
Transcriptomic Analysis Reveals Differential Expression of Genes between Lung Capillary and Post Capillary Venules in Abdominal Sepsis



Abstract
:1. Introduction
2. Results
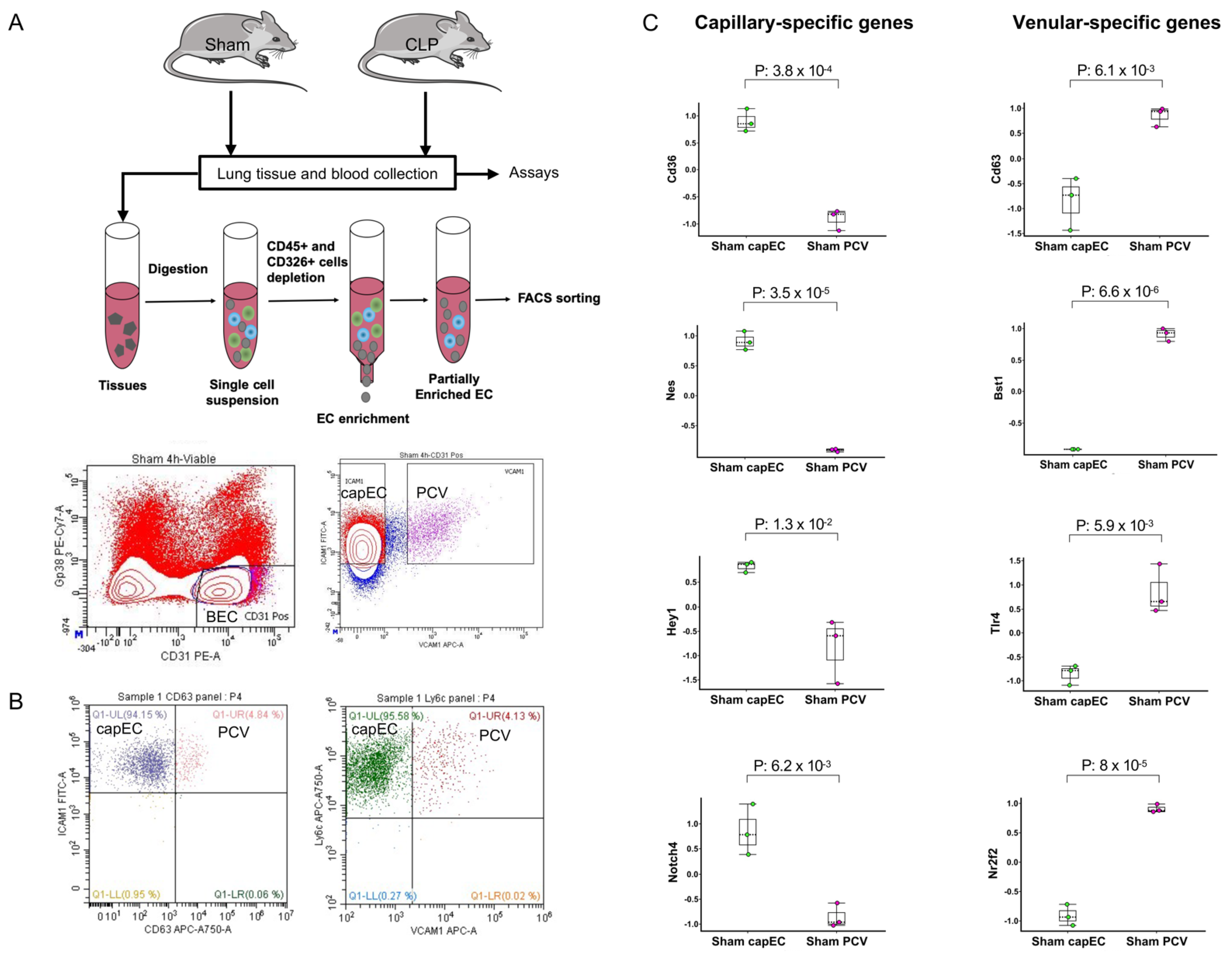
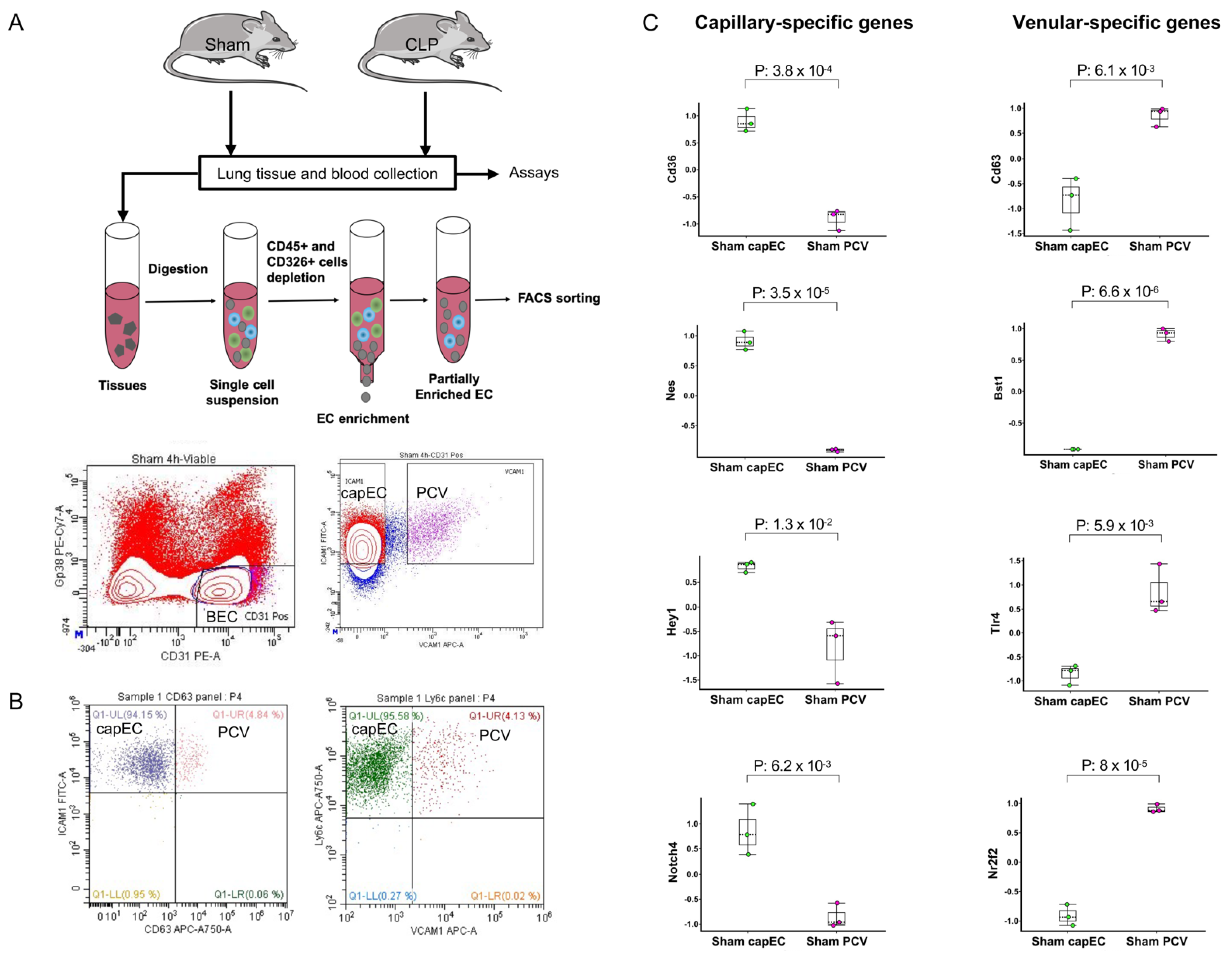
2.1. Lung capEC and PCV Are Transcriptionally Different
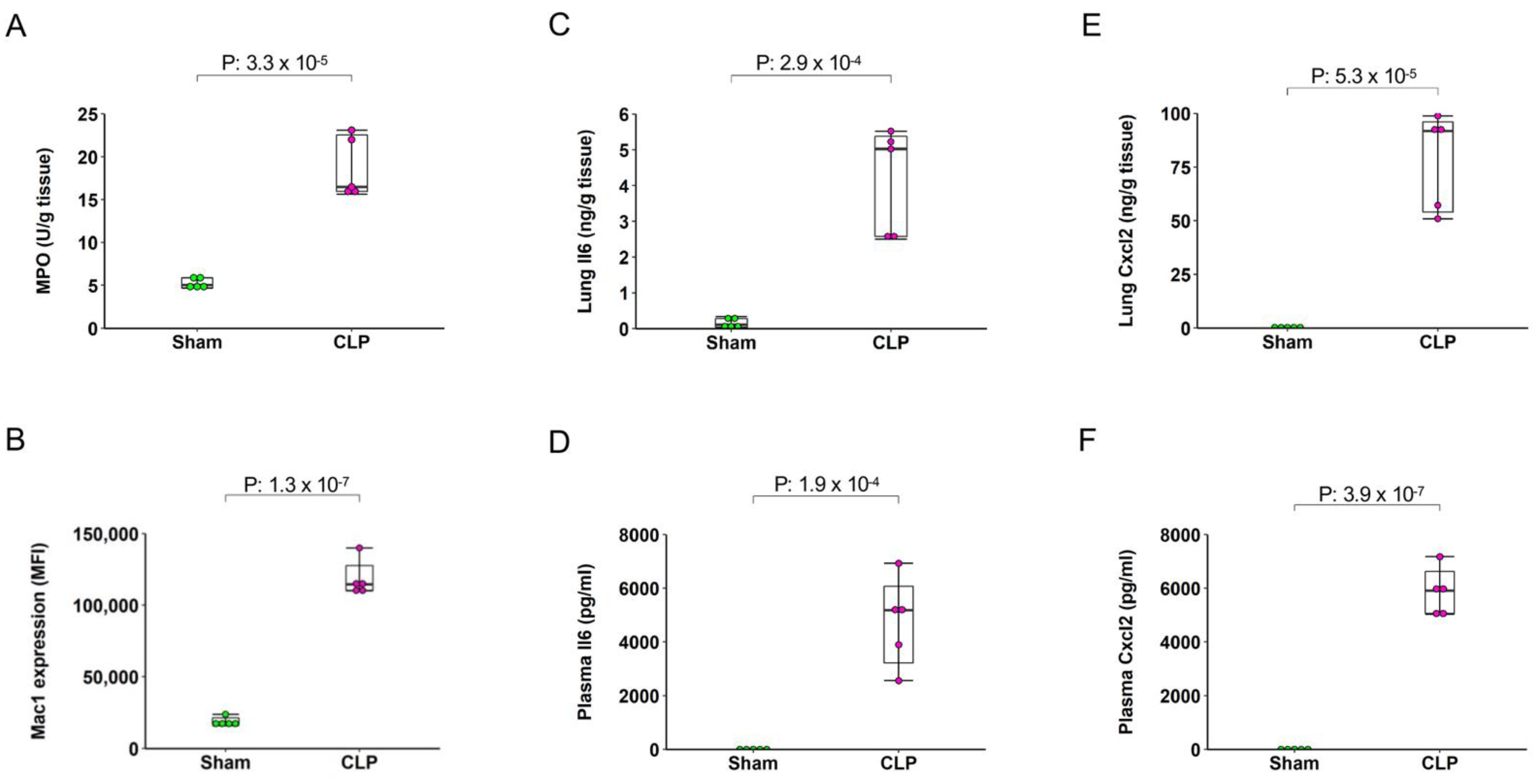
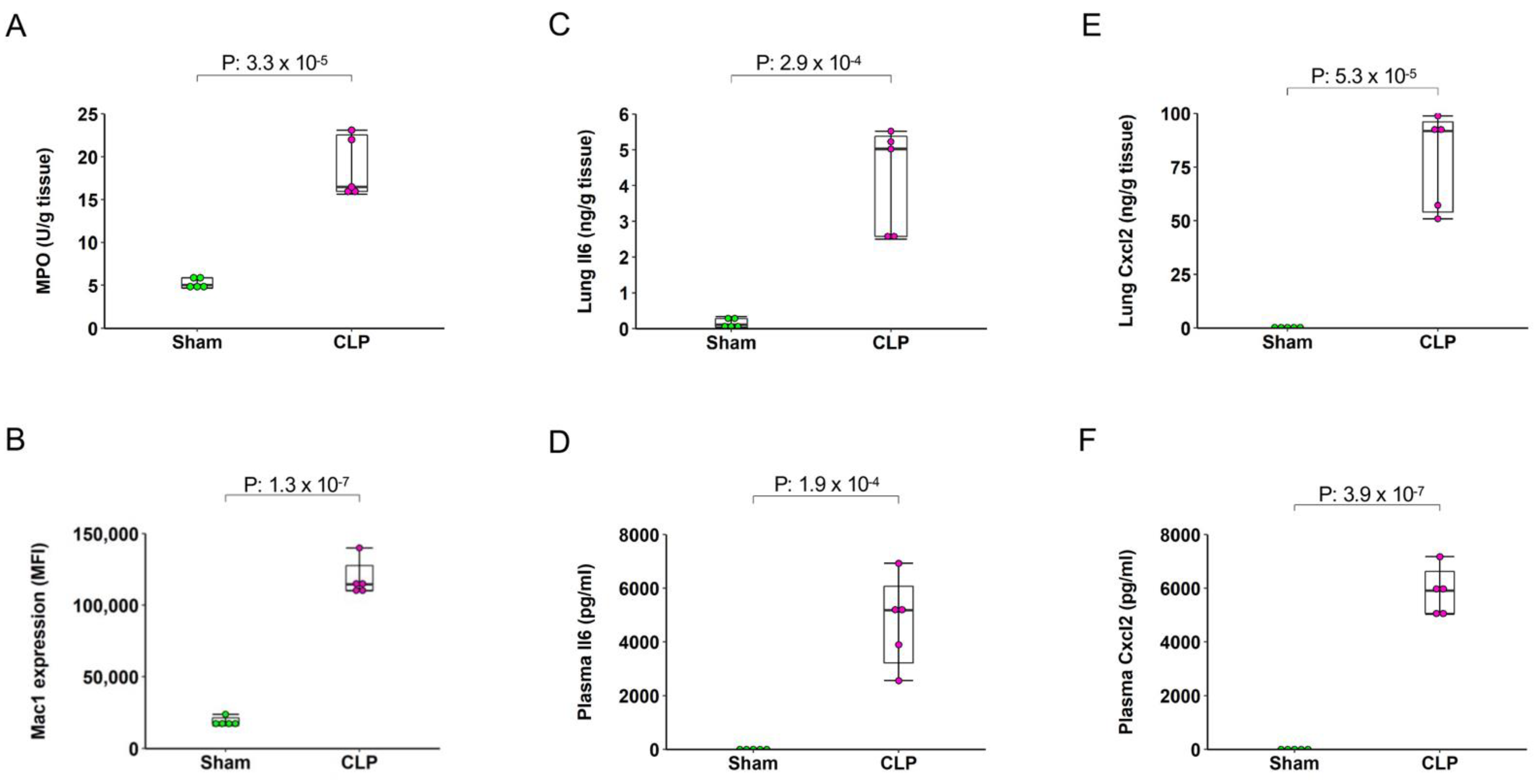
2.2. Sepsis Induces Pulmonary and Systemic Inflammation
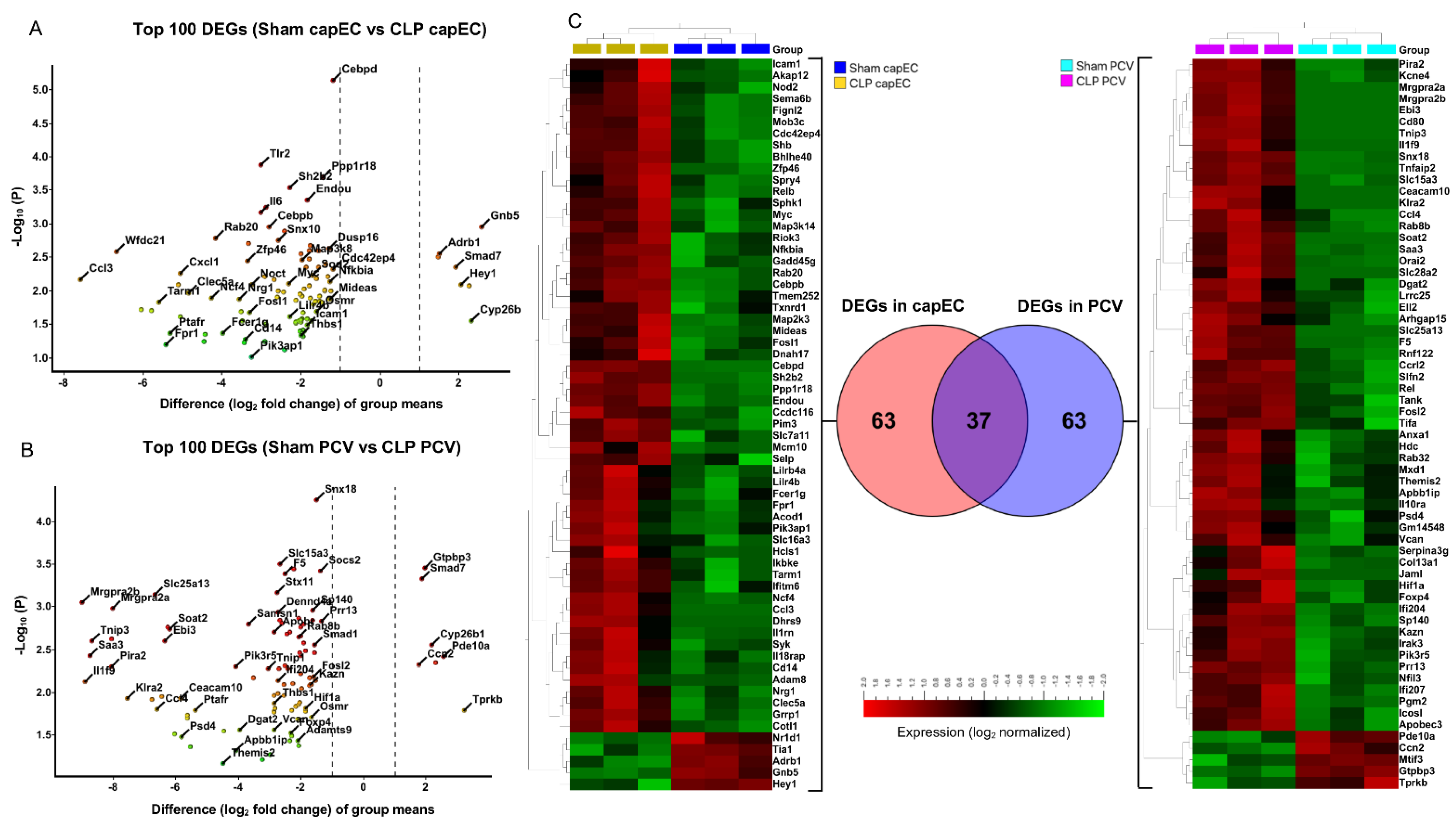
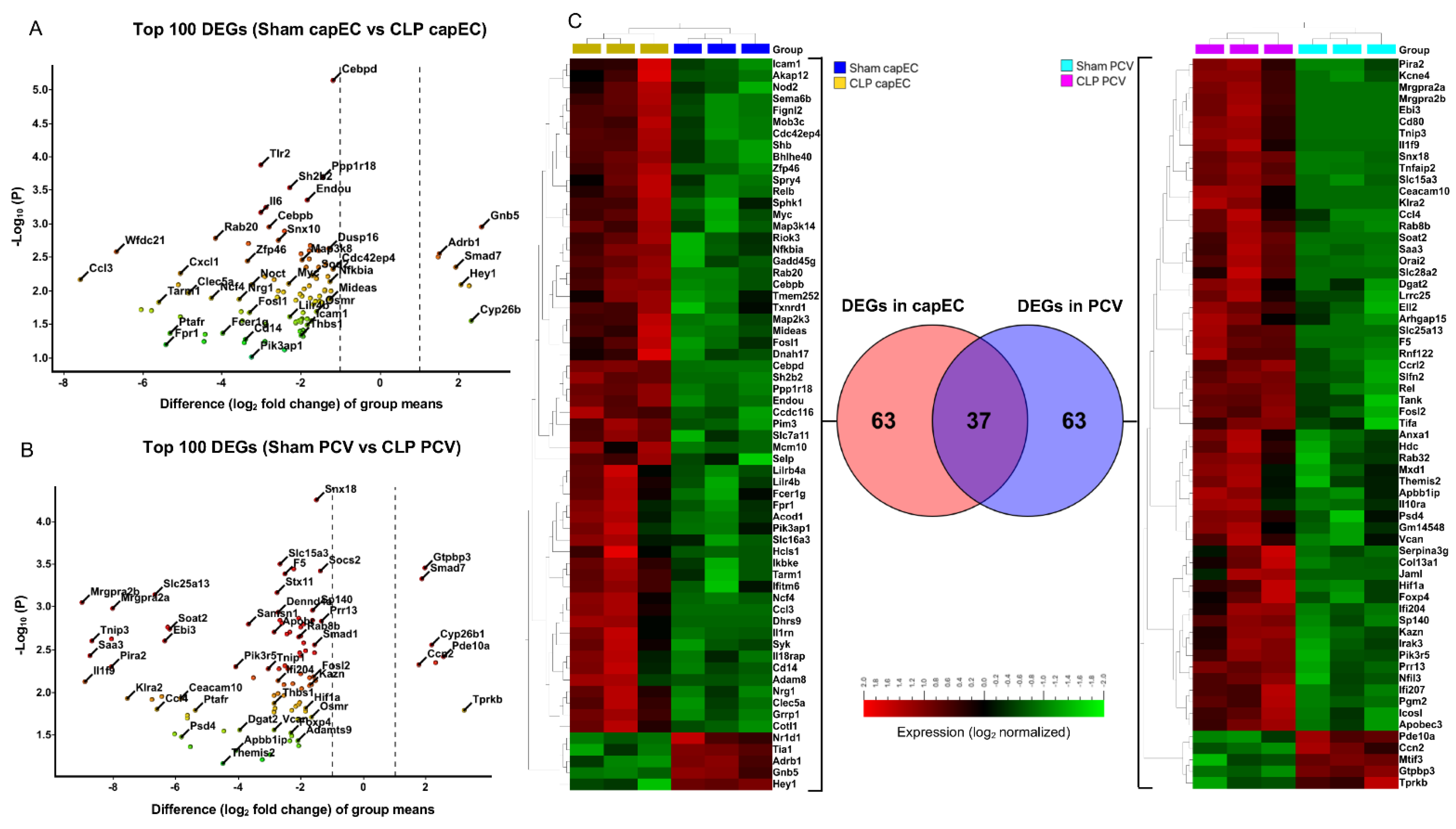
2.3. Differential Gene Expression between capEC and PCV during Sepsis
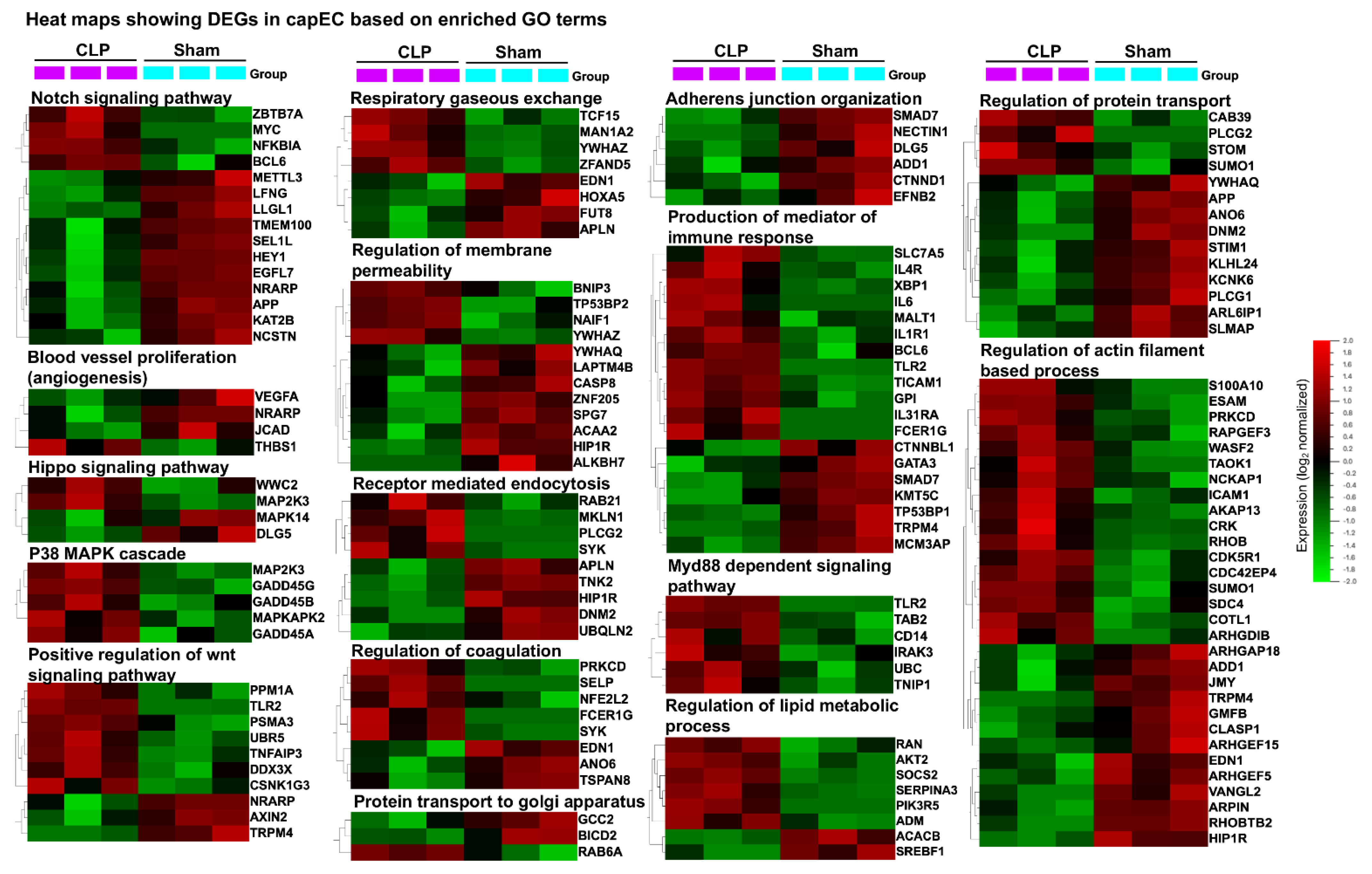
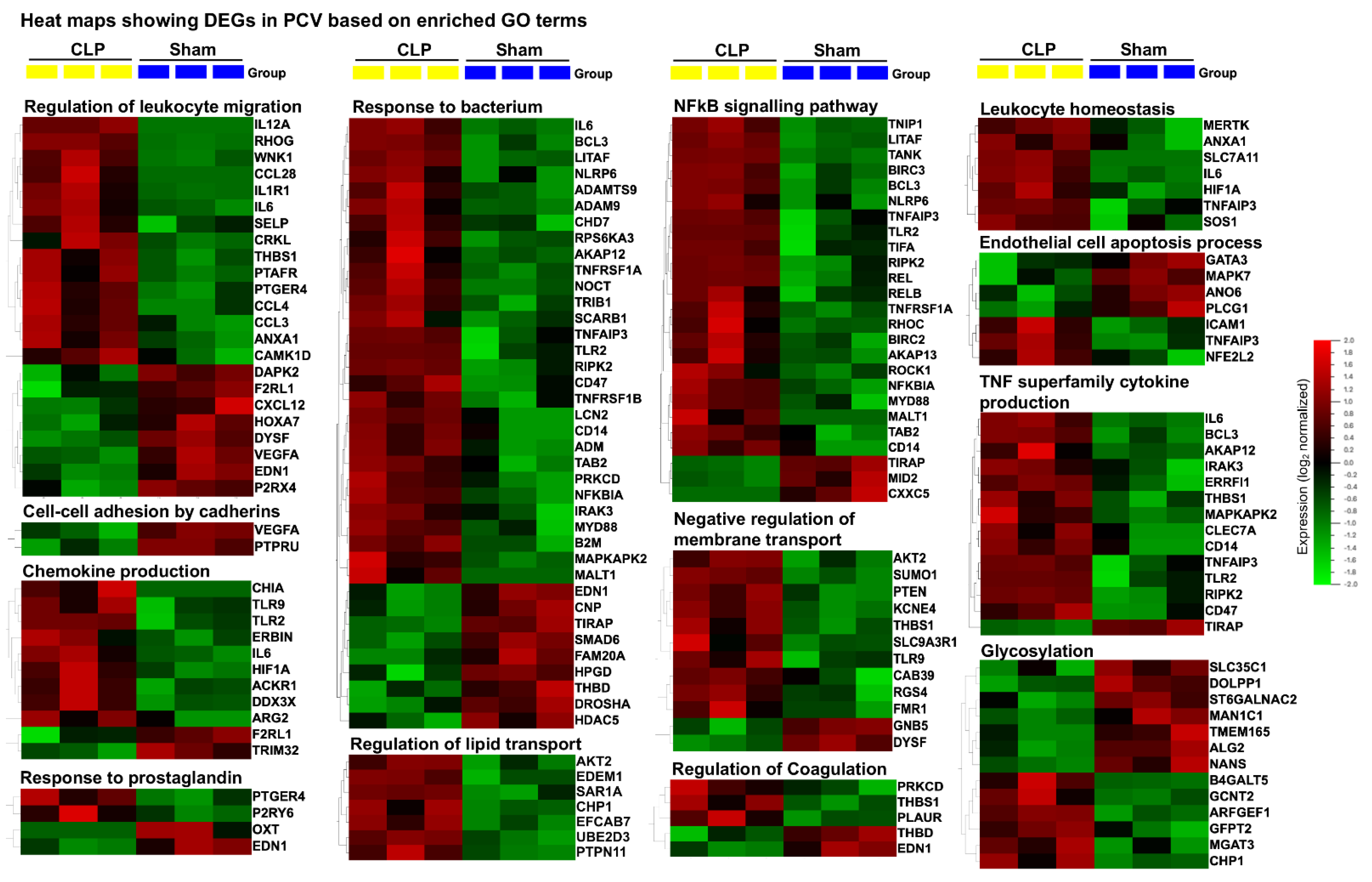
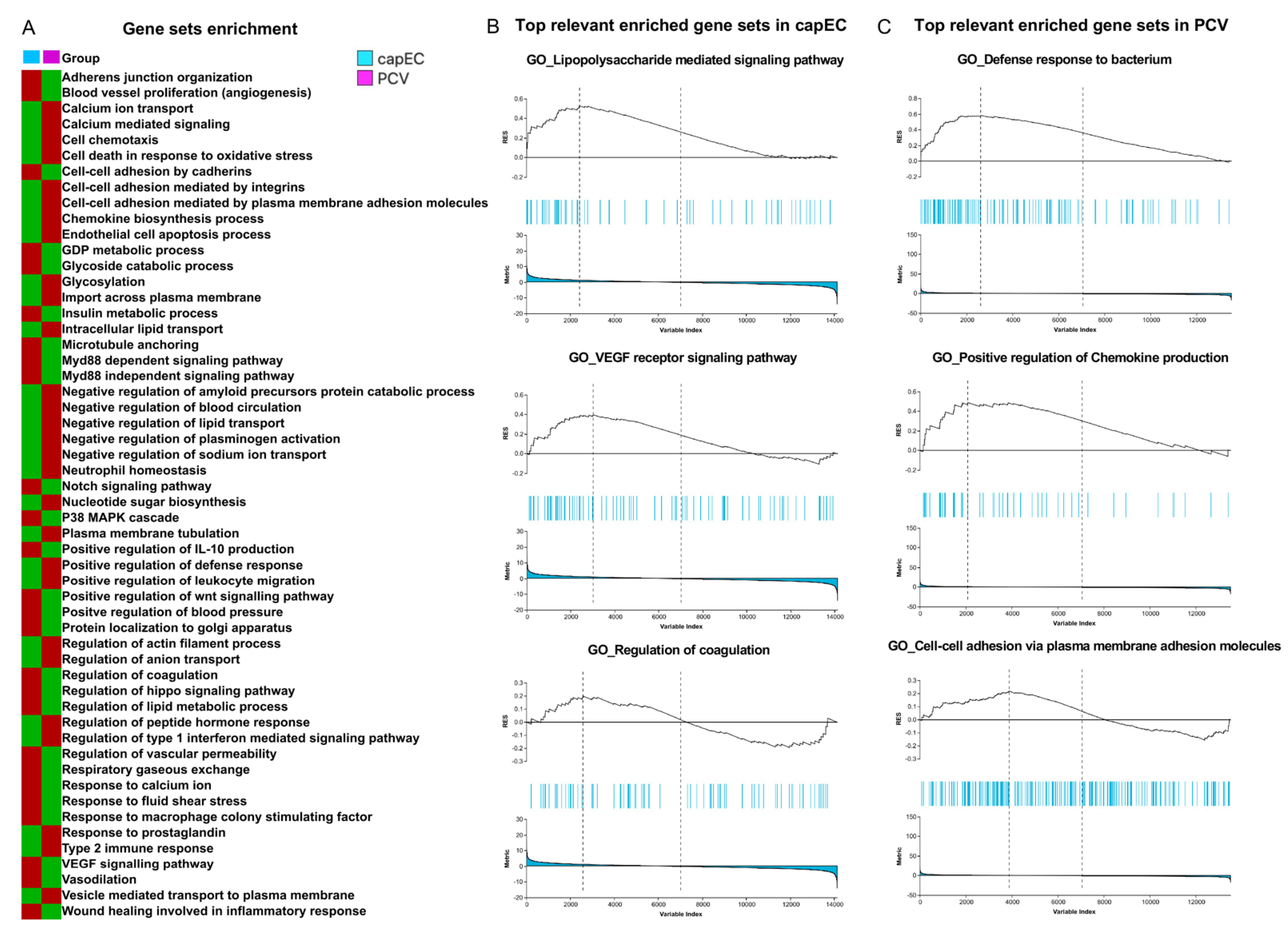
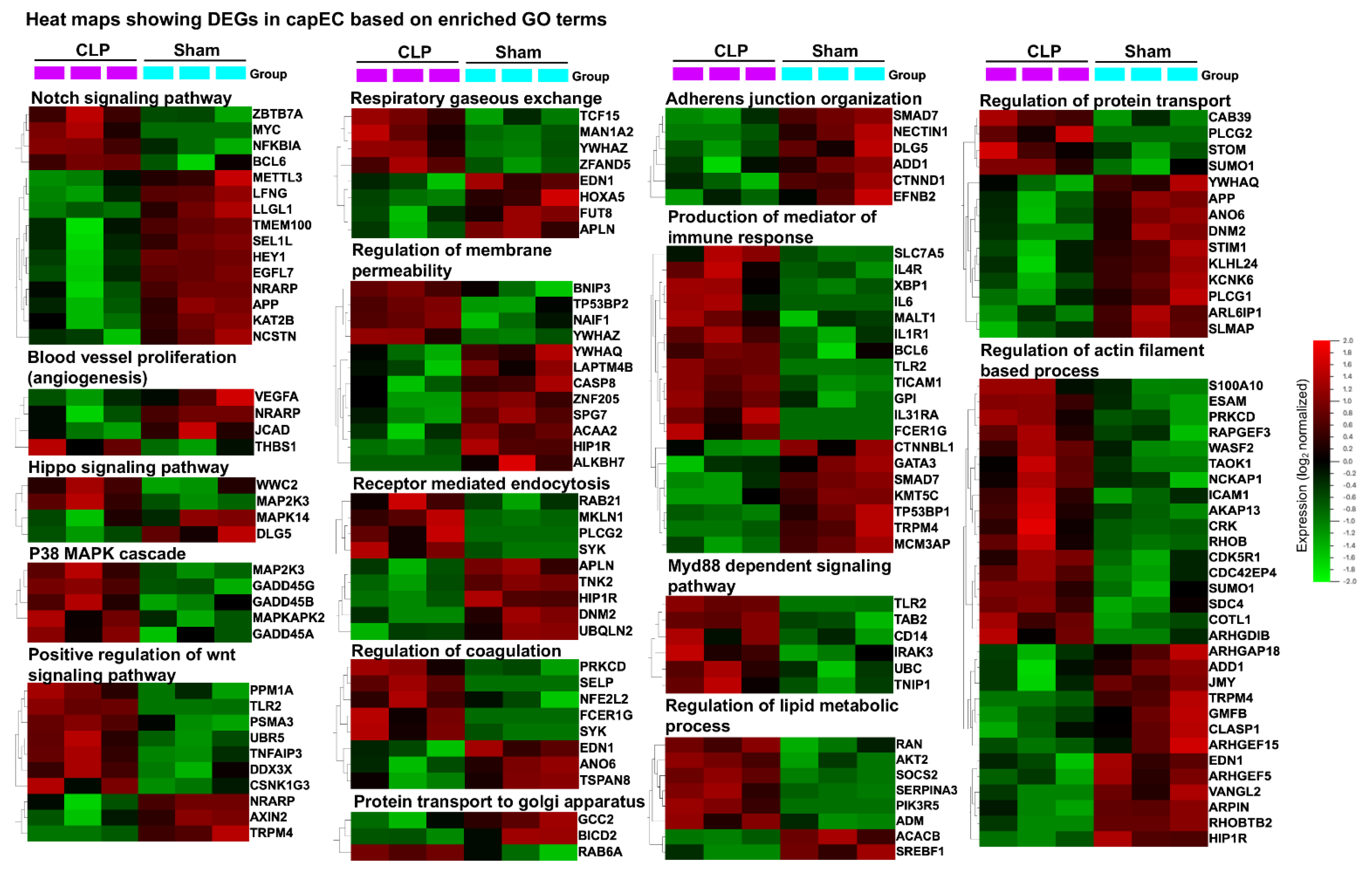
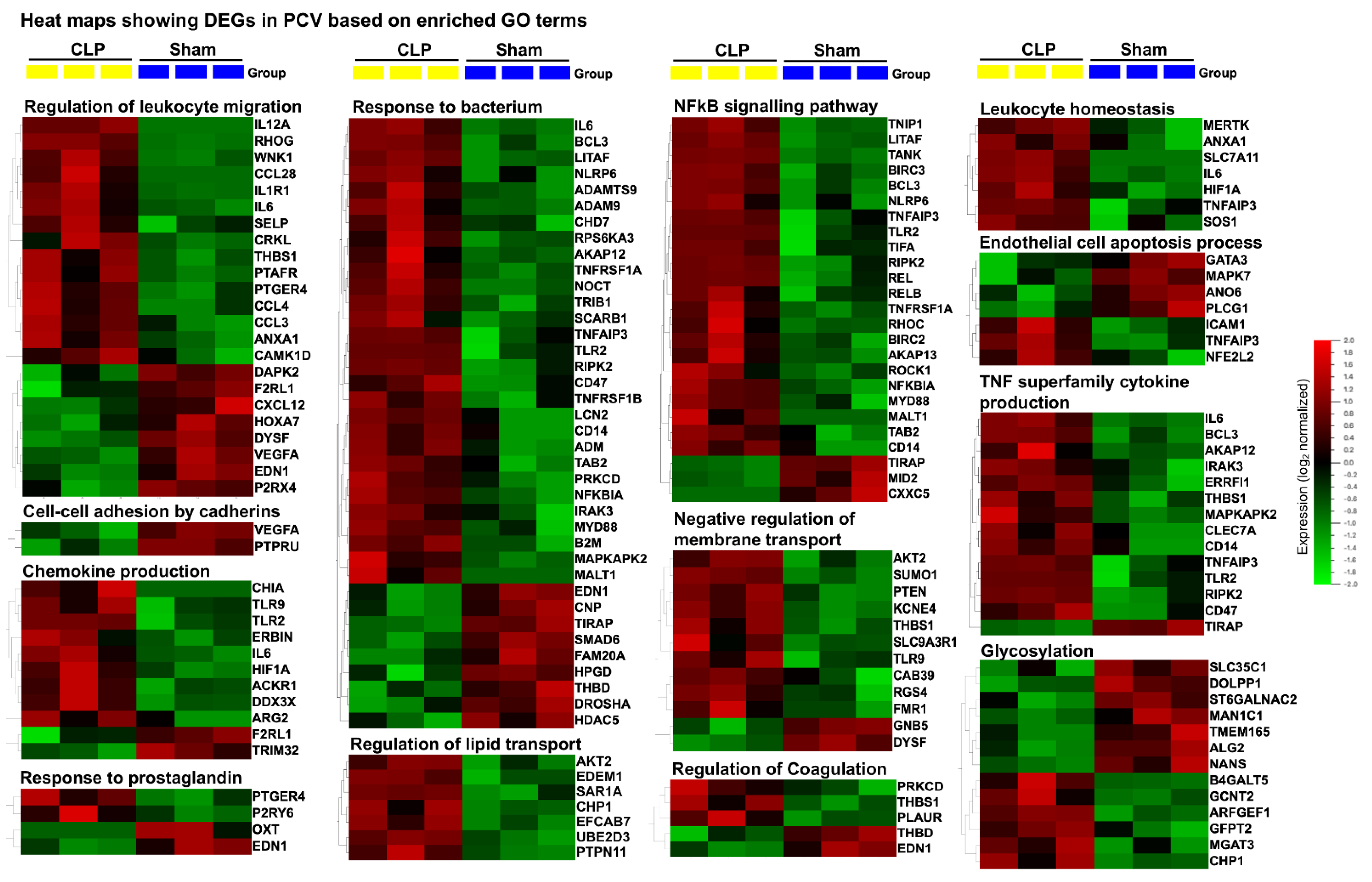
2.4. Distinct Gene Sets and Pathways Enrichment between capEC and PCV
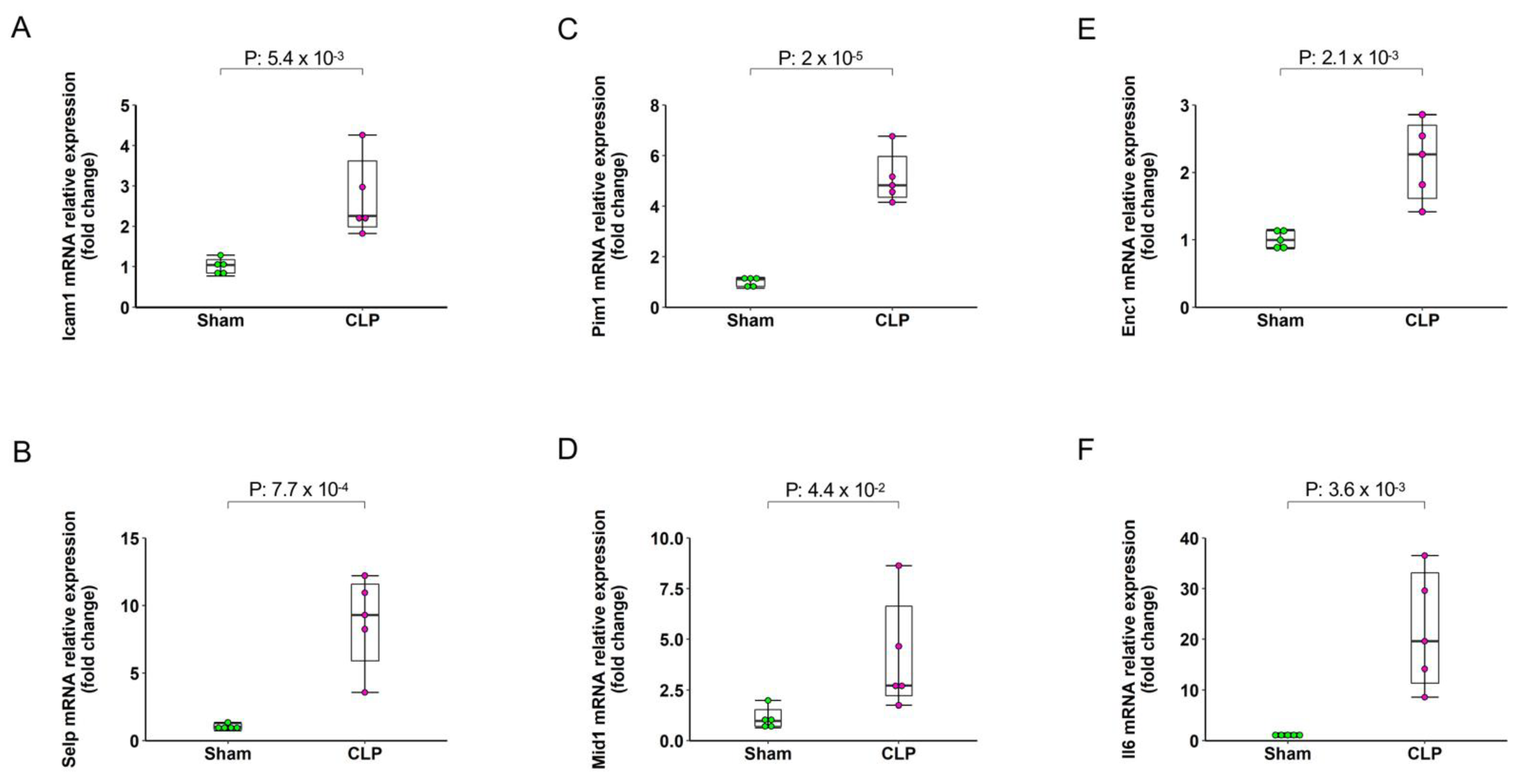
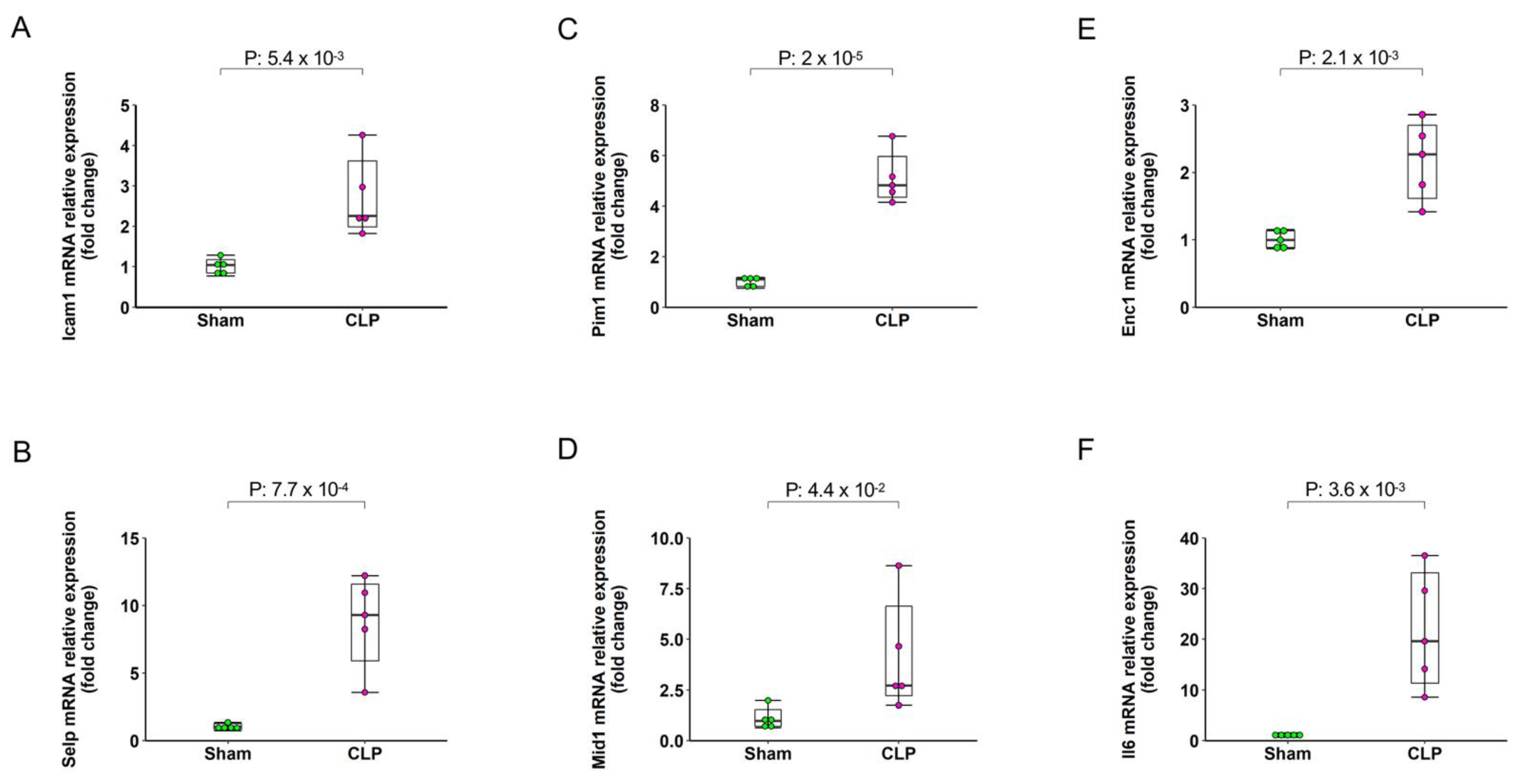
2.5. Validation by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Endothelial Subsets Sorting and Total RNA Extraction
4.3. RNA Sequencing and Data Analysis
4.4. Flow Cytometry
4.5. Myeloperoxidase (MPO) Assay
4.6. ELISA
4.7. Gene Sets Enrichment Analysis and KEGG Pathway Analysis
4.8. Validation by qRT-PCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Robson, W.P.; Daniel, R. The Sepsis Six: Helping patients to survive sepsis. Br. J. Nurs. 2008, 17, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascon, G.A.; Hernandez, G.; Murray, P.; De Backer, D.; Workgroup, A.X. The Endothelium in Sepsis. Shock 2016, 45, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef]
- Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef]
- Ploppa, A.; Schmidt, V.; Hientz, A.; Reutershan, J.; Haeberle, H.A.; Nohe, B. Mechanisms of leukocyte distribution during sepsis: An experimental study on the interdependence of cell activation, shear stress and endothelial injury. Crit. Care 2010, 14, R201. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Roller, J.; Menger, M.D.; Thorlacius, H. Sepsis-induced leukocyte adhesion in the pulmonary microvasculature in vivo is mediated by CD11a and CD11b. Eur. J. Pharmacol. 2013, 702, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kubes, P. Molecular mechanisms of leukocyte recruitment: Organ-specific mechanisms of action. Thromb. Haemost. 2003, 89, 213–220. [Google Scholar]
