
Growth Factors, PI3K/AKT/mTOR and MAPK Signaling Pathways in Colorectal Cancer Pathogenesis: Where Are We Now?

 and
and 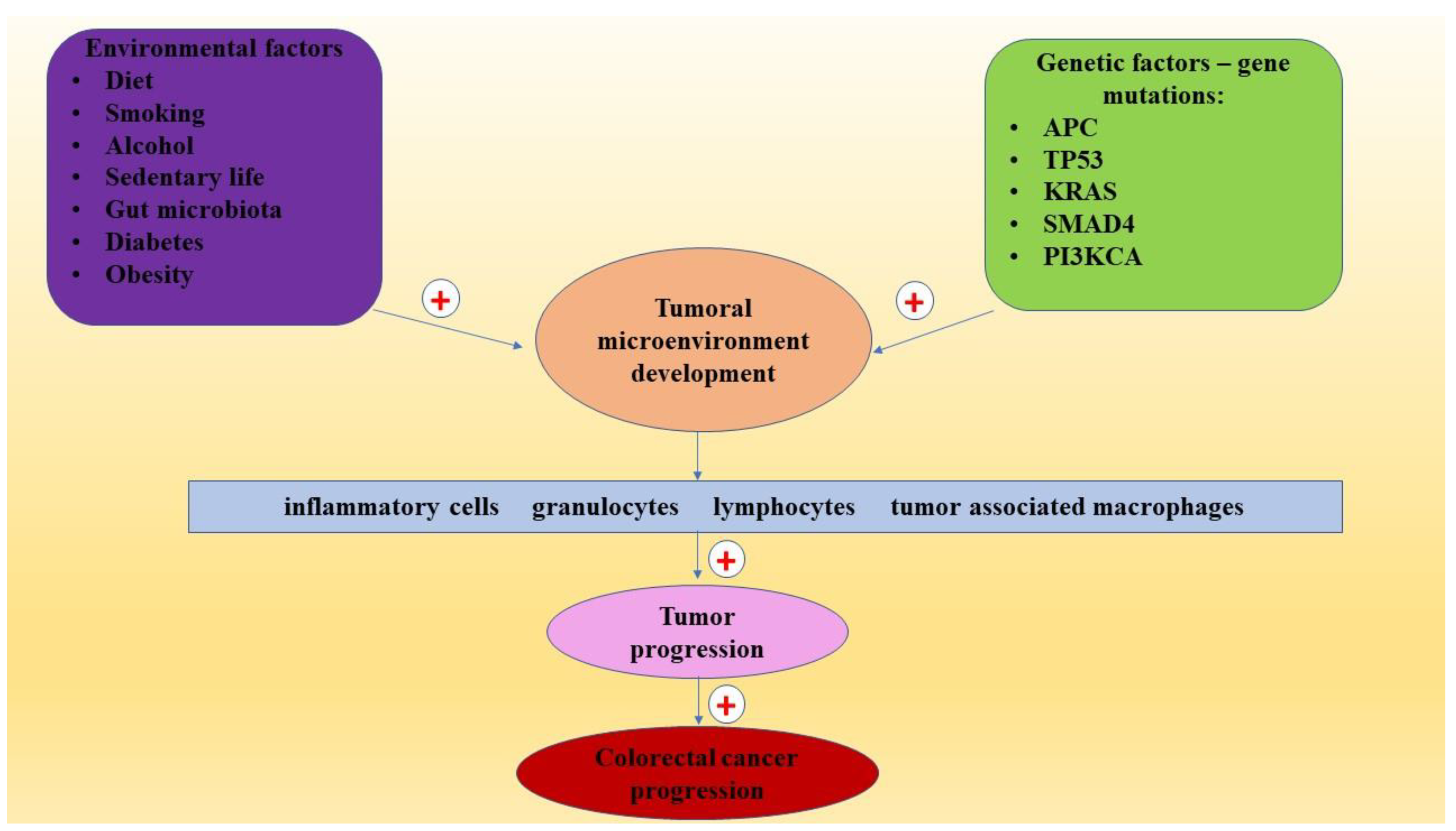{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Risk Factors in CRC
3. CRC Pathogenesis
3.1. CRC and Insulin-Like Growth Factor Family (IGF)
3.2. CRC and Epidermal Growth Factor (EGF)
3.3. Colorectal Cancer and Vascular Endothelial Growth Factor (VEGF)
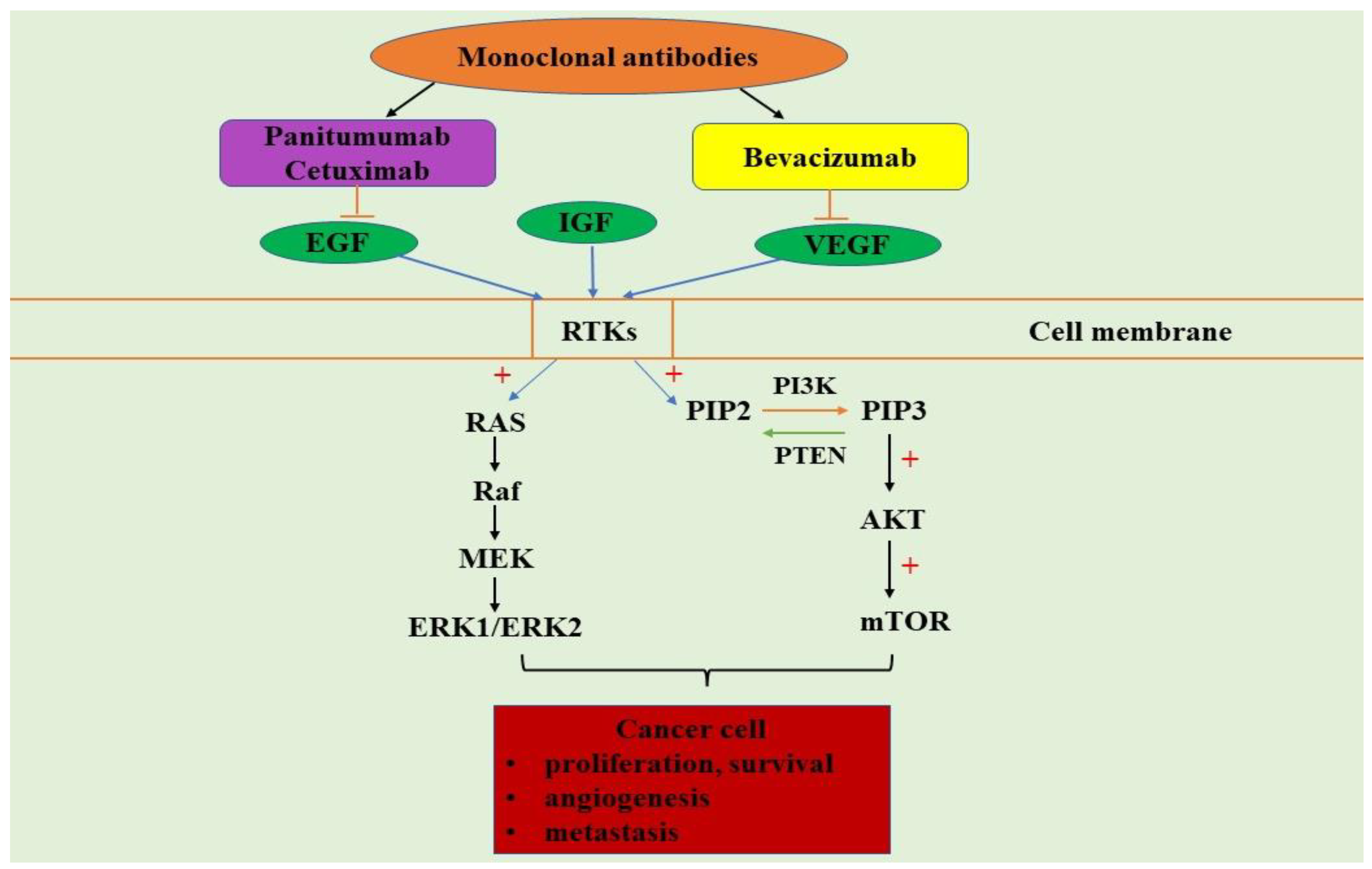
4. PI3K/AKT/mTOR and MAPK Signaling Pathways in Colorectal Cancer
5. PI3K/AKT/mTOR and MAPK Signaling Pathways Inhibitors
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 5, 15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balhareth, A.; Aldossary, M.Y.; McNamara, D. Impact of physical activity and diet on colorectal cancer survivors’ quality of life: A systematic review. World J. Surg. Oncol. 2019, 31, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Yu, M.; Qin, J.; Luo, Y.; Zhong, M. LACTB regulates PIK3R3 to promote autophagy and inhibit EMT and proliferation through the PI3K/AKT/mTOR signaling pathway in colorectal cancer. Cancer Manag. Res. 2020, 30, 5181–5200. [Google Scholar] [CrossRef]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 26, 1490–1502. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Einarsdottir, H.M.; Smaradottir, A.; Gunnlaugsson, A.; Halfdanarson, T.R. Colorectal cancer—Review. Laeknabladid 2014, 100, 75–82. [Google Scholar]
- Navarro, M.; Nicolas, A.; Ferrandez, A.; Lanas, A. Colorectal cancer population screening programs worldwide in 2016: An update. World J. Gastroenterol. 2017, 28, 3632–3642. [Google Scholar] [CrossRef]
- Onyoh, E.F.; Hsu, W.F.; Chang, L.C.; Lee, Y.C.; Wu, M.S.; Chiu, H.M. The rise of colorectal cancer in Asia: Epidemiology, screening, and management. Curr. Gastroenterol. Rep. 2019, 10, 36. [Google Scholar] [CrossRef]
- Bardou, M.; Barkun, A.N.; Martel, M. Obesity and colorectal cancer. Gut 2013, 62, 933–947. [Google Scholar] [CrossRef]
- Schulpen, M.; van den Brandt, P.A. Mediterranean diet adherence and risk of colorectal cancer: The prospective Netherlands Cohort Study. Eur. J. Epidemiol. 2020, 35, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.G.; Marsoni, S.; Bardelli, A.; Siena, S. Early-onset colorectal cancer in young individuals. Mol. Oncol. 2019, 13, 109–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connell, L.C.; Mota, J.M.; Braghiroli, M.I.; Hoff, P.M.; Connell, L.C.; Mota, J.M.; Braghiroli, M.I.; Hoff, P.M. The rising incidence of younger patients with colorectal cancer: Questions about screening, biology, and treatment. Curr. Treat. Options Oncol. 2017, 18, 23. [Google Scholar] [CrossRef]
- Weinberg, B.A.; Marshall, J.L.; Salem, M.E. The Growing challenge of young adults with colorectal cancer. Oncology 2017, 15, 381–389. [Google Scholar]
- O’Sullivan, D.E.; Hilsden, R.J.; Ruan, Y.; Forbes, N.; Heitman, S.J.; Brenner, D.R. The incidence of young-onset colorectal cancer in Canada continues to increase. Cancer Epidemiol. 2020, 69, 101828. [Google Scholar] [CrossRef]
- Siegel, R.; Desantis, C.; Jemal, A. Colorectal cancer statistics—2014. CA Cancer J. Clin. 2014, 64, 104–117. [Google Scholar] [CrossRef]
- Jeong, M.A.; Kang, H.W. Early-onset colorectal cancer. Korean J. Gastroenterol. 2019, 25, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Abualkhair, W.H.; Zhou, M.; Ahnen, D.; Yu, Q.; Wu, X.C.; Karlitz, J.J. Trends in incidence of early-onset colorectal cancer in the United States among those approaching screening age. JAMA Netw. Open 2020, 3, e1920407. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.E.; Paik, H.Y.; Yoon, H.; Lee, J.E.; Kim, N.; Sung, M.K. Sex- and gender-specific disparities in colorectal cancer risk. World J. Gastroenterol. 2015, 7, 5167–5175. [Google Scholar] [CrossRef]
- Modest, D.P.; Pant, S.; Sartore-Bianchi, A. Treatment sequencing in metastatic colorectal cancer. Eur. J. Cancer 2019, 109, 70–83. [Google Scholar] [CrossRef]
- Fornasier, G.; Francescon, S.; Baldo, P. An update of efficacy and safety of cetuximab in metastatic colorectal cancer: A narrative review. Adv. Ther. 2018, 35, 1497–1509. [Google Scholar] [CrossRef]
- Dzunic, M.; Petkovic, I.; Cvetanovic, A.; Vrbic, S.; Pejcic, I. Current and future targets and therapies in metastatic colorectal cancer. J. BUON 2019, 24, 1785–1792. [Google Scholar]
- Dienstmann, R.; Salazar, R.; Tabernero, J. Molecular subtypes and the evolution of treatment decisions in metastatic colorectal cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 23, 231–238. [Google Scholar] [CrossRef]
- Meng, S.; Jian, Z.; Yan, X.; Li, J.; Zhang, R. LncRNA SNHG6 inhibits cell proliferation and metastasis by targeting ETS1 via the PI3K/AKT/mTOR pathway in colorectal cancer. Mol. Med. Rep. 2019, 20, 2541–2548. [Google Scholar] [CrossRef] [Green Version]
- Cornish, A.J.; Tomlinson, I.P.M.; Houlston, R.S. Mendelian randomisation: A powerful and inexpensive method for identifying and excluding non-genetic risk factors for colorectal cancer. Mol. Asp. Med. 2019, 69, 41–47. [Google Scholar] [CrossRef]
- Shaw, E.; Farris, M.S.; Stone, C.R.; Derksen, J.W.G.; Johnson, R.; Hilsden, R.J.; Friedenreich, C.M.; Brenner, D.R. Effects of physical activity on colorectal cancer risk among family history and body mass index subgroups: A systematic review and meta-analysis. BMC Cancer 2018, 11, 71. [Google Scholar] [CrossRef]
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 19, 967–976. [Google Scholar]
- Song, M.; Garrett, W.S.; Chan, A.T. Nutrients, foods, and colorectal cancer prevention. Gastroenterology 2015, 148, 1244–1260. [Google Scholar] [CrossRef] [Green Version]
