
Adaption and Degradation Strategies of Methylotrophic 1,4-Dioxane Degrading Strain Xanthobacter sp. YN2 Revealed by Transcriptome-Scale Analysis


Abstract
:1. Introduction
2. Results
2.1. RNA-Sequencing Results and Assembly
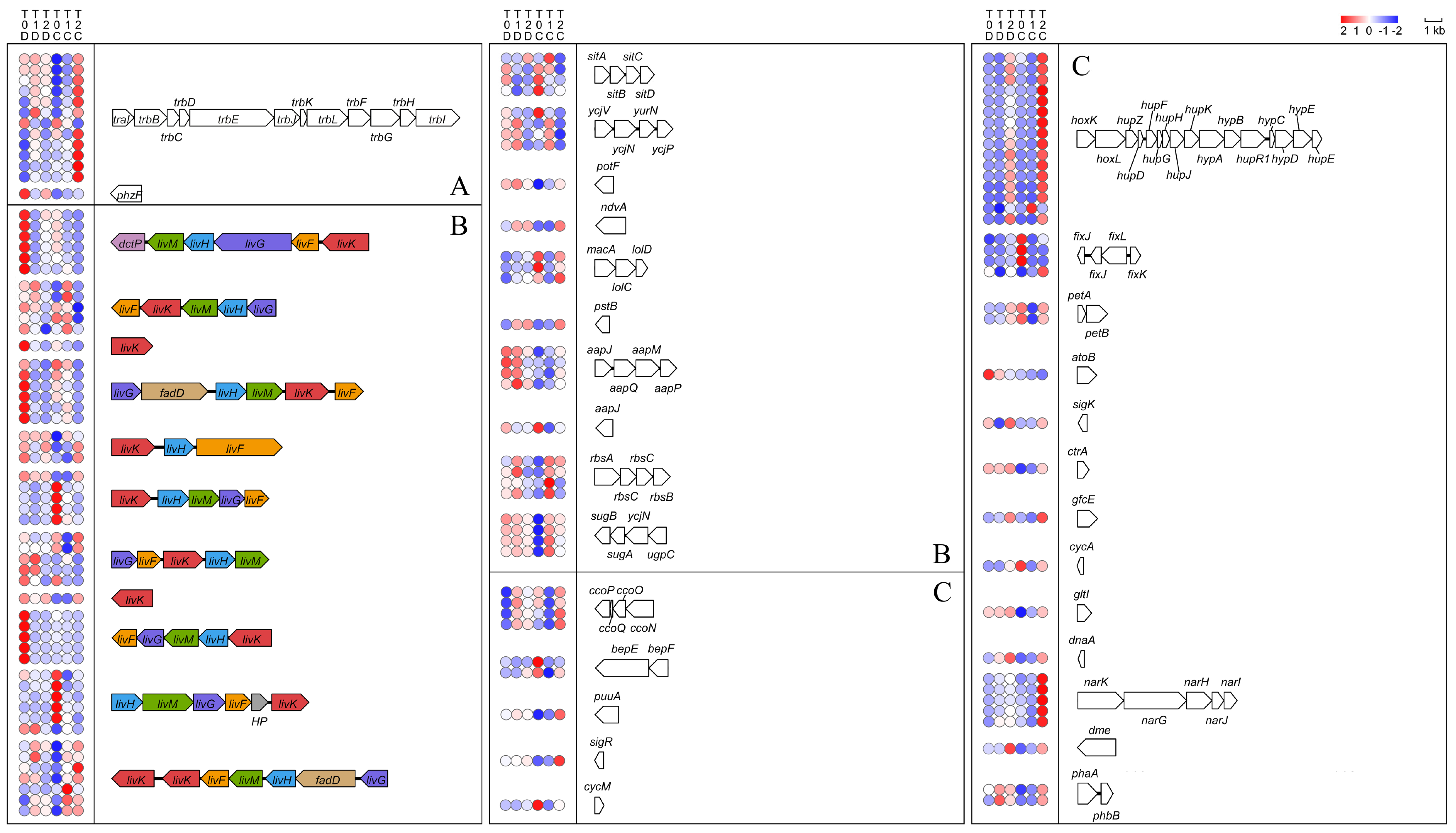
2.2. Essential Pathways Related to Metabolism of and Response to Dioxane
2.3. Genes Involved in Metabolism from Dioxane to Glyoxylate
2.4. Genes Involved in Metabolism of Glyoxylate
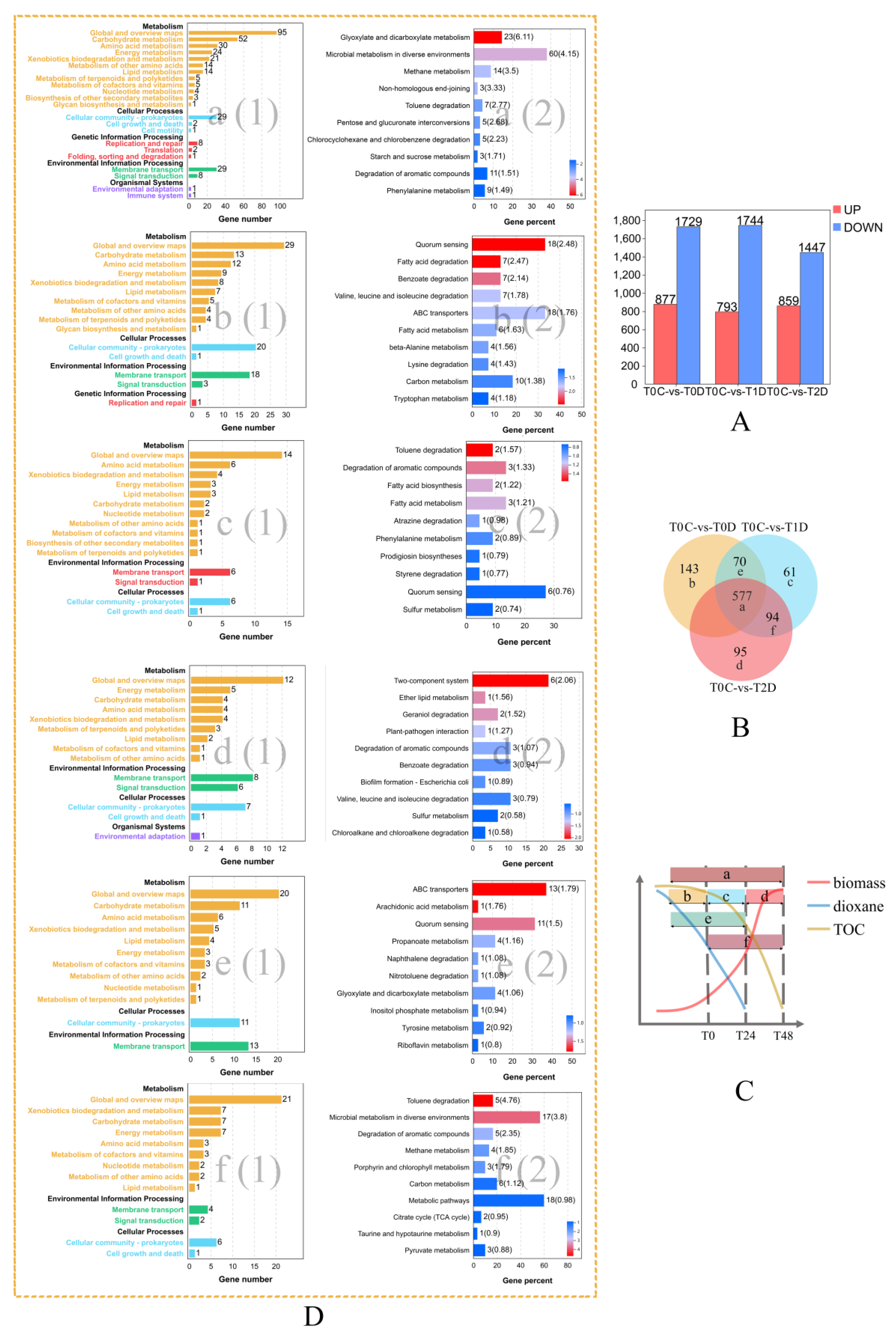
2.5. Dynamic Transcriptome through Three Stages of Dioxane Degradation
2.5.1. Genes Involved in Quorum Sensing
2.5.2. Genes Encoding Transporters
2.5.3. Genes Encoding Two-Component Systems
2.5.4. Genes Involved in Other Important Cellular Functions
3. Discussion
4. Materials and Methods
4.1. Chemicals and Culture Media
4.2. Experimental Setup
4.3. RNA Extraction, cDNA Library Construction, and Sequencing
4.4. Differentially Expressed Genes Analysis
4.5. Gene Expression Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohr, T.K.; Stickney, J.A.; DiGuiseppi, W.H. Environmental Investigation and Remediation: 1, 4-Dioxane and Other Solvent Stabilizers; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Selcoe, B.; Kappleman, W.B. Risk Assessment. In Environmental Investigation and Remediation; Mohr, T., Stickney, J., Eds.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Steffan, R.J.; McClay, K.R.; Masuda, H.; Zylstra, G.J. ER-1422: Biodegradation of 1, 4-Dioxane; Steffan, R.J., McClay, K.R., Masuda, H., Zylstra, G.J., Eds.; Shaw Environmental Inc.: Lawrenceville, NJ, USA, 2007. [Google Scholar]
- Krystal, J.G.; Pollitt Jae-Hong, K.; Jordan, P.; Menachem, E.; Yawei, Z.; Georgia, C.; Brenna, H.; Ines, Z.; Huang, H.; Nicole, C.D.; et al. 1,4-Dioxane as an emerging water contaminant: State of the science and evaluation of research needs. Sci. Total Environ. 2019, 690, 853–866. [Google Scholar] [CrossRef]
- Grostern, A.; Sales, C.M.; Zhuang, W.Q.; Erbilgin, O.; Alvarez-Cohen, L. Glyoxylate metabolism is a key feature of the metabolic degradation of 1,4-dioxane by Pseudonocardia dioxanivorans strain CB1190. Appl. Environ. Microbiol. 2012, 78, 3298–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolan, S.K.; Welch, M. The Glyoxylate Shunt, 60 Years On. Annu. Rev. Microbiol. 2018, 72, 309–330. [Google Scholar] [CrossRef]
- Beeckmans, S.; Xie, J.P. Glyoxylate Cycle. In Encyclopedia of Microbiology; Schaechter, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar] [CrossRef]
- Kathrin, S.; Rémi, P.; Patrick, K.; Philipp, C.; Nathanaël, D.; Stéphane, M.; Jean-Charles, P.; Julia, A.V. The Ethylmalonyl-CoA Pathway Is Used in Place of the Glyoxylate Cycle by Methylobacterium extorquens AM1 during Growth on Acetate*. J. Biol. Chem. 2012, 287, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Alber, B.E. Biotechnological potential of the ethylmalonyl-CoA pathway. Appl. Microbiol. Biotechnol. 2011, 89, 17–25. [Google Scholar] [CrossRef]
