
Synthetic Retinoids as Potential Therapeutics in Prostate Cancer—An Update of the Last Decade of Research: A Review

 , , , and
, , , and
Abstract
:1. Introduction
1.1. Overview
1.2. Chemistry of Retinoids
1.3. Retinoid Receptors
2. Materials and Methods
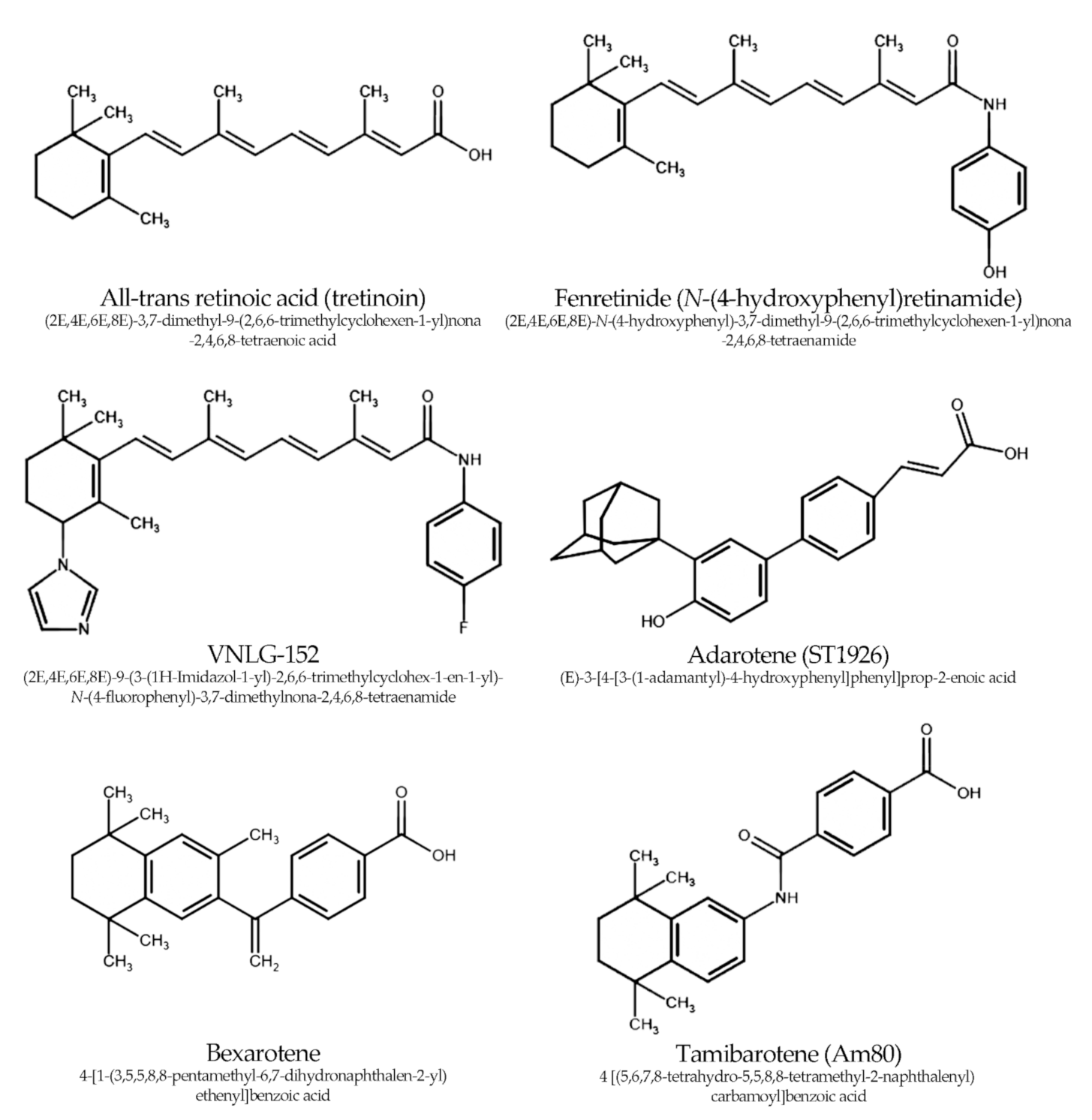
3. Synthetic Retinoids
3.1. Fenretinide—Experimental Studies
- DHODH-dependent generation of ROS (provisionally the exclusive mechanism for generation of >90% of ROS caused by fenretinide), resulting in increased apoptosis;
- RAR/RXR dependent down-regulation of the Wnt pathway (through a decrease in phosphorylated Akt and GSK-3β, followed by increased degradation of β-catenin), leading to reduced cell growth, migration, invasion, and neoangiogenesis.
