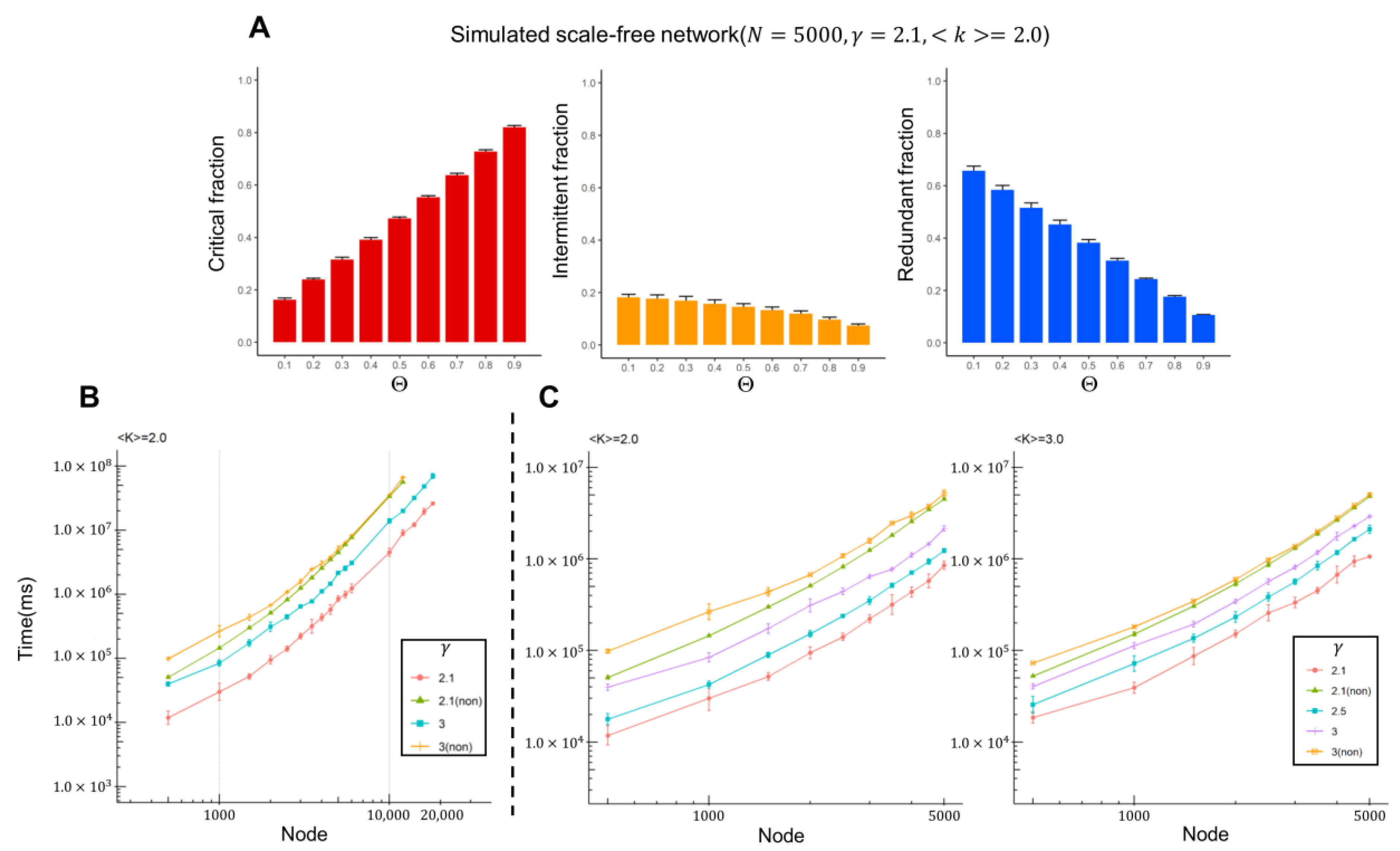
- We validated the algorithm performance using artificially constructed weighted scale-free networks.