- Azeem, S.; Gillani, S.W.; Siddiqui, A.; Jandrajupalli, S.B.; Poh, V.; Syed, S.S.A. Diet and colorectal cancer risk in Asia—A systematic review. Asian Pac. J. Cancer Prev. 2015, 16, 5389–5396. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.C.; Prochaska, J.D.; Yu, X.; Kaul, S. An examination between census tract unhealthy food availability and colorectal cancer incidence. Cancer Epidemiol. 2020, 67, 101761. [Google Scholar] [CrossRef]
- Diallo, A.; Deschasaux, M.; Latino-Martel, P.; Hercberg, S.; Galan, P.; Fassier, P.; Allès, B.; Guéraud, F.; Pierre, F.H.; Touvier, M. Red and processed meat intake and cancer risk: Results from the prospective nutrinnet-sante cohort study. Int. J. Cancer 2018, 15, 230–237. [Google Scholar] [CrossRef]
- Martin, O.C.B.; Olier, M.; Ellero-Simatos, S.; Naud, N.; Dupuy, J.; Huc, L.; Taché, S.; Graillot, V.; Levêque, M.; Bézirard, V.; et al. Haem iron reshapes colonic luminal environment: Impact on mucosal homeostasis and microbiome through aldehyde formation. Microbiome 2019, 6, 72. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.D.; Lloyd, S.K. Association between red meat consumption and colon cancer: A systematic review of experimental results. Exp. Biol. Med. 2017, 242, 813–839. [Google Scholar] [CrossRef] [Green Version]
- Ekine-Afolabi, B.A.; Njan, A.A.; Rotimi SO, R.I.A.; Elbehi, A.M.; Cash, E.; Adeyeye, A. The impact of diet on the involvement of non-coding RNAs, extracellular vesicles, and gut microbiome-virome in colorectal cancer initiation and progression. Front. Oncol. 2020, 14, 583372. [Google Scholar] [CrossRef]
- Sánchez-Alcoholado, L.; Ordóñez, R.; Otero, A.; Plaza-Andrade, I.; Laborda-Illanes, A.; Medina, J.A.; Ramos-Molina, B.; Gómez-Millán, J.; Queipo-Ortuño, M.I. Gut microbiota-mediated inflammation and gut permeability in patients with obesity and colorectal cancer. Int. J. Mol. Sci. 2020, 16, 6782. [Google Scholar] [CrossRef]
- Farinetti, A.; Zurlo, V.; Manenti, A.; Coppi, F.; Mattioli, A.V. Mediterranean diet and colorectal cancer: A systematic review. Nutrition 2017, 43, 83–88. [Google Scholar] [CrossRef]
- Cai, L.; Bennedsen, A.L.B.; Qvortrup, C.; Gögenur, I. Increasing incidence of colorectal cancer in young patients. Ugeskr. Laeger. 2019, 30, V09190524. [Google Scholar]
- Gram, I.T.; Park, S.Y.; Wilkens, L.R.; Haiman, C.A.; Le Marchand, L. Smoking-related risks of colorectal cancer by anatomical subsite and sex. Am. J. Epidemiol. 2020, 1, 543–553. [Google Scholar] [CrossRef]
- Jung, Y.S.; Kim, N.H.; Yang, H.J.; Park, S.K.; Park, J.H.; Park, D.I.; Sohn, C.I. The impact of passive smoking on the risk of colorectal neoplasia in never, former, and current smokers. J. Gastroenterol. Hepatol. 2018, 3, 1023–1030. [Google Scholar] [CrossRef]
- Van Blarigan, E.L.; Meyerhardt, J.A. Role of physical activity and diet after colorectal cancer diagnosis. J. Clin. Oncol. 2015, 1, 1825–1834. [Google Scholar] [CrossRef] [Green Version]
- Baena, R.; Salinas, P. Diet and colorectal cancer. Maturitas 2015, 80, 258–264. [Google Scholar] [CrossRef]
- Ustundag, H.; Zengin, N.; Andsoy, I.I.; Gul, A. Awareness of health sciences students about colorectal cancer risk factors. Eur. J. Cancer Care 2019, 28, e13016. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, K.E.; Murphy, N.; Key, T.J. Diet and colorectal cancer in UK Biobank: A prospective study. Int. J. Epidemiol. 2020, 1, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Na, H.K.; Lee, J.Y. Molecular basis of alcohol-related gastric and colon cancer. Int. J. Mol. Sci. 2017, 24, 1116. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Jain, S. Rising incidence of colorectal cancer in patients younger than age 50 in Hawai’i. Hawaii J. Med. Public Health 2019, 78, 195–199. [Google Scholar]
- Cirillo, F.; Catellani, C.; Sartori, C.; Lazzeroni, P.; Amarri, S.; Street, M.E. Obesity, insulin resistance, and colorectal cancer: Could miRNA dysregulation play a role? Int. J. Mol. Sci. 2019, 14, 2922. [Google Scholar] [CrossRef] [Green Version]
- Pietrzyk, L.; Torres, A.; Maciejewski, R.; Torres, K. Obesity and obese-related chronic low-grade inflammation in promotion of colorectal cancer development. Asian Pac. J. Cancer Prev. 2015, 6, 4161–4168. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Useros, J.; Garcia-Foncillas, J. Obesity and colorectal cancer: Molecular features of adipose tissue. J. Transl. Med. 2016, 22, 21. [Google Scholar] [CrossRef] [Green Version]
- Ahechu, P.; Zozaya, G.; Martí, P.; Hernández-Lizoáin, J.L.; Baixauli, J.; Unamuno, X.; Frühbeck, G.; Catalán, V. NLRP3 inflammasome: A possible link between obesity-associated low-grade chronic inflammation and colorectal cancer development. Front. Immunol. 2018, 11, 2918. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, S.; Li, J.; Bao, W.; Zhang, P.; Huang, Y.; Ling, P.; Wang, Y.; Zhao, Q. Effects of high-fat diet-induced adipokines and cytokines on colorectal cancer development. FEBS Open Bio 2019, 9, 2117–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltani, G.; Poursheikhani, A.; Yassi, M.; Hayatbakhsh, A.; Kerachian, M.; Kerachian, M.A. Obesity, diabetes and the risk of colorectal adenoma and cancer. BMC Endocr. Disord. 2019, 29, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Gao, Z.; Huang, L.; Qin, H. Gut microbiota and colorectal cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yu, J. The association of diet, gut microbiota and colorectal cancer: What we eat may imply what we get. Protein Cell 2018, 9, 474–487. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Zhuang, J.; Wu, Y.; Wu, W.; Yang, X. Progress in research on colorectal cancer-related microorganisms and metabolites. Cancer Manag. Res. 2020, 21, 8703–8720. [Google Scholar] [CrossRef]
- Ganesan, K.; Jayachandran, M.; Xu, B. Diet-derived phytochemicals targeting colon cancer stem cells and microbiota in colorectal cancer. Int. J. Mol. Sci. 2020, 1, 3976. [Google Scholar] [CrossRef] [PubMed]