- Scarlatescu, E.; Juffermans, N.P.; Thachil, J. The current status of viscoelastic testing in septic coagulopathy. Thromb. Res. 2019, 183, 146–152. [Google Scholar] [CrossRef]
- Lupu, F.; Kinasewitz, G.; Dormer, K. The role of endothelial shear stress on haemodynamics, inflammation, coagulation and glycocalyx during sepsis. J. Cell Mol. Med. 2020, 24, 12258–12271. [Google Scholar] [CrossRef]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar] [CrossRef]
- Lehr, H.A.; Bittinger, F.; Kirkpatrick, C.J. Microcirculatory dysfunction in sepsis: A pathogenetic basis for therapy? J. Pathol. 2000, 190, 373–386. [Google Scholar] [CrossRef]
- Pries, A.R.; Secomb, T.W. Modeling structural adaptation of microcirculation. Microcirculation 2008, 15, 753–764. [Google Scholar] [CrossRef]
- Friederici, H.H. On the diaphragm across fenestrae of capillary endothelium. J. Ultrastruct. Res. 1969, 27, 373–375. [Google Scholar] [CrossRef]
- Lee, M.; Kiefel, H.; LaJevic, M.D.; Macauley, M.S.; Kawashima, H.; O′Hara, E.; Pan, J.; Paulson, J.C.; Butcher, E.C. Transcriptional programs of lymphoid tissue capillary and high endothelium reveal control mechanisms for lymphocyte homing. Nat. Immunol. 2014, 15, 982–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, E.L.; Ridger, V.; Ferraro, F.; Turmaine, M.; Saftig, P.; Cutler, D.F. CD63 is an essential cofactor to leukocyte recruitment by endothelial P-selectin. Blood 2011, 118, 4265–4273. [Google Scholar] [CrossRef] [Green Version]
- Brulois, K.; Rajaraman, A.; Szade, A.; Nordling, S.; Bogoslowski, A.; Dermadi, D.; Rahman, M.; Kiefel, H.; O′Hara, E.; Koning, J.J.; et al. A molecular map of murine lymph node blood vascular endothelium at single cell resolution. Nat. Commun. 2020, 11, 3798. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L.; Jones, G.; David, S.; Olariu, E.; Cadwell, K.K. Frequency and mortality of septic shock in Europe and North America: A systematic review and meta-analysis. Crit. Care 2019, 23, 196. [Google Scholar] [CrossRef] [Green Version]
- Freeman, B.D.; Natanson, C. Anti-inflammatory therapies in sepsis and septic shock. Expert Opin. Investig. Drugs 2000, 9, 1651–1663. [Google Scholar] [CrossRef]