- Ludmila, C.; Marina, G.K. Current Trends in Methylotrophy. Trends Microbiol. 2018, 26, 703–714. [Google Scholar] [CrossRef]
- Jens, S.; Martin, S.; Dirk, H.; Dieter, S.; Murillo Villela, F.; Achim, M.; Julia, A.V. Methanol-based industrial biotechnology: Current status and future perspectives of methylotrophic bacteria. Trends Biotechnol. 2009, 27, 107–115. [Google Scholar] [CrossRef]
- Kumar, M.; Tomar, R.S.; Lade, H.; Paul, D. Methylotrophic bacteria in sustainable agriculture. World J. Microbiol. Biotechnol. 2016, 32, 120. [Google Scholar] [CrossRef]
- Pfeifenschneider, J.; Brautaset, T.; Wendisch, V.F. Methanol as carbon substrate in the bio-economy: Metabolic engineering of aerobic methylotrophic bacteria for production of value-added chemicals. Biofuels Bioprod. Biorefin. 2017, 11, 719–731. [Google Scholar] [CrossRef]
- Strong, P.J.; Xie, S.; Clarke, W.P. Methane as a resource: Can the methanotrophs add value? Environ. Sci. Technol. 2015, 49, 4001–4018. [Google Scholar] [CrossRef] [PubMed]
- Kartik, D.; Suresh, R.S.; Kadiyala, V.; Mallavarapu, M. Mesorhizobium tamadayense MM3441: A novel methylotroph with a great potential in degrading N,N′-dimethylformamide. Int. Biodeterior. Biodegrad. 2020, 153, 105045. [Google Scholar] [CrossRef]
- Hamer, G. 6.53—BT Technology for the Control of Methane Emissions from Permafrost and Natural Gas Hydrates. In Comprehensive Biotechnology, 2nd ed.; Hamer, G., Ed.; Academic Press: Cambridge, MA, USA, 2011; pp. 701–707. [Google Scholar] [CrossRef]
- Peyraud, R.; Kiefer, P.; Christen, P.; Massou, S.; Portais, J.-C.; Vorholt, J. Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc. Natl. Acad. Sci. USA 2009, 106, 4846–4851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaszycki, P.; Koloczek, H. Biodegradation of formaldehyde and its derivatives in industrial wastewater with methylotrophic yeast Hansenula polymorpha and with the yeast-bioaugmented activated sludge. Biodegradation 2002, 13, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Paweł, K.; Henryk, K. Formaldehyde and methanol biodegradation with the methylotrophic yeast Hansenula polymorpha in a model wastewater system. Microbiol. Res. 2000, 154, 289–296. [Google Scholar] [CrossRef]
- Ni, S.; Boone, D.R. Catabolism of Dimethylsulfide and Methane Thiol by Methylotrophic Methanogens. In Biogeochemistry of Global Change: Radiatively Active Trace Gases Selected Papers from the Tenth International Symposium on Environmental Biogeochemistry, San Francisco, CA, USA, 19–24 August 1991; Oremland, R.S., Ed.; Springer: Boston, MA, USA, 1993; pp. 796–810. [Google Scholar] [CrossRef]