- Triantafillidis, J.K.; Nasioulas, G.; Kosmidis, P.A. Colorectal cancer and inflammatory bowel disease: Epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009, 29, 2727–2737. [Google Scholar] [PubMed]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61, 1500902. [Google Scholar] [CrossRef] [Green Version]
- Chapkin, R.S.; Navarro, S.L.; Hullar, M.A.J.; Lampe, J.W. Diet and gut microbes act coordinately to enhance programmed cell death and reduce colorectal cancer risk. Dig. Dis. Sci. 2020, 65, 840–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botteri, E.; Iodice, S.; Bagnardi, V.; Raimondi, S.; Lowenfels, A.B.; Maisonneuve, P. Smoking and colorectal cancer: A meta-analysis. JAMA 2008, 17, 2765–2778. [Google Scholar] [CrossRef]
- Li, S.; Ung, T.T.; Nguyen, T.T.; Sah, D.K.; Park, S.Y.; Jung, Y.D. Cholic Acid stimulates MMP-9 in human colon cancer cells via activation of MAPK, AP-1, and NF-kappaB Activity. Int. J. Mol. Sci. 2020, 12, 3420. [Google Scholar] [CrossRef]
- Kasprzak, A.; Adamek, A. Insulin-Like Growth Factor 2 (IGF2) signaling in colorectal cancer-from basic research to potential clinical applications. Int. J. Mol. Sci. 2019, 3, 4915. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.F.; Ibrahim, A.E.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [Green Version]
- La Vecchia, S.; Sebastián, C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippo, C.; Luceri, C.; Caderni, G.; Pacini, M.; Messerini, L.; Biggeri, A.; Mini, E.; Tonelli, F.; Cianchi, F.; Dolara, P. Mutations of the APC gene in human sporadic colorectal cancers. Scand. J. Gastroenterol. 2002, 37, 1048–1053. [Google Scholar] [CrossRef] [Green Version]
- Yu, I.S.; Cheung, W.Y. Epidermal growth factor receptor immunohistochemistry: New opportunities in metastatic colorectal cancer. J. Transl. Med. 2015, 7, 217. [Google Scholar]
- Wang, D.; Liang, W.; Duan, X.; Liu, L.; Shen, H.; Peng, Y.; Li, B. Detection of KRAS gene mutations in colorectal carcinoma: A study of 6 364 patients. Zhonghua Bing Li Xue Za Zhi 2014, 43, 583–587. [Google Scholar] [PubMed]
- Wang, D.; Zhang, Z.; Li, Y.; Xu, C.; Yu, Y.; Li, M.; Chen, C.; Zhang, X. Adenomatous polyposis coli gene mutations in 22 Chinese pedigrees with familial adenomatous polyposis. Med. Sci. Monit. 2019, 22, 3796–3803. [Google Scholar] [CrossRef]
- Nallamilli, B.R.R.; Hegde, M. Detecting APC Gene mutations in familial adenomatous polyposis (FAP). Curr. Protoc. Hum. Genet. 2017, 11, 10–18. [Google Scholar] [CrossRef]
- Wachsmannova, L.; Mego, M.; Stevurkova, V.; Zajac, V.; Ciernikova, S. Novel strategies for comprehensive mutation screening of the APC gene. Neoplasma 2017, 64, 338–343. [Google Scholar] [CrossRef]
- Ye, Z.L.; Qiu, M.Z.; Tang, T.; Wang, F.; Zhou, Y.X.; Lei, M.J.; Guan, W.L.; He, C.Y. Gene mutation profiling in Chinese colorectal cancer patients and its association with clinicopathological characteristics and prognosis. Cancer Med. 2020, 9, 745–756. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2018, 8, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017, 1, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shi, Y.L.; Zhou, K.; Wang, L.L.; Yan, Z.X.; Liu, Y.L.; Xu, L.L.; Zhao, S.W.; Chu, H.L.; Shi, T.T.; et al. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 2018, 3, 739. [Google Scholar] [CrossRef]
- Afrin, S.; Giampieri, F.; Gasparrini, M.; Forbes-Hernández, T.Y.; Cianciosi, D.; Reboredo-Rodriguez, P.; Zhang, J.; Manna, P.P.; Daglia, M.; Atanasov, A.G.; et al. Dietary phytochemicals in colorectal cancer prevention and treatment: A focus on the molecular mechanisms involved. Biotechnol. Adv. 2020, 38, 107322. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 17, 3980–4000. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Wang, L.; Ren, Y.X.; Pang, Z.; Liu, Y.; Sun, X.D.; Tu, J.; Zhi, Z.; Qin, Y.; Sun, L.N.; et al. The circular RNA circ-ERBIN promotes growth and metastasis of colorectal cancer by miR-125a-5p and miR-138-5p/4EBP-1 mediated cap-independent HIF-1α translation. Mol. Cancer 2020, 23, 164. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Jung, B.H. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology 2015, 149, 1177–1190.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armengol, G.; Sarhadi, V.K.; Ghanbari, R.; Doghaei-Moghaddam, M.; Ansari, R.; Sotoudeh, M.; Puolakkainen, P.; Kokkola, A.; Malekzadeh, R.; Knuutila, S. Driver gene mutations in stools of colorectal carcinoma patients detected by targeted next-generation sequencing. J. Mol. Diagn. 2016, 18, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Timar, J.; Kashofer, K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020, 39, 1029–1038. [Google Scholar] [CrossRef]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 15, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Mo, H.Y.; Lee, J.H.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Frameshift mutations and loss of expression of CLCA4 gene are frequent in colorectal cancers with microsatellite instability. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Flashner-Abramson, E.; Shalapour, S.; Zhong, Z.; Taniguchi, K.; Levitzki, A.; Karin, M. Targeting colorectal cancer via its microenvironment by inhibiting IGF-1 receptor-insulin receptor substrate and STAT3 signaling. Oncogene 2016, 19, 2634–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, W.; Zhou, Q.; Chen, C.; Yuan, W.; Liu, J.; Li, X.; Sun, Z. Roles of circRNAs in the tumour microenvironment. Mol. Cancer 2020, 23, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Li, C.; Wang, J.; Xue, L. Myeloid-derived suppressor cells: Roles in the tumor microenvironment and tumor radiotherapy. Int. J. Cancer 2019, 144, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019, 20, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzat-Saplacan, R.M.; Balacescu, L.; Gherman, C.; Chira, R.I.; Craiu, A.; Mircea, P.A.; Lisencu, C.; Balacescu, O. The role of PDGFs and PDGFRs in colorectal cancer. Mediators Inflamm. 