- Ni, J.; Lin, M.; Jin, Y.; Li, J.; Guo, Y.; Zhou, J.; Hong, G.; Zhao, G.; Lu, Z. Gas6 Attenuates Sepsis-Induced Tight Junction Injury and Vascular Endothelial Hyperpermeability via the Axl/NF-kappaB Signaling Pathway. Front. Pharmacol. 2019, 10, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.T.; Tauseef, M.; Yue, L.; Bonini, M.G.; Gothert, J.; Shen, T.L.; Guan, J.L.; Predescu, S.; Sadikot, R.; Mehta, D. Conditional deletion of FAK in mice endothelium disrupts lung vascular barrier function due to destabilization of RhoA and Rac1 activities. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L291–L300. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.T.; Xiong, S.; Ye, Z.; Hong, Z.; Di, A.; Tsang, K.M.; Gao, X.; An, S.; Mittal, M.; Vogel, S.M.; et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Invest. 2017, 127, 4124–4135. [Google Scholar] [CrossRef]
- Jurasz, P.; Yurkova, N.; Kirshenbaum, L.; Stewart, D.J. VEGF masks BNIP3-mediated apoptosis of hypoxic endothelial cells. Angiogenesis 2011, 14, 199–207. [Google Scholar] [CrossRef]
- Lv, B.; Shi, T.; Wang, X.; Song, Q.; Zhang, Y.; Shen, Y.; Ma, D.; Lou, Y. Overexpression of the novel human gene, nuclear apoptosis-inducing factor 1, induces apoptosis. Int. J. Biochem. Cell Biol. 2006, 38, 671–683. [Google Scholar] [CrossRef]
- Yang, Y.; Fang, X.; Yang, R.; Yu, H.; Jiang, P.; Sun, B.; Zhao, Z. MiR-152 Regulates Apoptosis and Triglyceride Production in MECs via Targeting ACAA2 and HSD17B12 Genes. Sci. Rep. 2018, 8, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komarova, Y.; Malik, A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef]
- Vouret-Craviari, V.; Boquet, P.; Pouyssegur, J.; Van Obberghen-Schilling, E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: Role of Rho proteins in endothelial barrier function. Mol. Biol. Cell 1998, 9, 2639–2653. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Menden, H.L.; Korfhagen, T.R.; Kume, T.; Sampath, V. Endothelial immune activation programmes cell-fate decisions and angiogenesis by inducing angiogenesis regulator DLL4 through TLR4-ERK-FOXC2 signalling. J. Physiol. 