- Paolo Catarina, C.P.; Ana Rita, F.; Pedro, M.-F. Novel pollutant-resistant methylotrophic bacteria for use in bioremediation. FEMS Microbiol. Lett. 2004, 234, 75–80. [Google Scholar] [CrossRef]
- Mishra, V.K.; Shukla, R. Metal uptake potential of four methylotrophic bacterial strains from coal mine spoil, exploring a new possible agent for bioremediation. Environ. Technol. Innov. 2018, 11, 174–186. [Google Scholar] [CrossRef]
- Barbara, C.H.; Friedrich, S.; Babel, W. Bioremediation of acid mine water using facultatively methylotrophic metal-tolerant sulfate-reducing bacteria. Microbiol. Res. 1997, 152, 65–73. [Google Scholar] [CrossRef]
- Cue, D.; Lam, H.; Dillingham, R.L.; Hanson, R.S.; Flickinger, M.C. Genetic manipulation of Bacillus methanolicus, a gram-positive, thermotolerant methylotroph. Appl. Environ. Microbiol. 1997, 63, 1406–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.; Wang, Y.N.; Yang, J.X.; Guo, H.J.; Su, D.L.; Yu, L. Degradation of 1,4-Dioxane by Xanthobacter sp. YN2. Curr. Microbiol. 2021, 78, 992–1005. [Google Scholar] [CrossRef]
- Paul, J.T. Glutathione-dependent detoxification of α-oxoaldehydes by the glyoxalase system: Involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem.-Biol. Interact. 1998, 111–112, 137–151. [Google Scholar] [CrossRef]
- Smith, C.V.; Huang C-c Miczak, A.; Russell, D.G.; Sacchettini, J.C.; Höner zu Bentrup, K. Biochemical and Structural Studies of Malate Synthase from Mycobacterium tuberculosis. J. Biol. Chem. 2003, 278, 1735–1743. [Google Scholar] [CrossRef] [Green Version]
- Anstrom, D.M.; Kallio, K.; Remington, S.J. Structure of the Escherichia coli malate synthase G:pyruvate:acetyl-coenzyme A abortive ternary complex at 1.95 Å resolution. Protein Sci. 2003, 12, 1822–1832. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Lemire, J.; Mailloux, R.J.; Chénier, D.; Hamel, R.; Appanna, V.D. An ATP and oxalate generating variant tricarboxylic acid cycle counters aluminum toxicity in Pseudomonas fluorescens. PLoS ONE 2009, 4, e7344. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.; Li, P.L.; Zhang, L.; Piper, K.R.; Cook, D.M.; Tate, M.E.; Farrand, S.K. TraI, a LuxI homologue, is responsible for production of conjugation factor, the Ti plasmid N-acylhomoserine lactone autoinducer. Proc. Natl. Acad. Sci. USA 1994, 91, 4639–4643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gerven, N.; Waksman, G.; Remaut, H. Chapter 2—Pili and Flagella: Biology, Structure, and Biotechnological Applications. Prog. Mol. Biol. Transl. Sci. 2011, 103, 21–72. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, D.; Chang, Y.W.; Jeong, K.C.; Vogel, J.P.; Jensen, G.J. In situ structure of the Legionella Dot/Icm type IV secretion system by electron cryotomography. EMBO Rep. 2017, 18, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Cameron, T.A.; Roper, M.; Zambryski, P.C. Quantitative image analysis and modeling indicate the Agrobacterium tumefaciens type IV secretion system is organized in a periodic pattern of foci. PLoS ONE 2012, 7, e42219. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.D.; Wagner, L.M.; Graddis, T.J.; Landick, R.; Antonucci, T.K.; Gibson, A.L.; Oxender, D.L. Nucleotide sequence and genetic characterization reveal six essential genes for the LIV-I and LS transport systems of Escherichia coli. J. Biol. Chem. 1990, 265, 11436–11443. [Google Scholar] [CrossRef]
- Koyanagi, T.; Katayama, T.; Suzuki, H.; Kumagai, H. Identification of the LIV-I/LS System as the Third Phenylalanine Transporter in Escherichia coli K-12. J. Bacteriol. 2004, 186, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Moussatova, A.; Kandt, C.; O’Mara, M.; Tieleman, D. ATP-binding cassette transporters in Escherichia coli. Biochim. Biophys. Acta 2008, 1778, 1757–1771. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J.F.; Castellani, L.G.; Draghi, W.O.; Pérez-Giménez, J.; Torres Tejerizo, G.A.; Pistorio, M. Proteomic Analysis of Rhizobium favelukesii LPU83 in Response to Acid Stress. J. Proteome Res. 2019, 18, 3615–3629. [Google Scholar] [CrossRef] [PubMed]
- Kehres, D.G.; Janakiraman, A.; Slauch, J.M.; Maguire, M.E. SitABCD is the alkaline Mn2+ transporter of Salmonella enterica serovar Typhimurium. J. Bacteriol. 2002, 184, 3159–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenech-Sanchez, A.; Benedí, V.J.; Martinez-Martinez, L.; Albertí, S. Evaluation of differential gene expression in susceptible and resistant clinical isolates of Klebsiella pneumoniae by DNA microarray analysis. Clin. Microbiol. Infect. 2006, 12, 936–940. [Google Scholar] [CrossRef] [Green Version]
- Menon, A.L.; Stults, L.W.; Robson, R.L.; Mortenson, L.E. Cloning, sequencing and characterization of the [NiFe]hydrogenase-encoding structural genes (hoxK and hoxG) from Azotobacter vinelandii. Gene 1990, 96, 67–74. [Google Scholar] [CrossRef]
- Du, L.; Stejskal, F.; Tibelius, K.H. Characterization of two genes (hupD and hupE) required for hydrogenase activity in Azotobacter chroococcum. FEMS Microbiol. Lett. 1992, 96, 93–101. [Google Scholar] [CrossRef]
- Tibelius, K.H.; Du, L.; Tito, D.; Stejskal, F. The Azotobacter chroococcum hydrogenase gene cluster: Sequences and genetic analysis of four accessory genes, hupA, hupB, hupY and hupC. Gene 1993, 127, 53–61. [Google Scholar] [CrossRef]
- Du, L.; Tibelius, K.H.; Souza, E.M.; Garg, R.P.; Yates, M.G. Sequences, organization and analysis of the hupZMNOQRTV genes from the Azotobacter chroococcum hydrogenase gene cluster. J. Mol. Biol. 1994, 243, 549–557. [Google Scholar] [CrossRef]