2017, 2017, 4708076. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.; Barnich, N.; Nguyen, H.T.T. Microbiota, inflammation and colorectal cancer. Int. J. Mol. Sci. 2017, 20, 1310. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zhu, C.; Chen, C.; Zong, Y.; Feng, H.; Liu, D.; Feng, W.; Zhao, J.; Lu, A. CCL19 suppresses angiogenesis through promoting miR-206 and inhibiting Met/ERK/Elk-1/HIF-1alpha/VEGF-A pathway in colorectal cancer. Cell Death Dis. 2018, 24, 974. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente López, M.; Landskron, G.; Parada, D.; Dubois-Camacho, K.; Simian, D.; Martinez, M.; Romero, D.; Roa, J.C.; Chahuán, I.; Gutiérrez, R.; et al. The relationship between chemokines CCL2, CCL3, and CCL4 with the tumor microenvironment and tumor-associated macrophage markers in colorectal cancer. Tumour Biol. 2018, 40, 1010428318810059. [Google Scholar] [CrossRef] [Green Version]
- Mola, S.; Pandolfo, C.; Sica, A.; Porta, C. The Macrophages-microbiota interplay in colorectal cancer (CRC)-related inflammation: Prognostic and therapeutic significance. Int. J. Mol. Sci. 2020, 18, 6866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Qiu, X.; Li, J.; Zheng, S.; Li, L.; Zhao, H. TGF-beta secreted by tumor-associated macrophages promotes proliferation and invasion of colorectal cancer via miR-34a-VEGF axis. Cell Cycle 2018, 17, 2766–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrosa, L.; Esposito, F.; Thomson, T.M.; Maurel, J. The tumor microenvironment in colorectal cancer therapy. Cancers 2019, 14, 1172. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Wang, S.; Sun, M.; Zhang, C.; Wei, C.; Yang, C.; Dou, R.; Liu, Q.; Xiong, B. MiR-195-5p/NOTCH2-mediated EMT modulates IL-4 secretion in colorectal cancer to affect M2-like TAM polarization. J. Hematol. Oncol. 2019, 26, 20. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP signaling in the tumor microenvironment of colorectal cancer. Int. J. Mol. Sci. 2019, 11, 6254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.J.; Li, W. MiR-10b suppresses the growth and metastasis of colorectal cancer cell by targeting FGF13. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 576–587. [Google Scholar]
- Wai Hon, K.; Zainal Abidin, S.A.; Othman, I.; Naidu, R. Insights into the role of microRNAs in colorectal cancer (CRC) metabolism. Cancers 2020, 31, 2462. [Google Scholar] [CrossRef]
- Hardbower, D.M.; Coburn, L.A.; Asim, M.; Singh, K.; Sierra, J.C.; Barry, D.P.; Gobert, A.P.; Piazuelo, M.B.; Washington, M.K.; Wilson, K.T. EGFR-mediated macrophage activation promotes colitis-associated tumorigenesis. Oncogene 2017, 6, 3807–3819. [Google Scholar] [CrossRef] [Green Version]
- D’Haene, N.; Koopmansch, C.; Van Eycke, Y.R.; Hulet, F.; Allard, J.; Bouri, S.; Rorive, S.; Remmelink, M.; Decaestecker, C.; Maris, C.; et al. The prognostic value of the combination of low VEGFR-1 and high VEGFR-2 expression in endothelial cells of colorectal cancer. Int. J. Mol. Sci. 2018, 9, 3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, S.Y.; Myung, S.J. Obesity and colorectal cancer. Korean J. Gastroenterol. 2012, 59, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Fettig, L.M.; Yee, D. Advances in insulin-like growth factor biology and -directed cancer therapeutics. Adv. Cancer Res. 2020, 147, 229–257. [Google Scholar] [PubMed]
- Codony-Servat, J.; Cuatrecasas, M.; Asensio, E.; Montironi, C.; Martínez-Cardús, A.; Marín-Aguilera, M.; Horndler, C.; Martínez-Balibrea, E.; Rubini, M.; Jares, P.; et al. Nuclear IGF-1R predicts chemotherapy and targeted therapy resistance in metastatic colorectal cancer. Br. J. Cancer 2017, 5, 1777–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiratsuchi, I.; Akagi, Y.; Kawahara, A.; Kinugasa, T.; Romeo, K.; Yoshida, T.; Ryu, Y.; Gotanda, Y.; Kage, M.; Shirouzu, K. Expression of IGF-1 and IGF-1R and their relation to clinicopathological factors in colorectal cancer. Anticancer Res. 2011, 31, 2541–2545. [Google Scholar] [PubMed]
- Ciulei, G.; Orasan, O.H.; Coste, S.C.; Cozma, A.; Negrean, V.; Procopciuc, L.M. Vitamin D and the insulin-like growth factor system: Implications for colorectal neoplasia. Eur. J. Clin. Invest. 2020, 50, 13265. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.A.; Zand, H.; Cheraghpour, M. The influence of curcumin on the downregulation of MYC, Insulin and IGF-1 receptors: A possible mechanism underlying the anti-growth and anti-migration in chemoresistant colorectal cancer cells. Medicina 2019, 3, 90. [Google Scholar] [CrossRef] [Green Version]
- Ramezani, P.; Abnous, K.; Taghdisi, S.M.; Zahiri, M.; Ramezani, M.; Alibolandi, M. Targeted MMP-2 responsive chimeric polymersomes for therapy against colorectal cancer Colloids Surf. B Biointerfaces 2020, 193, 111135. [Google Scholar] [CrossRef] [PubMed]
- Sipos, F.; Székely, H.; Kis, I.D.; Tulassay, Z.; Műzes, G. Relation of the IGF/IGF1R system to autophagy in colitis and colorectal cancer. World J. Gastroenterol. 2017, 14, 8109–8119. [Google Scholar] [CrossRef]
- Wang, S.Q.; Yang, X.Y.; Cui, S.X.; Gao, Z.H.; Qu, X.J. Heterozygous knockout insulin-like growth factor-1 receptor (IGF-1R) regulates mitochondrial functions and prevents colitis and colorectal cancer. Free Radic. Biol. Med. 2019, 134, 87–98. [Google Scholar] [CrossRef]
- Shali, H.; Ahmadi, M.; Kafil, H.S.; Dorosti, A.; Yousefi, M. IGF1R and c-met as therapeutic targets for colorectal cancer. Biomed. Pharmacother. 2016, 82, 528–536. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, X.; Du, L.L.; Wang, Y.; Liu, D.B.; Han, C.Z.; Jing, J.X.; Zhao, X.W.; Xu, X.Q. Possible roles of insulin, IGF-1 and IGFBPs in initiation and progression of colorectal cancer. World J. Gastroenterol. 2014, 14, 1608–1613. [Google Scholar] [CrossRef]