2018, 596, 1397–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Yang, W.L.; Ochani, M.; Wang, P. Mitigation of sepsis-induced inflammatory responses and organ injury through targeting Wnt/beta-catenin signaling. Sci. Rep. 2017, 7, 9235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Kim, K.; Sheng, Y.; Cho, J.; Qian, Z.; Zhao, Y.Y.; Hu, G.; Pan, D.; Malik, A.B.; Hu, G. YAP Controls Endothelial Activation and Vascular Inflammation Through TRAF6. Circ. Res. 2018, 123, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lao, Y.; Yi, J.; Yang, J.; He, S.; Chen, Y. SENP3 in monocytes/macrophages up-regulates tissue factor and mediates lipopolysaccharide-induced acute lung injury by enhancing JNK phosphorylation. J. Cell Mol. Med. 2020, 24, 5454–5462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Tamura, T.; Sawatsubashi, Y. Sepsis and disseminated intravascular coagulation. J. Intensive Care 2016, 4, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallet, B. Bench-to-bedside review: Endothelial cell dysfunction in severe sepsis: A role in organ dysfunction? Crit Care 2003, 7, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Whyte, C.S.; Morrow, G.B.; Mitchell, J.L.; Chowdary, P.; Mutch, N.J. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J. Thromb. Haemost. 2020, 18, 1548–1555. [Google Scholar] [CrossRef]
- Lehr, H.A.; Arfors, K.E. Mechanisms of tissue damage by leukocytes. Curr. Opin. Hematol. 1994, 1, 92–99. [Google Scholar] [PubMed]
- Bozza, F.A.; Shah, A.M.; Weyrich, A.S.; Zimmerman, G.A. Amicus or adversary: Platelets in lung biology, acute injury, and inflammation. Am. J. Respir. Cell Mol. Biol. 2009, 40, 123–134. [Google Scholar] [CrossRef]
- Mantovani, A.; Bussolino, F.; Dejana, E. Cytokine regulation of endothelial cell function. FASEB J. 1992, 6, 2591–2599. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Yi, W.J.; Wan, X.J.; Wang, J.; Tao, T.Z.; Li, J.B.; Wang, J.F.; Deng, X.M. Blockade of ICAM-1 improves the outcome of polymicrobial sepsis via modulating neutrophil migration and reversing immunosuppression. Mediators Inflamm. 2014, 2014, 195290. [Google Scholar] [CrossRef]
- Croner, R.S.; Lehmann, T.G.; Fallsehr, C.; Herfarth, C.; Klar, E.; Kirschfink, M. C1-inhibitor reduces hepatic leukocyte-endothelial interaction and the expression of VCAM-1 in LPS-induced sepsis in the rat. Microvasc. Res. 2004, 67, 182–191. [Google Scholar] [CrossRef]