- Hansel, A.; Axelsson, R.; Lindberg, P.; Troshina, O.Y.; Wünschiers, R.; Lindblad, P. Cloning and characterisation of a hyp gene cluster in the filamentous cyanobacterium Nostoc sp. strain PCC 73102. FEMS Microbiol. Lett. 2001, 201, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Carrica, M.d.C.; Fernandez, I.; Martí, M.A.; Paris, G.; Goldbaum, F.A. The NtrY/X two-component system of Brucella spp. acts as a redox sensor and regulates the expression of nitrogen respiration enzymes. Mol. Microbiol. 2012, 85, 39–50. [Google Scholar] [CrossRef]
- Wiegel, J. The genus Xanthobacter. In The Prokaryotes: Volume 5: Proteobacteria: Alpha and Beta Subclasses; Springer: New York, NY, USA, 2006; pp. 290–314. [Google Scholar] [CrossRef]
- Parales, R.; Ju, K.-S.; Rollefson, J.; Ditty, J. Bioavailability, chemotaxis, and transport of organic pollutants. In Microbial Biodegradation: Genomics and Molecular Biology; Parales, R., Ju, K.-S., Rollefson, J., Ditty, J., Eds.; Caister Academic Press: Norfolk, UK, 2008. [Google Scholar]
- Parales, R.; Adamus, J.; White, N.; May, H. Degradation of 1, 4-dioxane by an actinomycete in pure culture. Appl. Environ. Microbiol. 1994, 60, 4527–4530. [Google Scholar] [CrossRef] [Green Version]
- Sales, C.M. Functional Genomics of the Bacterial Degradation of the Emerging Water Contaminants: 1,4-Dioxane and N-Nitrosodimethylamine (NDMA). Ph.D. Thesis, UC Berkeley, Berkeley, CA, USA, 2012. [Google Scholar]
- Erb, T.J.; Frerichs-Revermann, L.; Fuchs, G.; Alber, B.E. The Apparent Malate Synthase Activity of Rhodobacter sphaeroides Is Due to Two Paralogous Enzymes, (3S)-Malyl-Coenzyme A (CoA)/β-Methylmalyl-CoA Lyase and (3S)-Malyl-CoA Thioesterase. J. Bacteriol. 2010, 192, 1249–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, J.; Quax, W.J. Chapter 7—Enzymatic Quorum Quenching in Biofilms. In Quorum Sensing; Vogel, J., Quax, W.J., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 173–193. [Google Scholar] [CrossRef]
- Laub, M.T.; Goulian, M. Specificity in Two-Component Signal Transduction Pathways. Annu. Rev. Genet. 2007, 41, 121–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H. Study on the Characteristics and Quorum Sensing of 1,4-Dioxane Degradation by A. Baumannii DD1. Master’s Thesis, Zhejiang Gongshang University, Hangzhou, China, 2015. [Google Scholar]
- Jakobsen, Ø.M.; Benichou, A.; Flickinger, M.C.; Valla, S.; Ellingsen, T.E.; Brautaset, T. Upregulated Transcription of Plasmid and Chromosomal Ribulose Monophosphate Pathway Genes Is Critical for Methanol Assimilation Rate and Methanol Tolerance in the Methylotrophic Bacterium Bacillus methanolicus. J. Bacteriol. 2006, 188, 3063–3072. [Google Scholar] [CrossRef] [Green Version]
- Kist, J.; Tate, R.L. Phylogeny of bacterial methylotrophy genes reveals robustness in Methylobacterium mxaF sequences and mxa operon construction. Soil Biol. Biochem. 2013, 59, 49–57. [Google Scholar] [CrossRef]