- (3)
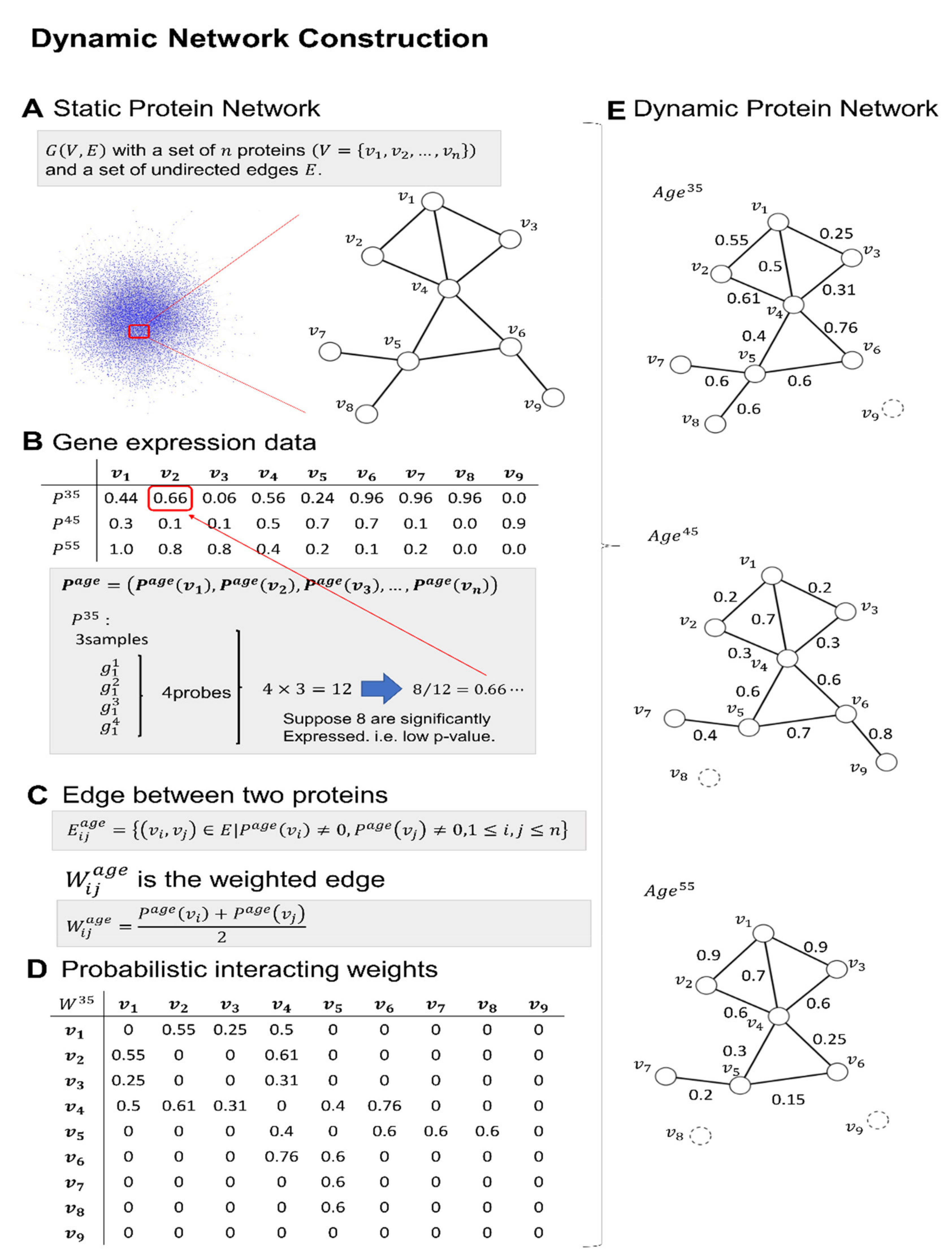
- To examine changes across time, the networks should not be a single snapshot, a time, or a convolution of different time steps. By using gene expression data, we constructed dynamic protein networks that were also weighted, which is probabilistic.
- (4)
- By following steps (1) and (3), we examined whether the CPMDS network-based model identified critical proteins that were also associated with known ageing genes. The findings show that the identified critical controllers were significantly enriched by well-known ageing genes collected from the GenAge database, which was one of the main results of the data analysis.