- Herter, J.M.; Rossaint, J.; Spieker, T.; Zarbock, A. Adhesion molecules involved in neutrophil recruitment during sepsis-induced acute kidney injury. J. Innate Immun. 2014, 6, 597–606. [Google Scholar] [CrossRef]
- Nolte, D.; Kuebler, W.M.; Muller, W.A.; Wolff, K.D.; Messmer, K. Attenuation of leukocyte sequestration by selective blockade of PECAM-1 or VCAM-1 in murine endotoxemia. Eur. Surg. Res. 2004, 36, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Matsukawa, A.; Lukacs, N.W.; Hogaboam, C.M.; Knibbs, R.N.; Bullard, D.C.; Kunkel, S.L.; Stoolman, L.M. Mice genetically lacking endothelial selectins are resistant to the lethality in septic peritonitis. Exp. Mol. Pathol. 2002, 72, 68–76. [Google Scholar] [CrossRef]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Coenen, D.M.; Mastenbroek, T.G.; Cosemans, J. Platelet interaction with activated endothelium: Mechanistic insights from microfluidics. Blood 2017, 130, 2819–2828. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.B. Glycan-dependent leukocyte adhesion and recruitment in inflammation. Curr. Opin. Cell Biol. 2003, 15, 531–538. [Google Scholar] [CrossRef]
- Aoki, T.; Suzuki, Y.; Nishio, K.; Suzuki, K.; Miyata, A.; Iigou, Y.; Serizawa, H.; Tsumura, H.; Ishimura, Y.; Suematsu, M.; et al. Role of CD18-ICAM-1 in the entrapment of stimulated leukocytes in alveolar capillaries of perfused rat lungs. Am. J. Physiol. 1997, 273, H2361–H2371. [Google Scholar] [CrossRef]
- Miranda, M.; Balarini, M.; Caixeta, D.; Bouskela, E. Microcirculatory dysfunction in sepsis: Pathophysiology, clinical monitoring, and potential therapies. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H24–H35. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.A.; Jones, W.K. NF-kappaB action in sepsis: The innate immune system and the heart. Front. Biosci. 2004, 9, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basilio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-kappaB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Hattori, Y.; Jesmin, S.; Gando, S. Nuclear factor-kappaB decoy oligodeoxynucleotides prevent acute lung injury in mice with cecal ligation and puncture-induced sepsis. Mol. Pharmacol. 2005, 67, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Chachami, G.; Simos, G. Specific Inhibition of HIF Activity: Can Peptides Lead the Way? Cancers 2021, 13, 410. [Google Scholar] [CrossRef]