- Gedalanga Phillip, B.; Pornwongthong, P.; Mora, R.; Chiang Sheau-Yun, D.; Baldwin, B.; Ogles, D.; Mahendra, S.; Kivisaar, M. Identification of Biomarker Genes To Predict Biodegradation of 1,4-Dioxane. Appl. Environ. Microbiol. 2014, 80, 3209–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marina, G.K.; Hristova Krassimira, R.; Lidstrom Mary, E.; Chistoserdova, L. Characterization of a Novel Methanol Dehydrogenase in Representatives of Burkholderiales: Implications for Environmental Detection of Methylotrophy and Evidence for Convergent Evolution. J. Bacteriol. 2008, 190, 3817–3823. [Google Scholar] [CrossRef] [Green Version]
- Mahendra, S.; Petzold, C.J.; Baidoo, E.E.; Keasling, J.D.; Alvarez-Cohen, L. Identification of the intermediates of in vivo oxidation of 1, 4-dioxane by monooxygenase-containing bacteria. Environ. Sci. Technol. 2007, 41, 7330–7336. [Google Scholar] [CrossRef]
- Notomista, E.; Lahm, A.; Di Donato, A.; Tramontano, A. Evolution of bacterial and archaeal multicomponent monooxygenases. J. Mol. Evol. 2003, 56, 435–445. [Google Scholar] [CrossRef]
- Boucher, Y.; Douady, C.J.; Papke, R.T.; Walsh, D.A.; Boudreau, M.E.; Nesbø, C.L.; Case, R.J.; Doolittle, W.F. Lateral Gene Transfer and the Origins of Prokaryotic Groups. Annu. Rev. Genet. 2003, 37, 283–328. [Google Scholar] [CrossRef]
- Hemme, C.L.; Deng, Y.; Gentry, T.J.; Fields, M.W.; Wu, L.; Barua, S.; Barry, K.; Tringe, S.G.; Watson, D.B.; He, Z.; et al. Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J. 2010, 4, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzat, J.L.; Hoostal, M.J. Evolutionary Analysis and Lateral Gene Transfer of Two-Component Regulatory Systems Associated with Heavy-Metal Tolerance in Bacteria. J. Mol. Evol. 2013, 76, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Gogarten, J.P.; Townsend, J.P. Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 2005, 3, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a T0C | T0D | T1C | T1D | T2C | T2D |
|---|---|---|---|---|---|---|
| Number of raw reads (×106) | 10.18 | 11.10 | 8.41 | 12.05 | 10.17 | 9.67 |
| Number of clean reads (×106) | 10.15 | 11.07 | 8.39 | 12.02 | 10.15 | 9.65 |
| Clean reads Q30 (%) | 94.20 | 94.38 | 94.43 | 94.39 | 94.99 | 94.63 |
| Clean reads ratio (%) | 99.69 | 99.71 | 99.79 | 99.78 | 99.81 | 99.77 |
| Mapping ratio (%) | 98.43 | 99.17 | 98.93 | 98.65 | 98.90 | 98.66 |
| Uniquely mapping ratio (%) | 81.87 | 80.25 | 74.62 | 75.26 | 78.08 | 77.89 |
| Gene ID | Gene Name | Gene Description | b Fold Change Compared with Citrate | |||||
|---|---|---|---|---|---|---|---|---|
| T0D | T1C | T2C | T1D | T2D | ||||
| Monooxygenase gene cluster (thmABCDEF) | GE02043 | thmA | monooxygenase α subunit | a1.31 | 1.63 | 1.28 | 1.62 | 1.39 |
| GE06236 | ||||||||
| GE02042 | thmB | monooxygenase γ subunit | 1.23 | 1.72 | 1.40 | 1.55 | 1.28 | |
| GE06235 | ||||||||
| GE02041 | thmC | ferredoxin | 1.03 | 1.44 | 1.30 | 1.24 | 1.08 | |