- Sandhu, M.S.; Dunger, D.B.; Giovannucci, E.L. Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. J. Natl. Cancer Inst. 2002, 3, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, X.; Chi, J.; Che, K.; Feng, Y.; Zhao, S.; Wang, Z.; Wang, Y. Expressions of IGF-1, ERK, GLUT4, IRS-1 in metabolic syndrome complicated with colorectal cancer and their associations with the clinical characteristics of CRC. Cancer Biomark. 2018, 21, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.; Gongoll, S.; Langner, C.; Mengel, M.; Piso, P.; Klempnauer, J.; Rüschoff, J.; Kreipe, H.; von Wasielewski, R. IGF-1R, IGF-1 and IGF-2 expression as potential prognostic and predictive markers in colorectal-cancer. Virchows Arch. 2003, 443, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Alagaratnam, S.; Loizidou, M.; Yang, S.Y.; Fuller, B.; Ramesh, B. Increased expression of IGF-1Ec with increasing colonic polyp dysplasia and colorectal cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2861–2870. [Google Scholar] [CrossRef]
- Pankaj, J.; Kumari, J.R.; Kim, W.; Lee, S.A. Insulin-like Growth Factor-1, IGF-binding Protein-3, C-peptide and colorectal cancer: A case-control study. Asian Pac. J. Cancer Prev. 2015, 16, 3735–3740. [Google Scholar] [PubMed] [Green Version]
- Hu, X.; Zheng, W.; Luo, Y.; Ou, X.; Song, L.; Zhang, S.; He, T.; Guo, Z.; Zhu, J.; Shi, H.; et al. Arca subcrenata polypeptides inhibit human colorectal cancer HT-29 cells growth via suppression of IGF-1R/Akt/mTOR signaling and ATP production. Nutr. Cancer 2020, 72, 260–272. [Google Scholar] [CrossRef]
- Daveri, E.; Adamo, A.M.; Alfine, E.; Zhu, W.; Oteiza, P.I. Hexameric procyanidins inhibit colorectal cancer cell growth through both redox and non-redox regulation of the epidermal growth factor signaling pathway. Redox Biol. 2021, 38, 101830. [Google Scholar] [CrossRef]
- Custodio, A.; Feliu, J. Prognostic and predictive biomarkers for epidermal growth factor receptor-targeted therapy in colorectal cancer: Beyond KRAS mutations. Crit. Rev. Oncol. Hematol. 2013, 85, 45–81. [Google Scholar] [CrossRef]
- Khan, K.; Valeri, N.; Dearman, C.; Rao, S.; Watkins, D.; Starling, N.; Chau, I.; Cunningham, D. Targeting EGFR pathway in metastatic colorectal cancer- tumour heterogeniety and convergent evolution. Crit. Rev. Oncol. Hematol. 2019, 143, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Muguruma, N.; Fujimoto, S.; Okada, Y.; Kida, Y.; Nakamura, F.; Tanaka, K.; Nakagawa, T.; Kitamura, S.; Okamoto, K.; et al. Epidermal growth factor receptor-targeted molecular imaging of colorectal tumors: Detection and treatment evaluation of tumors in animal models. Cancer Sci. 2019, 110, 1921–1930. [Google Scholar] [CrossRef] [Green Version]
- de Mello, R.A.; Marques, A.M.; Araújo, A. Epidermal growth factor receptor and metastatic colorectal cancer: Insights into target therapies. World J. Gastroenterol. 2013, 14, 6315–6318. [Google Scholar] [CrossRef]
- Grossmann, A.H.; Samowitz, W.S. Epidermal growth factor receptor pathway mutations and colorectal cancer therapy. Arch. Pathol. Lab. Med. 2011, 135, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Nappi, A.; Berretta, M.; Romano, C.; Tafuto, S.; Cassata, A.; Casaretti, R.; Silvestro, L.; Divitiis, C.; Alessandrini, L.; Fiorica, F.; et al. Metastatic colorectal cancer: Role of target therapies and future perspectives. Curr. Cancer Drug Targets 2018, 18, 421–429. [Google Scholar] [CrossRef]
- Yang, W.J.; Shen, X.J.; Ma, X.X.; Tan, Z.G.; Song, Y.; Guo, Y.T.; Yuan, M. Correlation of human epidermal growth factor receptor protein expression and colorectal cancer. World J. Gastroenterol. 2015, 28, 8687–8696. [Google Scholar] [CrossRef]
- Greally, M.; Kelly, C.M.; Cercek, A. HER2: An emerging target in colorectal cancer. Curr. Probl. Cancer 2018, 42, 560–571. [Google Scholar] [CrossRef]
- Lawan, A.I.; Ogunbiyi, J.O. Epidermal growth factor receptor expression of colorectal carcinoma in Nigerian patients. West. Afr. J. Med. 2020, 37, 100–105. [Google Scholar]
- Xu, W.; Jing, H.; Zhang, F.; Xu, W.; Jing, H.; Zhang, F. Epidermal growth factor receptor-targeted therapy in colorectal cancer. Front. Biosci. 2016, 1, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Nemanqani, D.M.; Aftab, K.; Al-Malki, S.H.; Al-Sufyani, W.M. Expression of epidermal growth factor receptor in colorectal adenocarcinoma and its correlation with clinicopathological factors. J. Coll. Phys. Surg. Pak. 2018, 28, 527–531. [Google Scholar] [CrossRef]
- Yun, S.; Kwak, Y.; Nam, S.K.; Seo, A.N.; Oh, H.K.; Kim, D.W.; Kang, S.B.; Lee, H.S. Ligand-independent epidermal growth factor receptor overexpression correlates with poor prognosis in colorectal cancer. Cancer Res. Treat. 2018, 50, 1351–1361. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Mysliwiec, M.; Sierko, E.; Sobierska, M.; Kruszewska, J.; Lipska, A.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Elevated microparticles, thrombin-antithrombin and VEGF Levels in colorectal cancer patients undergoing chemotherapy. Pathol. Oncol. Res. 2020, 26, 2499–2507. [Google Scholar] [CrossRef]
- Wu, Q.B.; Chen, J.; Zhu, J.W.; Yin, X.; You, H.Y.; Lin, Y.R.; Zhu, H.Q. MicroRNA-125 inhibits RKO colorectal cancer cell growth by targeting VEGF. Int. J. Mol. Med. 2018, 42, 665–673. [Google Scholar] [CrossRef]