- Inamoto, I.; Shin, J.A. Peptide therapeutics that directly target transcription factors. Pept. Sci. 2019, 111, e24048. [Google Scholar] [CrossRef] [Green Version]
- Du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, M.; Ode, Y.; Zhou, M.; Ochani, M.; Holodick, N.E.; Rothstein, T.L.; Wang, P. B-1a cells protect mice from sepsis-induced acute lung injury. Mol. Med. 2018, 24, 26. [Google Scholar] [CrossRef] [Green Version]
- Asaduzzaman, M.; Lavasani, S.; Rahman, M.; Zhang, S.; Braun, O.O.; Jeppsson, B.; Thorlacius, H. Platelets support pulmonary recruitment of neutrophils in abdominal sepsis. Crit. Care Med. 2009, 37, 1389–1396. [Google Scholar] [CrossRef]
- Schierwagen, C.; Bylund-Fellenius, A.C.; Lundberg, C. Improved method for quantification of tissue PMN accumulation measured by myeloperoxidase activity. J. Pharmacol. Methods 1990, 23, 179–186. [Google Scholar] [CrossRef]
- Mullane, K.M.; Kraemer, R.; Smith, B. Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J. Pharmacol. Methods 1985, 14, 157–167. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, B.T.; Huang da, W.; Tan, Q.; Guo, Y.; Bour, S.; Liu, D.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID Knowledgebase: A gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinform. 2007, 8, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M. Molecular network analysis of diseases and drugs in KEGG. Methods Mol. Biol. 2013, 939, 263–275. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathway Term | Gene Count | p-Value | Fold Enrichment | Bonferroni | FDR |
|---|---|---|---|---|---|
| mmu04668: TNF signaling pathway | 22 | 4.5 × 10−12 | 6.8 | 1.0 × 10−9 | 8.5 × 10−10 |
| mmu04621: NOD-like receptor signaling pathway | 10 | 3.7 × 10−5 | 5.9 | 8.4 × 10−3 | 1.7 × 10−3 |
| mmu04620: Toll-like receptor signaling pathway | 13 | 3.8 × 10−5 | 4.3 | 8.5 × 10−3 | 1.7 × 10−3 |
| mmu04210: Apoptosis | 10 | 5.7 × 10−5 | 5.6 | 0.012 | 2.1 × 10−3 |
| mmu04064: NF-kappa B signaling pathway | 12 | 1.2 × 10−4 | 4.1 | 0.027 | 3.3 × 10−3 |
| mmu04060: Cytokine-cytokine receptor interaction | 18 | 7.8 × 10−4 | 2.5 | 0.161 | 0.011 |
| mmu04010: MAPK signaling pathway | 17 | 3.0 × 10−3 | 2.2 | 0.49 | 0.028 |