| GE06234 | ||||||||
| GE02040 | thmD | coupling/effector protein | 1.09 | 1.51 | 1.36 | 1.40 | 1.24 | |
| GE06233 | ||||||||
| GE02039 | thmE | monooxygenase β subunit | 1.17 | 1.64 | 1.53 | 1.64 | 1.44 | |
| GE06232 | ||||||||
| GE02038 | thmF | ferredoxin oxidoreductase | 0.75 | 0.92 | 1.05 | 0.89 | 0.87 | |
| GE06231 | ||||||||
| Alcohol dehydrogenase encoding genes | GE01514 | xoxF | alcohol dehydrogenase | 1.02 | 0.60 | 2.00 | 1.09 | 2.49 |
| GE02036 | ybdR | alcohol dehydrogenase | 1.23 | 1.70 | 1.50 | 1.74 | 1.50 | |
| GE03038 | adh | alcohol dehydrogenase | 0.88 | 0.65 | 1.71 | 1.71 | 1.41 | |
| GE06014 | adh | alcohol dehydrogenase | 1.34 | 1.52 | 1.34 | 1.45 | 1.35 | |
| GE06229 | ybdR | alcohol dehydrogenase | 1.23 | 1.70 | 1.50 | 1.74 | 1.50 | |
| Methanol dehydrogenase gene cluster | GE04892 | moxR | ATPase | 8.00 | 5.94 | 9.99 | 8.92 | 15.94 |
| GE04893 | moxI | methanol dehydrogenase | 41.95 | 34.47 | 48.91 | 63.09 | 75.12 | |
| GE04894 | moxG | cytochrome c-L | 50.96 | 8.24 | 13.87 | 13.13 | 22.92 | |
| GE04895 | moxJ | methanol oxidation system protein | 14.51 | 11.39 | 19.71 | 21.19 | 35.92 | |
| GE04896 | moxF | methanol dehydrogenase | 79.15 | 72.69 | 108.39 | 135.63 | 169.09 | |
| GE04898 | moxY | methanol utilization control sensor protein | 0.78 | 0.43 | 0.92 | 0.74 | 1.71 | |
| GE04899 | moxX | methanol utilization control regulatory protein | 0.96 | 0.76 | 1.27 | 1.38 | 2.10 | |
| Glyoxalase encoding genes | GE00986 | glyoxalase | 5.86 | 6.84 | 8.06 | 6.21 | 6.77 | |
| GE04331 | 2.23 | 1.37 | 1.17 | 2.45 | 1.76 | |||
| GE04345 | 1.23 | 0.34 | 0.35 | 0.50 | 0.42 | |||
| Aldehyde dehydrogenase encoding genes | GE02965 | aldA | aldehyde dehydrogenase | 1.82 | 1.55 | 1.63 | 1.55 | 1.94 |
| GE05991 | aldHT | aldehyde dehydrogenase | 3.20 | 3.08 | 3.21 | 3.00 | 2.84 | |
| GE05994 | ald | aldehyde dehydrogenase | 4.15 | 4.27 | 3.94 | 3.78 | 3.85 | |
| Glycolate oxidase gene cluster 1 | GE01453 | glcD | glycolate oxidase subunit | 2.87 | 2.94 | 2.15 | 2.99 | 2.95 |
| GE01454 | glcE | 2-hydroxy-acid oxidase | 2.55 | 2.33 | 1.81 | 2.54 | 2.34 | |
| GE01455 | glcF | 2-hydroxy-acid oxidase | 2.22 | 2.70 | 2.00 | 2.40 | 2.35 | |
| GE01457 | glcB | malate synthase | 8.26 | 4.29 | 6.51 | 5.87 | 6.71 | |
| Glycolate oxidase gene cluster 2 | GE05986 | glcF | 2-hydroxy-acid oxidase | 1.59 | 1.99 | 1.57 | 1.32 | 1.45 |
| GE05987 | glcE | 2-hydroxy-acid oxidase | 1.68 | 2.38 | 1.82 | 1.45 | 1.70 | |
| GE05988 | glcD | FAD-binding protein | 2.95 | 3.40 | 2.41 | 2.53 | 2.39 | |
| Gene ID | Gene Name | Gene Description | b Fold Change Compared with Citrate | ||||
|---|---|---|---|---|---|---|---|
| T0D | T1C | T2C | T1D | T2D | |||
| Glyoxylate degradation gene cluster | |||||||
| GE02332 | glxR | Tartronate semialdehyde reductase | a11.89 | 10.98 | 11.89 | 13.30 | 12.95 |