- (5)
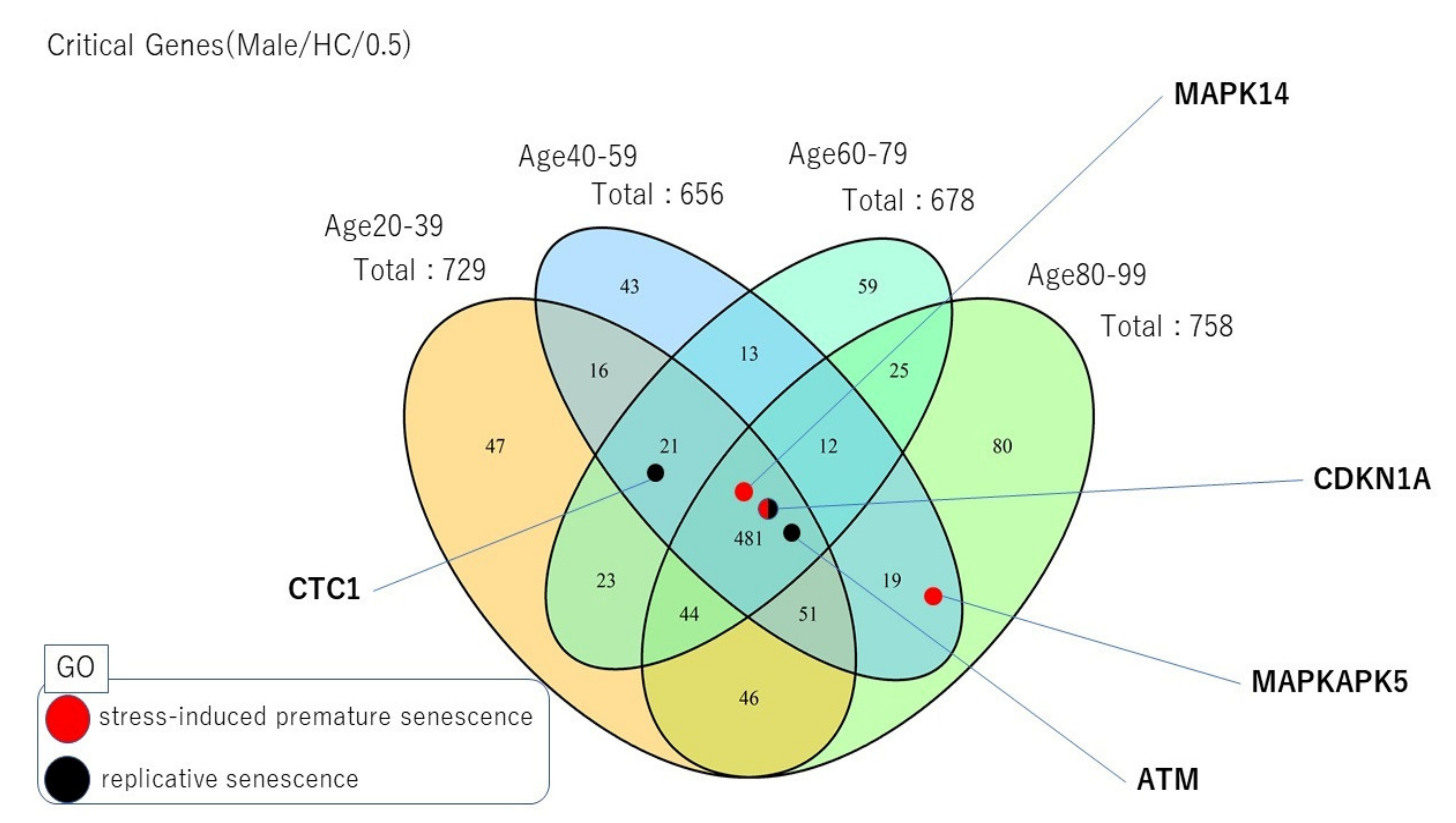
- Critical ageing genes are also proteins enriched in many gene ontology (GO) annotations, showing the biological importance of these proteins. In particular, the enrichment observed in the replicative and premature senescence biological processes with critical proteins for male samples in HC brain regions led to the identification of possible new ageing-gene candidates.