| mmu04115: p53 signaling pathway | 8 | 3.4 × 10−3 | 4.0 | 0.53 | 0.028 |
| mmu04350: TGF-beta signaling pathway | 9 | 3.4 × 10−3 | 3.5 | 0.54 | 0.028 |
| mmu04062: Chemokine signaling pathway | 14 | 5.2 × 10−3 | 2.4 | 0.68 | 0.037 |
| KEGG Pathway Term | Gene Count | p-Value | Fold Enrichment | Bonferroni | FDR |
|---|---|---|---|---|---|
| mmu04668: TNF signaling pathway | 20 | 1.1 × 10−10 | 6.5 | 2.5 × 10−8 | 2.27 × 10−8 |
| mmu04064: NF-kappa B signaling pathway | 14 | 2.5 × 10−6 | 5.1 | 5.6 × 10−4 | 1.2 × 10−4 |
| mmu04620: Toll-like receptor signaling pathway | 14 | 4.1 × 10−6 | 4.9 | 8.8 × 10−4 | 1.5 × 10−4 |
| mmu04621: NOD-like receptor signaling pathway | 10 | 2.4 × 10−5 | 6.2 | 5.3 × 10−3 | 6.7 × 10−4 |
| mmu04060: Cytokine-cytokine receptor interaction | 20 | 4.3 × 10−5 | 2.9 | 9.3 × 10−3 | 1.0 × 10−3 |
| mmu04210: Apoptosis | 8 | 1.3 × 10−3 | 4.7 | 0.249 | 0.016 |
| mmu04514: Cell adhesion molecules (CAMs) | 13 | 1.8 × 10−3 | 2.8 | 0.335 | 0.022 |
| mmu04630: JAK-STAT signaling pathway | 11 | 7.3 × 10−3 | 2.7 | 0.794 | 0.065 |
| mmu04066: HIF-1 signaling pathway | 9 | 7.5 × 10−3 | 3.1 | 0.805 | 0.065 |
| mmu04350: TGF-beta signaling pathway | 8 | 9.4 × 10−3 | 3.3 | 0.871 | 0.072 |
| GSEA Name of TFT | DEGs (p < 0.05) |
|---|---|
| capEC | |
| GSEA_CREL_01 | BCL3, BIRC3, CASKIN1, CDC42SE1, CLDN5, CLOCK, COL11A2, CTDSP1, CYP2D6, FUT7, GPBP1, IER3, IER5, ITPKC, MAP3K8, MIDEAS, MOB3C, NFKBIA, PIK3R3, RAP2C, RBMS1, REL, RELB, S100A10, SDC4, STAT6, TACC1, TCF7L2, UBE2D3, UPF2, YWHAQ, YWHAZ, ZFAND3, ZNF593 |
| GSEA_NFKAPPAB65_01 | BCL3, BIRC3, CDC42SE1, CLDN5, CLOCK, COL11A2, CTDSP1, CYP2D6, FGF17, FUT7, GPBP1, HSP90B1, IER3, IER5, ILK, ITPKC, MAP3K8, MIDEAS, MOB3C, NFKBIA, NFKBIB, RAP2C, RBMS1, REL, RELB, RND1, SDC4, STAT6, UBE2D3, UPF2, YWHAQ, YWHAZ |
| GSEA_ZNF791_TARGET_GENES | ADA, BRWD1, CWC25, CYP2D6, FES, GIT2, RND1, SDC4, STAT6, ZNF827 |
| GSEA_TTGCWCAAY_CEBPB_02 | BNC2, CEBPB, HOXA5, KLF5, MIDEAS, NFKBIE, NRP2, RHOB, SLC7A11, TRIB1, VCAN |
| MIR649 | ACER3, BICRAL, CACNA1H, CCDC6, GAPVD1, INPP5K, KPNA1, MEX3C, MOB1B, NUFIP2, RFX7, SENP5, STT3A, ZNF281 |
| PCV | |
| GSEA_GATA6_01 | ACKR1, BCL6, CTCF, ECHDC2, ERRFI1, FBRS, FOSL2, GPBP1, IGFBP5, KIZ, LHX6, LIMS1, LIX1, MID1, NOCT, NRAS, PCDH9, PFKFB1, PLAGL2, PSD4, SALL2, SLC39A14, SMAD5, TACC1, TGIF1, TGIF2, TPH2, UBE2H, WAPL, XPO6 |