- Lan, J.; Li, H.; Luo, X.; Hu, J.; Wang, G. BRG1 promotes VEGF-A expression and angiogenesis in human colorectal cancer cells. Exp. Cell Res. 2017, 15, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Coşkun, Ö.; Öztopuz, Ö.; Özkan, Ö.F. Determination of IL-6, TNF-alpha and VEGF levels in the serums of patients with colorectal cancer. Cell Mol. Biol. 2017, 20, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Mathonnet, M.; Perraud, A.; Christou, N.; Akil, H.; Melin, C.; Battu, S.; Jauberteau, M.O.; Denizot, Y. Hallmarks in colorectal cancer: Angiogenesis and cancer stem-like cells. World J. Gastroenterol. 2014, 21, 4189–4196. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Fan, F.; Wang, R.; Ye, X.; Xia, L.; Boulbes, D.; Ellis, L.M. Intracrine VEGF signalling mediates colorectal cancer cell migration and invasion. Br. J. Cancer 2017, 5, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, S.Y.; Mohammed, H.L.; Ibrahim, H.M.; Mohamed, E.M.; Salah, M. Role of VEGF, CD105, and CD31 in the prognosis of colorectal cancer cases. J. Gastrointest. Cancer 2019, 50, 23–34. [Google Scholar] [CrossRef]
- Dinami, R.; Porru, M.; Amoreo, C.A.; Sperduti, I.; Mottolese, M.; Buglioni, S.; Marinelli, D.; Maugeri-Saccà, M.; Sacconi, A.; Blandino, G.; et al. TRF2 and VEGF-A: An unknown relationship with prognostic impact on survival of colorectal cancer patients. J. Exp. Clin. Cancer Res. 2020, 15, 111. [Google Scholar] [CrossRef]
- Karpuz, T.; Araz, M.; Korkmaz, L.; Kılınc, I.; Findik, S.; Karaagaç, M.; Eryilmaz, M.K.; Artac, M. The prognostic value of serum semaphorin3A and VEGF Levels in patients with metastatic colorectal cancer. J. Gastrointest. Cancer 2020, 51, 491–497. [Google Scholar] [CrossRef]
- Jannuzzi, A.T.; Özhan, G.; Yanar, H.T.; Alpertunga, B. VEGF gene polymorphisms and susceptibility to colorectal cancer. Genet. Test. Mol. Biomark. 2015, 19, 133–137. [Google Scholar] [CrossRef]
- Chen, X.; Xu, X.; Pan, B.; Zeng, K.; Xu, M.; Liu, X.; He, B.; Pan, Y.; Sun, H.; Wang, S. miR-150-5p suppresses tumor progression by targeting VEGFA in colorectal cancer. Aging 2018, 26, 3421–3437. [Google Scholar] [CrossRef]
- Kim, Y.I.; Jeong, S.; Jung, K.O.; Song, M.G.; Lee, C.H.; Chung, S.J.; Park, J.Y.; Cha, M.G.; Lee, S.G.; Jun, B.H.; et al. Simultaneous detection of EGFR and VEGF in colorectal cancer using fluorescence-Raman Endoscopy. Sci. Rep. 2017, 21, 1035. [Google Scholar] [CrossRef]
- Herichova, I.; Reis, R.; Hasakova, K.; Vician, M.; Zeman, M. Sex-dependent regulation of estrogen receptor beta in human colorectal cancer tissue and its relationship with clock genes and VEGF-A expression. Physiol. Res. 2019, 20, S297–S305. [Google Scholar] [CrossRef]
- Narayanankutty, A. PI3K/ Akt/ mTOR pathway as a therapeutic target for colorectal cancer: A review of preclinical and clinical evidence. Curr. Drug Targets 2019, 20, 217–1226. [Google Scholar] [CrossRef] [PubMed]
- Moafian, Z.; Maghrouni, A.; Soltani, A.; Hashemy, S.I. Cross-talk between non-coding RNAs and PI3K/AKT/mTOR pathway in colorectal cancer. Mol. Biol. Rep. 2021, 48, 4797–4811. [Google Scholar] [CrossRef]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Lech, G.; Słotwiński, R.; Słodkowski, M.; Krasnodębski, I.W. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J. Gastroenterol. 2016, 7, 1745–1755. [Google Scholar] [CrossRef]
- Martinelli, E.; Ciardiello, D.; Martini, G.; Troiani, T.; Cardone, C.; Vitiello, P.P.; Normanno, N.; Rachiglio, A.M.; Maiello, E.; Latiano, T.; et al. Implementing anti-epidermal growth factor receptor (EGFR) therapy in metastatic colorectal cancer: Challenges and future perspectives. Ann. Oncol. 2020, 31, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Pape, J.; Magdeldin, T.; Stamati, K.; Nyga, A.; Loizidou, M.; Emberton, M.; Cheema, U. Cancer-associated fibroblasts mediate cancer progression and remodel the tumouroid stroma. Br. J. Cancer 2020, 123, 1178–1190. [Google Scholar] [CrossRef]
- Yao, W.; Lin, Z.; Shi, P.; Chen, B.; Wang, G.; Huang, J.; Sui, Y.; Liu, Q.; Li, S.; Lin, X.; et al. Delicaflavone induces ROS-mediated apoptosis and inhibits PI3K/AKT/mTOR and Ras/MEK/Erk signaling pathways in colorectal cancer cells. Biochem. Pharmacol. 2020, 171, 113680. [Google Scholar] [CrossRef]
- Hybel, T.E.; Dietrichs, D.; Sahana, J.; Corydon, T.J.; Nassef, M.Z.; Wehland, M.; Krüger, M.; Magnusson, N.E.; Bauer, J.; Utpatel, K.; et al. Simulated microgravity influences VEGF, MAPK, and PAM signaling in prostate cancer cells. Int. J. Mol. Sci. 2020, 13, 1263. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Huang, W.; Liu, X.; Liu, X.; Chen, N.; Xu, Q.; Hu, Y.; Song, W.; Zhou, J. IMPDH2 promotes colorectal cancer progression through activation of the PI3K/AKT/mTOR and PI3K/AKT/FOXO1 signaling pathways. J. Exp. Clin. Cancer Res. 2018, 5, 304. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Xiao, Y.; Song, Y.; Yuan, H.; Luo, J.; Xu, W. FAT4 regulates the EMT and autophagy in colorectal cancer cells in part via the PI3K-AKT signaling axis. J. Exp. Clin. Cancer Res. 2019, 4, 112. [Google Scholar] [CrossRef] [Green Version]
- Pandurangan, A.K. Potential targets for prevention of colorectal cancer: A focus on PI3K/Akt/mTOR and Wnt pathways. Asian Pac. J. Cancer Prev. 2013, 14, 2201–2205. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Jiang, X.; Zhang, Q.; Ma, J.; Cheng, R.; Yong, H.; Shi, H.; Zhou, X.; Ge, L.; Gao, G. Naringin inhibits colorectal cancer cell growth by repressing the PI3K/AKT/mTOR signaling pathway. Exp. Ther. Med. 2020, 19, 3798–3804. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 13, 735. [Google Scholar] [CrossRef] [Green Version]
- Winder, T.; Lenz, H.J. Vascular endothelial growth factor and epidermal growth factor signaling pathways as therapeutic targets for colorectal cancer. Gastroenterology 2010, 138, 2163–2176. [Google Scholar] [CrossRef]
- Yarom, N.; Jonker, D.J. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov. Med. 2011, 11, 95–105. [Google Scholar]
- Foroughi, S.; Tie, J.; Gibbs, P.; Burgess, A.W. Epidermal growth factor receptor ligands: Targets for optimizing treatment of metastatic colorectal cancer. Growth Factors 2019, 37, 209–225. [Google Scholar] [CrossRef]