2. Results
2.1. Computational Results from Artificial Scale-Free Networks
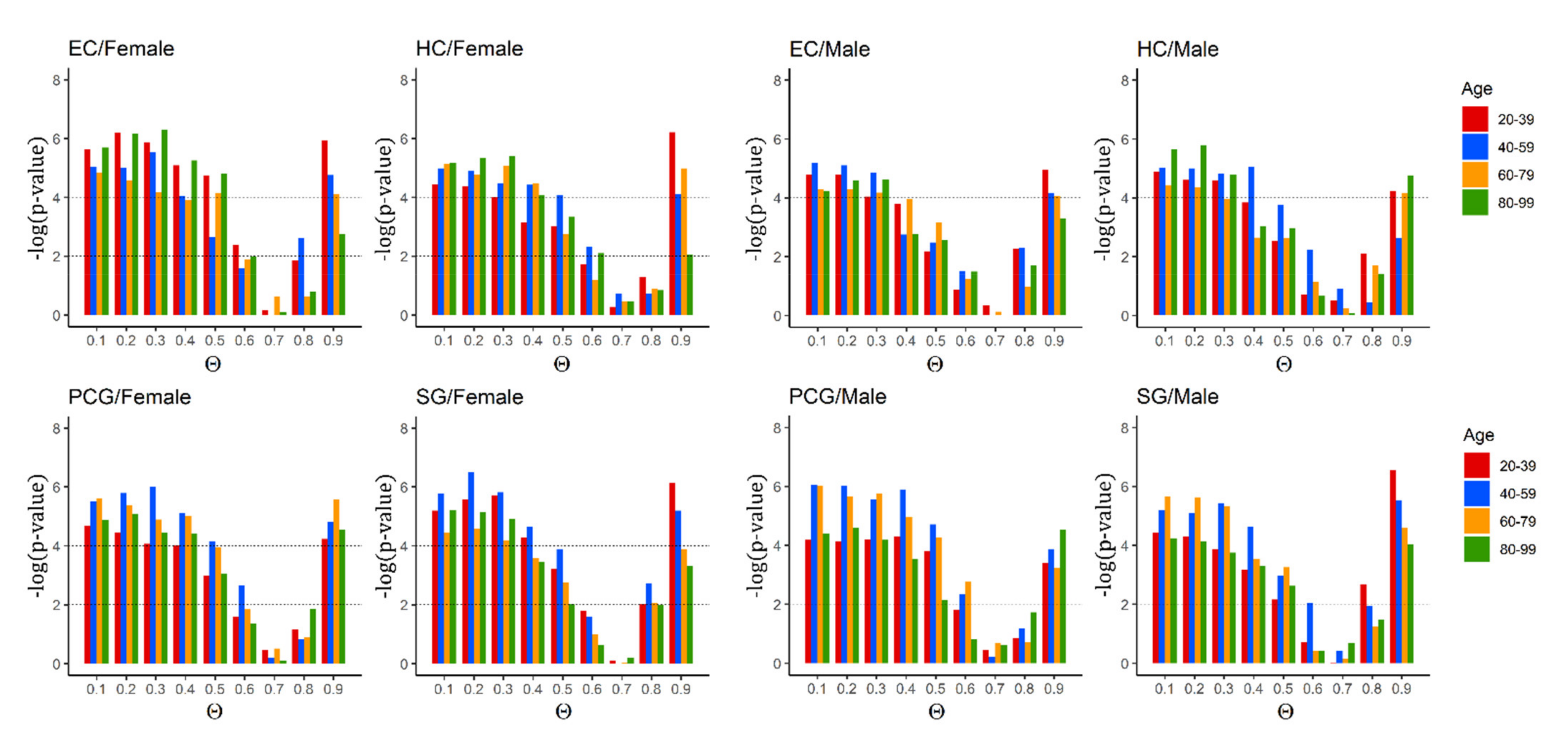
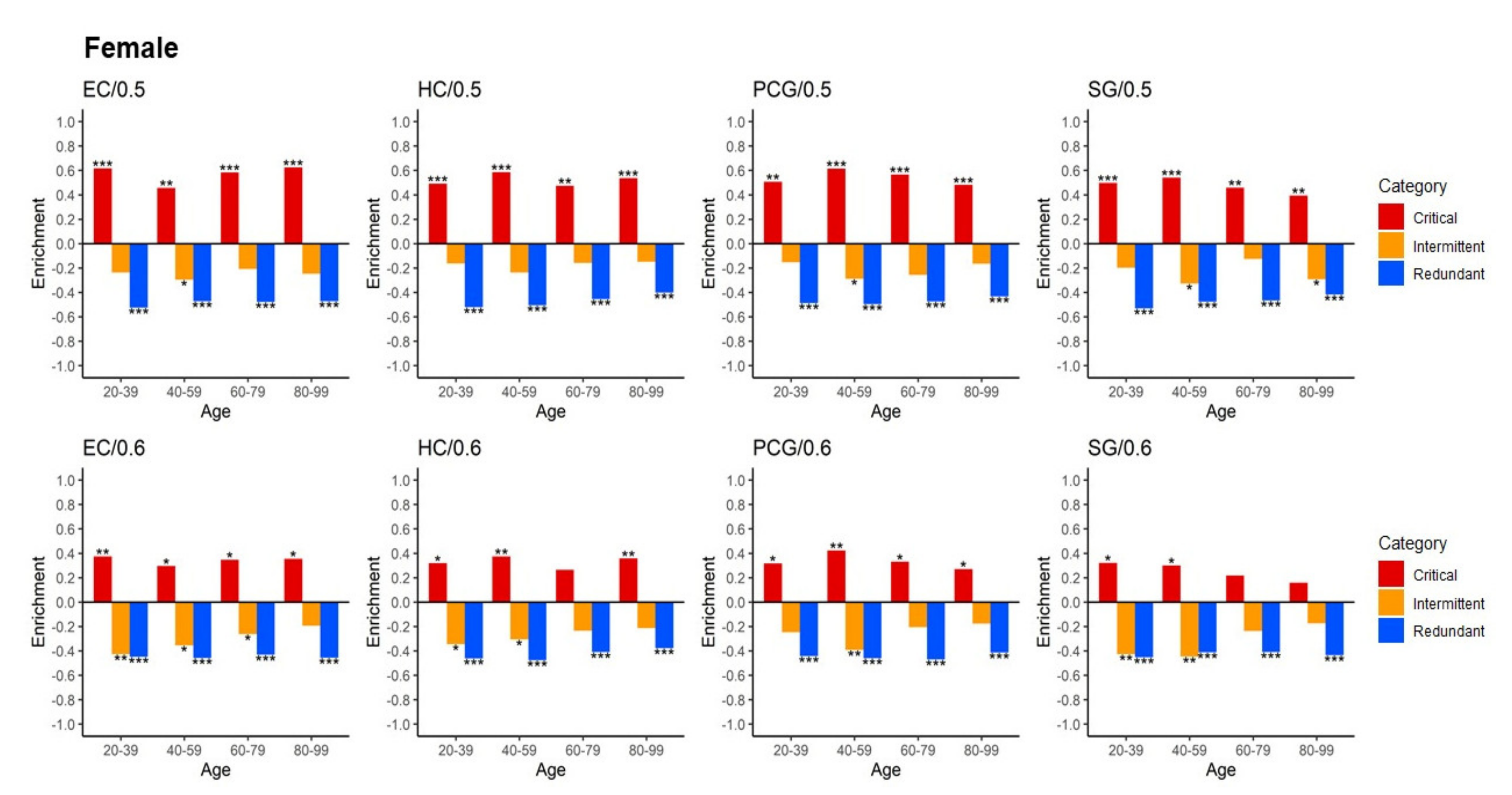
2.2. Critical Control Proteins Are Significantly Enriched and Associated with Ageing Genes across the Lifespan
2.3. Identified Critical Ageing Proteins Are Dynamically Assigned across Ageing