| GSEA_TTGCWCAAY_CEBPB_02 | CSDE1, DBH, GSR, IDH1, IP6K2, MIDEAS, NFKBIE, RHOB, TRIB1, VCAN |
| GSEA_MIR4769_3P | BCL6, CCDC6, ERBIN, NFKBIE, PAK2, PTEN, WAPL |
| GSEA_MIR362_5P | ATF1, CPEB4, EDEM1, IDH1, LARP1B, PEG10, PLAGL2, RBM27, RPS6KB1, SNX18, SYNC, ADAM9, ATF1 |
| GSEA_CCTGAGT_MIR510 | CAPZA2, IP6K2, WAPL, ZHX2 |
| Mouse Gene Name | Primer Sequence (5′ -> 3′) | Length | Tm | Location | |
|---|---|---|---|---|---|
| Icam1 | Forward Primer | GTGATGCTCAGGTATCCATCCA | 22 | 61.3 | 74–95 |
| Reverse Primer | CACAGTTCTCAAAGCACAGCG | 21 | 62.0 | 286–266 | |
| Mid1 | Forward Primer | CTGTGACGGCACCTGTCTC | 19 | 62.3 | 1293–1311 |
| Reverse Primer | AAACGGCTGACTGTTGGTCTT | 21 | 62.2 | 1497–1477 | |
| Pim1 | Forward Primer | CTGGAGTCGCAGTACCAGG | 19 | 61.4 | 100–118 |
| Reverse Primer | CAGTTCTCCCCAATCGGAAATC | 22 | 60.7 | 240–219 | |
| Enc1 | Forward Primer | CTGTTTCATAAGTCCTCCTACGC | 23 | 60.4 | 64–86 |
| Reverse Primer | CACCACTGAACATGGCTTCG | 20 | 61.3 | 232–213 | |
| Il6 | Forward Primer | CCAAGAGGTGAGTGCTTCCC | 20 | 62.2 | 462–481 |
| Reverse Primer | CTGTTGTTCAGACTCTCTCCCT | 22 | 60.8 | 579–558 | |
| Selp | Forward Primer | CATCTGGTTCAGTGCTTTGATCT | 23 | 60.5 | 69–91 |
| Reverse Primer | ACCCGTGAGTTATTCCATGAGT | 22 | 60.8 | 173–152 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.; Ding, Z.; Rönnow, C.-F.; Thorlacius, H. Transcriptomic Analysis Reveals Differential Expression of Genes between Lung Capillary and Post Capillary Venules in Abdominal Sepsis. Int. J. Mol. Sci. 2021, 22, 10181. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910181
Rahman M, Ding Z, Rönnow C-F, Thorlacius H. Transcriptomic Analysis Reveals Differential Expression of Genes between Lung Capillary and Post Capillary Venules in Abdominal Sepsis. International Journal of Molecular Sciences. 2021; 22(19):10181. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910181
Chicago/Turabian StyleRahman, Milladur, Zhiyi Ding, Carl-Fredrik Rönnow, and Henrik Thorlacius. 2021. "Transcriptomic Analysis Reveals Differential Expression of Genes between Lung Capillary and Post Capillary Venules in Abdominal Sepsis" International Journal of Molecular Sciences 22, no. 19: 10181. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910181