- Martini, G.; Troiani, T.; Cardone, C.; Vitiello, P.; Sforza, V.; Ciardiello, D.; Napolitano, S.; Della Corte, C.M.; Morgillo, F.; Raucci, A.; et al. Present and future of metastatic colorectal cancer treatment: A review of new candidate targets. World J. Gastroenterol. 2017, 14, 4675–4688. [Google Scholar] [CrossRef]
- Weinberg, B.A.; Hartley, M.L.; Salem, M.E. Precision medicine in metastatic colorectal cancer: Relevant carcinogenic pathways and targets-part 1: Biologic therapies targeting the epidermal growth factor receptor and vascular endothelial growth factor. Oncology 2017, 15, 539–548. [Google Scholar]
- Price, T.J.; Tang, M.; Gibbs, P.; Haller, D.G.; Peeters, M.; Arnold, D.; Segelov, E.; Roy, A.; Tebbutt, N.; Pavlakis, N.; et al. Targeted therapy for metastatic colorectal cancer. Expert Rev. Anticancer Ther. 2018, 18, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Martini, G.; Ciardiello, D.; Vitiello, P.P.; Napolitano, S.; Cardone, C.; Cuomo, A.; Troiani, T.; Ciardiello, F.; Martinelli, E. Resistance to anti-epidermal growth factor receptor in metastatic colorectal cancer: What does still need to be addressed? Cancer Treat. Rev. 2020, 86, 102023. [Google Scholar] [CrossRef] [PubMed]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canavese, M.; Ngo, D.T.; Maddern, G.J.; Hardingham, J.E.; Price, T.J.; Hauben, E. Biology and therapeutic implications of VEGF-A splice isoforms and single-nucleotide polymorphisms in colorectal cancer. Int. J. Cancer 2017, 5, 2183–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, S.; Matrone, N.; Muddassir, A.L.; Martini, G.; Sorokin, A.; De Falco, V.; Giunta, E.F.; Ciardiello, D. Triple blockade of EGFR, MEK and PD-L1 has antitumor activity in colorectal cancer models with constitutive activation of MAPK signaling and PD-L1 overexpression. J. Exp. Clin. Cancer Res. 2019, 16, 492. [Google Scholar] [CrossRef] [Green Version]
- Itatani, Y.; Yamamoto, T.; Zhong, C.; Molinolo, A.A.; Ruppel, J.; Hegde, P.; Taketo, M.M.; Ferrara, N. Suppressing neutrophil-dependent angiogenesis abrogates resistance to anti-VEGF antibody in a genetic model of colorectal cancer. Proc. Natl. Acad. Sci. USA 2020, 1, 21598–21608. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Marisi, G.; Scarpi, E.; Passardi, A.; Nanni, O.; Ragazzini, A.; Valgiusti, M.; Casadei-Gardini, A.; Neri, L.M.; Frassineti, G.L.; Amadori, D.; et al. Circulating VEGF and eNOS variations as predictors of outcome in metastatic colorectal cancer patients receiving bevacizumab. Sci. Rep. 2017, 2, 1293. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.; Kline, C.L.; Zhou, L.; Khazak, V.; El-Deiry, W.S. Anti-tumor effects of ONC201 in combination with VEGF-inhibitors significantly impacts colorectal cancer growth and survival in vivo through complementary non-overlapping mechanisms. J. Exp. Clin. Cancer Res. 2018, 22, 11. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Li, X. Fruquintinib and its use in the treatment of metastatic colorectal cancer. Future Oncol. 2019, 15, 2571–2576. [Google Scholar] [CrossRef]
- Abdalla, A.N.; Malki, W.H.; Qattan, A.; Shahid, I.; Hossain, M.A.; Ahmed, M. Chemosensitization of HT29 and HT29-5FU cell lines by a combination of a multi-tyrosine kinase inhibitor and 5FU downregulates ABCC1 and inhibits PIK3CA in light of their importance in Saudi colorectal cancer. Molecules 2021, 11, 334. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Suyama, K.; Baba, H. Recent advances in targeting the EGFR signaling pathway for the treatment of metastatic colorectal cancer. Int. J. Mol. Sci. 2017, 2, 752. [Google Scholar] [CrossRef] [Green Version]
- Pranteda, A.; Piastra, V.; Stramucci, L.; Fratantonio, D.; Bossi, G. The p38 MAPK signaling activation in colorectal cancer upon therapeutic treatments. Int. J. Mol. Sci. 2020, 16, 2773. [Google Scholar] [CrossRef]
- Grossi, V.; Peserico, A.; Tezil, T.; Simone, C. p38alpha MAPK pathway: A key factor in colorectal cancer therapy and chemoresistance. World J. Gastroenterol. 2014, 7, 9744–9758. [Google Scholar] [CrossRef]
- Wang, J.; Liang, D.; Zhang, X.P.; He, C.F.; Cao, L.; Zhang, S.Q.; Xiao, X.; Li, S.J.; Cao, Y.X. Novel PI3K/Akt/mTOR signaling inhibitor, W922, prevents colorectal cancer growth via the regulation of autophagy. Int. J. Oncol. 2021, 58, 70–82. [Google Scholar] [CrossRef]
- Wei, H.; Dong, C.; Shen, Z. Kallikrein-related peptidase (KLK10) cessation blunts colorectal cancer cell growth and glucose metabolism by regulating the PI3K/Akt/mTOR pathway. Neoplasma 2020, 67, 889–897. [Google Scholar] [CrossRef]
- Helmy, M.W.; Ghoneim, A.I.; Katary, M.A.; Elmahdy, R.K. The synergistic anti-proliferative effect of the combination of diosmin and BEZ-235 (dactolisib) on the HCT-116 colorectal cancer cell line occurs through inhibition of the PI3K/Akt/mTOR/NF-kappaB axis. Mol. Biol. Rep. 2020, 47, 2217–2230. [Google Scholar] [CrossRef]
- Li, S.; Wang, X.; Wang, G.; Shi, P.; Lin, S.; Xu, D.; Chen, B.; Liu, A.; Huang, L.; Lin, X.; et al. Ethyl acetate extract of selaginella doederleinii hieron induces cell autophagic death and apoptosis in colorectal cancer via PI3K-Akt-mTOR and AMPKalpha-signaling pathways. Front. Pharmacol. 2020, 19, 565090. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Mun, J.G.; Jeon, H.D.; Kee, J.Y.; Hong, S.H. Betulin inhibits lung metastasis by inducing cell cycle arrest, autophagy, and apoptosis of metastatic colorectal cancer cells. Nutrients 2019, 26, 66. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhang, Z.; Jiang, G.; Sun, H.; Yu, D. Nobiletin sensitizes colorectal cancer cells to oxaliplatin by PI3K/Akt/MTOR pathway. Front. Biosci. 2019, 1, 303–312. [Google Scholar]