2.4. Unique Critical Control Proteins across Lifespans
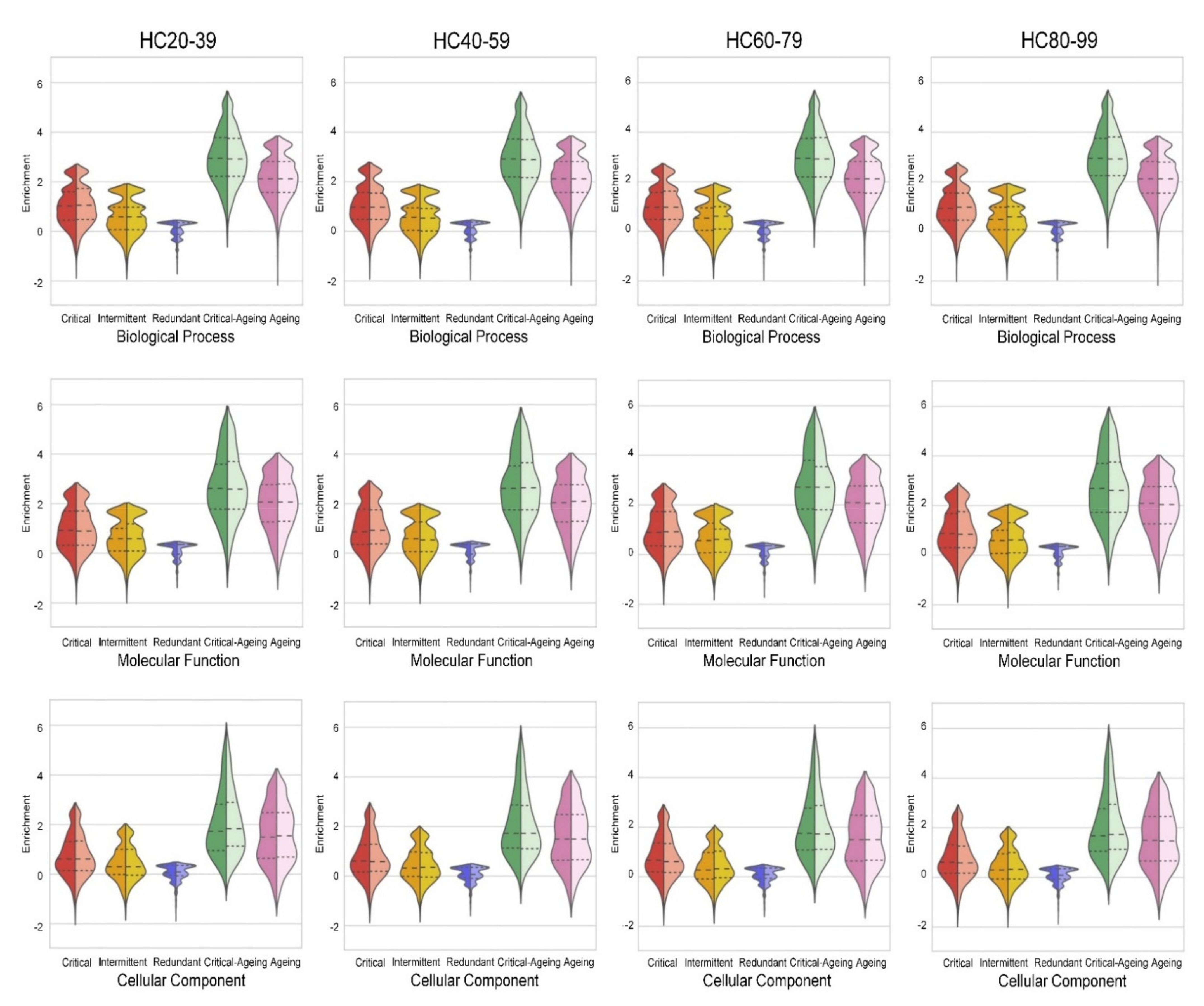
2.5. Ageing Proteins Identified as Critical Controllers Are Enriched in Gene Ontology Functional Categories
3. Discussion
4. Methods
4.1. Gene Expression Data at Different Ages
4.2. Static Protein Interaction Network
4.3. Ageing Genes Database
4.4. Construction of the Dynamic Weighted Protein Interaction Network
4.5. Standard Probabilistic Control Model
4.6. ILP-Formalized PMDS Problem
4.7. Critical Probabilistic Control Model (CPMDS) and Its Efficient Algorithm
- We applied Proposition 1 for each node.