| GE02333 | hyi | Hydroxypyruvate isomerase | 13.11 | 11.9 | 15.14 | 15.50 | 15.48 |
| GE02334 | gcl | Glyoxylate carboligase | 28.46 | 21.81 | 20.80 | 27.91 | 25.94 |
| Other genes related to glyoxylate degradation | |||||||
| GE05377 | gck | glycerate 2-kinase | 2.59 | 1.44 | 2.35 | 2.10 | 2.27 |
| Gene related to the ethylmalonyl–CoA pathway | |||||||
| GE05373 | phaA | Acetyl–CoA acetyltransferase | 2.31 | 1.52 | 2.99 | 2.97 | 2.71 |
| GE03297 | 1.26 | 0.96 | 0.93 | 1.11 | 1.04 | ||
| GE05374 | phbB | Acetoacetyl–CoA reductase | 1.16 | 1.06 | 1.84 | 2.08 | 1.64 |
| GE01950 | croR | 3-Hydroxybutyryl–CoA dehydratase | 1.49 | 0.95 | 1.77 | 1.57 | 1.61 |
| GE00219 | ccr | Crotonyl–CoA reductase | 2.13 | 0.78 | 1.53 | 1.44 | 1.42 |
| GE04789 | yqjC | Methylmalonyl–CoA epimerase | 0.51 | 0.40 | 0.51 | 0.42 | 0.51 |
| GE00217 | meaA | Ethylmalonyl–CoA mutase | 1.43 | 0.81 | 1.04 | 1.26 | 1.08 |
| GE01348 | yngJ | (2S)-Methylsuccinyl–CoA dehydrogenase | 1.61 | 0.63 | 1.10 | 1.16 | 1.08 |
| GE00952 | mch | 2-Methylfumaryl–CoA hydratase | 2.08 | 0.79 | 1.52 | 1.74 | 1.48 |
| GE03891 | mcl | Malyl–CoA lyase | 1.56 | 0.55 | 1.14 | 1.03 | 1.00 |
| GE00948 | mcl2 | (3S)-malyl–CoA thioesterase | 0.93 | 0.34 | 0.21 | 0.65 | 0.61 |
| GE03545 | pccA | Propionyl–CoA carboxylase alpha chain | 0.96 | 0.75 | 1.30 | 0.84 | 0.94 |
| GE03546 | pccB | Propionyl–CoA carboxylase beta chain | 1.99 | 1.20 | 2.53 | 1.46 | 1.53 |
| GE03765 | 0.21 | 0.92 | 2.00 | 1.80 | 1.68 | ||
| GE03541 | mutB | Methylmalonyl–CoA mutase | 1.13 | 1.20 | 1.24 | 1.17 | 1.20 |
| GE03543 | mutA | 0.80 | 0.71 | 0.95 | 0.73 | 0.81 | |
| GE05659 | bhbA | 0.43 | 0.67 | 0.51 | 0.52 | 0.59 | |
| GE05660 | meaA | 0.57 | 0.69 | 0.42 | 0.45 | 0.57 | |
| GE05792 | scpA | 0.55 | 0.68 | 0.29 | 0.27 | 0.25 | |
| GE05793 | bhbA | 0.77 | 0.67 | 0.34 | 0.32 | 0.31 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Ma, F.; Yang, J.; Guo, H.; Su, D.; Yu, L. Adaption and Degradation Strategies of Methylotrophic 1,4-Dioxane Degrading Strain Xanthobacter sp. YN2 Revealed by Transcriptome-Scale Analysis. Int. J. Mol. Sci. 2021, 22, 10435. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910435
Wang Y, Ma F, Yang J, Guo H, Su D, Yu L. Adaption and Degradation Strategies of Methylotrophic 1,4-Dioxane Degrading Strain Xanthobacter sp. YN2 Revealed by Transcriptome-Scale Analysis. International Journal of Molecular Sciences. 2021; 22(19):10435. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910435
Chicago/Turabian StyleWang, Yingning, Fang Ma, Jixian Yang, Haijuan Guo, Delin Su, and Lan Yu. 2021. "Adaption and Degradation Strategies of Methylotrophic 1,4-Dioxane Degrading Strain Xanthobacter sp. YN2 Revealed by Transcriptome-Scale Analysis" International Journal of Molecular Sciences 22, no. 19: 10435. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910435