- Shahi Thakuri, P.; Luker, G.D.; Tavana, H. Cyclical treatment of colorectal tumor spheroids induces resistance to MEK inhibitors. Transl. Oncol. 2019, 12, 404–416. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, R.H.; Shen, L.; Xu, J.; Bai, Y.; Yang, L.; Deng, Y.; Chen, Z.D.; Zhong, H.; et al. Effect of fruquintinib vs placebo on overall survival in patients with previously treated metastatic colorectal cancer: The FRESCO randomized clinical trial. JAMA 2018, 26, 486–2496. [Google Scholar] [CrossRef]
- Arqués, O.; Chicote, I.; Puig, I.; Tenbaum, S.P.; Argilés, G.; Dienstmann, R.; Fernández, N.; Caratù, G.; Matito, J.; Silberschmidt, D.; et al. Tankyrase inhibition blocks Wnt/beta-catenin pathway and reverts resistance to PI3K and AKT inhibitors in the treatment of colorectal cancer. Clin. Cancer Res. 2016, 1, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Wanigasooriya, K.; Tyler, R.; Barros-Silva, J.D.; Sinha, Y.; Ismail, T.; Beggs, A.D. Radiosensitising cancer using phosphatidylinositol-3-Kinase (PI3K), protein kinase B (AKT) or mammalian target of rapamycin (mTOR) inhibitors. Cancers 2020, 18, 1278. [Google Scholar] [CrossRef]
- Ganesan, P.; Janku, F.; Naing, A.; Hong, D.S.; Tsimberidou, A.M.; Falchook, G.S.; Wheler, J.J.; Piha-Paul, S.A.; Fu, S.; Stepanek, V.M.; et al. Target-based therapeutic matching in early-phase clinical trials in patients with advanced colorectal cancer and PIK3CA mutations. Mol. Cancer Ther. 2013, 12, 2857–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Laguna, I.; Hong, D.S.; Janku, F.; Nguyen, L.M.; Falchook, G.S.; Fu, S.; Wheler, J.J.; Luthra, R.; Naing, A.; Wang, X.; et al. KRASness and PIK3CAness in patients with advanced colorectal cancer: Outcome after treatment with early-phase trials with targeted pathway inhibitors. PLoS ONE 2012, 7, e38033. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Braden, A.M.; Kolesar, J.M.; Eickhoff, J.C.; Bailey, H.H.; Heideman, J.; Liu, G.; Wisinski, K.B. A phase I study of tivantinib in combination with temsirolimus in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.; Tabernero, J.; Hwang, J.; Bajetta, E.; Sharma, S.; Del Prete, S.A.; Arrowsmith, E.R.; Ryan, D.P.; Sedova, M.; Jin, J.; et al. Phase II study of everolimus in patients with metastatic colorectal adenocarcinoma previously treated with bevacizumab-, fluoropyrimidine-, oxaliplatin-, and irinotecan-based regimens. Clin. Cancer Res. 2013, 15, 3987–3995. [Google Scholar] [CrossRef] [Green Version]
- Van Geel, R.M.J.M.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Brummelen, E.M.J.; Huijberts, S.; van Herpen, C.; Desar, I.; Opdam, F.; van Geel, R.; Marchetti, S.; Steeghs, N.; Monkhorst, K.; Thijssen, B.; et al. Phase I study of afatinib and selumetinib in patients with KRAS-mutated colorectal, non-small cell lung, and pancreatic cancer. Oncologist 2021, 26, 290–e545. [Google Scholar] [CrossRef]
- Folprecht, G.; Tabernero, J.; Köhne, C.H.; Zacharchuk, C.; Paz-Ares, L.; Rojo, F.; Quinn, S.; Casado, E.; Salazar, R.; Abbas, R.; et al. Phase I pharmacokinetic/pharmacodynamic study of EKB-569, an irreversible inhibitor of the epidermal growth factor receptor tyrosine kinase, in combination with irinotecan, 5-fluorouracil, and leucovorin (FOLFIRI) in first-line treatment of patients with metastatic colorectal cancer. Clin. Cancer Res. 2008, 1, 215–223. [Google Scholar]
- Tabernero, J.; Cervantes, A.; Rivera, F.; Martinelli, E.; Rojo, F.; von Heydebreck, A.; Macarulla, T.; Rodriguez-Braun, E.; Eugenia Vega-Villegas, M.; Senger, S.; et al. Pharmacogenomic and pharmacoproteomic studies of cetuximab in metastatic colorectal cancer: Biomarker analysis of a phase I dose-escalation study. J. Clin. Oncol. 2010, 1, 1181–1189. [Google Scholar] [CrossRef]
- Antoniotti, C.; Borelli, B.; Rossini, D.; Pietrantonio, F.; Morano, F.; Salvatore, L.; Lonardi, S.; Marmorino, F.; Tamberi, S.; Corallo, S.; et al. AtezoTRIBE: A randomised phase II study of FOLFOXIRI plus bevacizumab alone or in combination with atezolizumab as initial therapy for patients with unresectable metastatic colorectal cancer. BMC Cancer 2020, 22, 683. [Google Scholar] [CrossRef] [PubMed]
- Damato, A.; Iachetta, F.; Antonuzzo, L.; Nasti, G.; Bergamo, F.; Bordonaro, R.; Maiello, E.; Zaniboni, A.; Tonini, G.; Romagnani, A.; et al. Phase II study on first-line treatment of NIVolumab in combination with folfoxiri/bevacizumab in patients with advanced colorectal cancer RAS or BRAF mutated—NIVACOR trial (GOIRC-03-2018). BMC Cancer 2020, 31, 822. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; Vander Walde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Mileo, A.M.; Nisticò, P.; Miccadei, S. Polyphenols: Immunomodulatory and Therapeutic Implication in colorectal cancer. Front. Immunol. 2019, 11, 729. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefani, C.; Miricescu, D.; Stanescu-Spinu, I.-I.; Nica, R.I.; Greabu, M.; Totan, A.R.; Jinga, M. Growth Factors, PI3K/AKT/mTOR and MAPK Signaling Pathways in Colorectal Cancer Pathogenesis: Where Are We Now? Int. J. Mol. Sci. 2021, 22, 10260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910260
Stefani C, Miricescu D, Stanescu-Spinu I-I, Nica RI, Greabu M, Totan AR, Jinga M. Growth Factors, PI3K/AKT/mTOR and MAPK Signaling Pathways in Colorectal Cancer Pathogenesis: Where Are We Now? International Journal of Molecular Sciences. 2021; 22(19):10260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910260
Chicago/Turabian StyleStefani, Constantin, Daniela Miricescu, Iulia-Ioana Stanescu-Spinu, Remus Iulian Nica, Maria Greabu, Alexandra Ripszky Totan, and Mariana Jinga. 2021. "Growth Factors, PI3K/AKT/mTOR and MAPK Signaling Pathways in Colorectal Cancer Pathogenesis: Where Are We Now?" International Journal of Molecular Sciences 22, no. 19: 10260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910260