- We applied Proposition 2 for each remaining node.
- We applied Proposition 3 for each remaining node.
- 4.
4.8. Enrichment Calculation for Control Categories
4.9. Statistical Significance Tests
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Szilard, L. On the Nature of the Aging Process. Proc. Natl. Acad. Sci. USA 1959, 45, 30–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The genetics of human ageing. Nat. Rev. Genet. 2020, 21, 88–101. [Google Scholar] [CrossRef]
- Berchtold, N.C.; Cribbs, D.H.; Zielke, H.R.; Cotman, C.W.; Coleman, P.D.; Rogers, J.; Head, E.; Kim, R.; Beach, T.; Miller, C.; et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. USA 2008, 105, 15605–15610. [Google Scholar] [CrossRef] [Green Version]
- Pakkenberg, B. Aging and the human neocortex. Exp. Gerontol. 2003, 38, 95–99. [Google Scholar] [CrossRef]
- Berchtold, N.C.; Coleman, P.D.; Cribbs, D.H.; Rogers, J.; Gillen, D.L.; Cotman, C.W. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1653–1661. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Slotine, J.-J.; Barabási, A.-L. Controllability of complex networks. Nature 2011, 473, 167–173. [Google Scholar] [CrossRef]
- Nacher, J.C.; Akutsu, T. Dominating scale-free networks with variable scaling exponent: Heterogeneous networks are not difficult to control. New J. Phys. 2012, 14, 073005. [Google Scholar] [CrossRef]
- Vinayagam, A.; Gibson, T.E.; Barabasi, A.; Lee, H.-J.; Yilmazel, B.; Roesel, C.; Hu, Y.; Kwon, Y.; Sharma, A.; Liu, Y.-Y.; et al. Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets. Proc. Natl. Acad. Sci. USA 2016, 113, 4976–4981. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.; Vértes, P.E.; Towlson, E.K.; Chew, Y.L.; Walker, D.S.; Schafer, W.; Barabasi, A. Network control principles predict neuron function in the Caenorhabditis elegans connectome. Nat. Cell Biol. 2017, 550, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Nacher, J.C.; Akutsu, T. Minimum dominating set-based methods for analyzing biological networks. Methods 2016, 102, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Kang, U.; Chang, H.; Cho, K.-H. The Hidden Control Architecture of Complex Brain Networks. Science 2019, 13, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuchty, S. Controllability in protein interaction networks. Proc. Natl. Acad. Sci. USA 2014, 111, 7156–7160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagami, H.; Akutsu, T.; Maegawa, S.; Hosokawa, H.; Nacher, J.C. Determining Associations between Human Diseases and non-coding RNAs with Critical Roles in Network Control. Sci. Rep. 2015, 5, 14577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.G. Co-controllability of drug-disease-gene network. New J. Phys. 2015, 17, 85009. [Google Scholar] [CrossRef] [Green Version]
- Basler, G.; Nikoloski, Z.; Larhlimi, A.; Barabási, A.-L.; Liu, Y.-Y. Control of fluxes in metabolic networks. Genome Res. 2016, 26, 956–968. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, J.-M.; Otokuni, H.; Akutsu, T.; Nacher, J.C. Probabilistic controllability approach to metabolic fluxes in normal and cancer tissues. Nat. Commun. 2019, 10, 2725. [Google Scholar] [CrossRef]
- Jia, T.; Liu, Y.Y.; Csóka, E.; Pósfai, M.; Slotine, J.J.; Barabási, A.L. Emergence of bimodality in controlling complex networks. Nat. Commun. 2013, 4, 2002. [Google Scholar] [CrossRef] [PubMed]
- Nacher, J.C.; Akutsu, T. Analysis of critical and redundant nodes in controlling directed and undirected complex networks using dominating sets. J. Complex Netw. 2014, 2, 394–412. [Google Scholar] [CrossRef]
- Ishitsuka, M.; Akutsu, T.; Nacher, J.C. Critical controllability in proteome-wide protein interaction network integrating transcriptome. Sci. Rep. 2016, 6, 23541. [Google Scholar] [CrossRef] [Green Version]
- Ishitsuka, M.; Akutsu, T.; Nacher, J.C. Critical controllability analysis of directed biological networks using efficient graph reduction. Sci. Rep. 2017, 7, 14361. [Google Scholar] [CrossRef]
- Viger, F.; Latapy, M. Efficient and Simple Generation of Random Simple Connected Graphs with Prescribed Degree Sequence. In Proceedings of the 11th International Conference Computing and Combinatorics Conference, Kunming, China, 16–19 August 2005. [Google Scholar]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Cristofalo, V.J.; Lorenzini, A.; Allen, R.; Torres, C.; Tresini, M. Replicative senescence: A critical review. Mech. Ageing Dev. 2004, 125, 827–848. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.-P.; Morin, G.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Bär, C.; Blasco, M.A. Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Research 2016, 5, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S. Telomeres in health and disease. J. Oral Maxillofac. Pathol. 2017, 21, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Epel, E.S.; Blackburn, E.H.; Lin, J.; Dhabhar, F.S.; Adler, N.E.; Morrow, J.D.; Cawthon, R.M. Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. USA 2004, 101, 17312–17315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zglinicki, T.; Martin-Ruiz, C. Telomeres as Biomarkers for Ageing and Age-Related Diseases. Curr. Mol. Med. 2005, 5, 197–203. [Google Scholar] [CrossRef]
- Whittemore, K.; Vera, E.; Martínez-Nevado, E.; Sanpera, C.; Blasco, M.A. Telomere shortening rate predicts species life span. Proc. Natl. Acad. Sci. USA 2019, 116, 15122–15127. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analysis. Curr. Protoc. Bioinform. 2016, 54, 1–33. [Google Scholar] [CrossRef]
- Shin, J.W.; Kwon, S.H.; Choi, J.Y.; Na, J.I.; Huh, C.H.; Choi, H.R.; Park, K.C. Molecular Mechanisms of Dermal Aging and Antiaging Approaches. Int. J. Mol. Sci. 2019, 20, 2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labat-Robert, J. Cell–matrix interactions in aging: Role of receptors and matricryptins. Ageing Res. Rev. 2004, 3, 233–247. [Google Scholar] [CrossRef]
- Gautier, L.; Møller, M.; Friis-Hansen, L.; Knudsen, S. Alternative mapping of probes to genes for Affymetrix chips. BMC Bioinform. 2004, 5, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Mausel, P.; Brondízio, E.; Moran, E. Change detection techniques. Int. J. Remote Sens. 2004, 25, 2365–2401. [Google Scholar] [CrossRef]
- Faisal, F.E.; Milenković, T. Dynamic networks reveal key players in aging. Bioinformatics 2014, 30, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y. Investigation into Aging Mechanism with Topology Analysis of Dynamic Networks. Master’s Thesis, Kyoto University, Kyoto, Japan, 2017. [Google Scholar]
- Das, J.; Yu, H. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 2012, 6, 92. [Google Scholar] [CrossRef] [Green Version]
- Tacutu, R.; Thornton, D.; Johnson, E.; Budovsky, A.; Barardo, D.; Craig, T.; Diana, E.; Lehmann, G.; Toren, D.; Wang, J.; et al. Human Ageing Genomic Resources: New and updated databases. Nucleic Acids Res. 2018, 46, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Nacher, J.C.; Akutsu, T. Structurally robust control of complex networks. Phys. Rev. E 2015, 91, 012826. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, E.; Akutsu, T.; Nacher, J.C. Probabilistic Critical Controllability Analysis of Protein Interaction Networks Integrating Normal Brain Ageing Gene Expression Profiles. Int. J. Mol. Sci. 2021, 22, 9891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189891
Yamaguchi E, Akutsu T, Nacher JC. Probabilistic Critical Controllability Analysis of Protein Interaction Networks Integrating Normal Brain Ageing Gene Expression Profiles. International Journal of Molecular Sciences. 2021; 22(18):9891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189891
Chicago/Turabian StyleYamaguchi, Eimi, Tatsuya Akutsu, and Jose C. Nacher. 2021. "Probabilistic Critical Controllability Analysis of Protein Interaction Networks Integrating Normal Brain Ageing Gene Expression Profiles" International Journal of Molecular Sciences 22, no. 18: 9891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189891