3.2. Fenretinide—Clinical Studies
3.3. RAMBAs
- It induces the degradation of AR (and preferentially its AR-V7 splice variant, responsible for resistance to treatment with enzalutamide and abiraterone);
- It impairs the process of protein synthesis by promoting Mnk1 degradation and then blocks the phosphorylation of eIF4E (and possibly other proteins important for translation, for example, mTOR);
- It reverses the molecular changes responsible for the epithelial-mesenchymal transition, showing the capacity to reduce tumor invasion and metastases in vivo.
3.4. Bexarotene
- inhibition of the expression of CyD1 and E2 expression;
- TR4 antagonism followed by inhibition of the lincRNA-p21/HIF-1α/VEGF downstream pathway.
3.5. Other Synthetic Retinoids
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4E-BP1 | 4E-binding protein |
| 4HPR | N-(4-hydroxyphenyl) retinamide |
| 5-AzadCyD | 5-aza-2′-deoxycytidine |
| AR | androgen receptor |
| ATRA | all-trans-retinoic acid |
| AnRE | antioxidant response element |
| Bcl-2 | B-cell lymphoma |
| BRCA1/2 | breast cancer 1/2 |
| CyB | cyclin B |
| CyD1 | cyclin D1 |
| CI | combination index |
| CRPC | castration resistant prostate cancer |
| DHODH | dihydroorotate dehydrogenase |
| DNA | deoxyribonucleic acid |
| E-cad | E-cadherin |
| ERG | ETS-related gene |
| ETS | erythroblast transformation specific |
| GSK-3β | glycogen kinase synthase-3β |
| HDAC | histone deacetylase |
| HIF-1α | hypoxia inducible factor-1α |
| IC50 | half-maximal inhibitory concentration |
| IGF-1 | insulin-like growth factor-1 |
| MMP | matrix metalloprotease |
| Mnk1/2 | mitogen-activated protein kinase interacting kinases 1 and 2 |
| N-cad | N-cadherin |
| Nrf2 | nuclear factor-erythroid 2 p45-related factor 2 |
| PARP | poly(ADP-ribose) polymerase |
| PC | prostate cancer |
| PI3K | phosphatidylinositol 3-kinase |
| PPAR | peroxisome proliferator-activated receptor |
| RAMBA | retinoid acid metabolism blocking agent |
| RAR | retinoid acid receptor |
| RARE | retinoid acid response element |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| RXR | retinoid X receptor |
| SAHA | suberoylanilide hydroxamic acid |
| SIRT | sirtuin |
| SOD | superoxide dismutase |
| TR | thyroid receptor |
| TR4 | testicular nuclear receptor 4 |
| VDR | vitamin D receptor |
| VEGF | vascular endothelial growth factor |
| eIF4E | eukaryotic translation initiation factor 4E |
| mTOR | mammalian target of rapamycin |
References
- Rawla, P. Epidemiology of prostate cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Zeigler-Johnson, C.M.; Rennert, H.; Mittal, R.D.; Jalloh, M.; Sachdeva, R.; Malkowicz, S.B.; Mandhani, A.; Mittal, B.; Gueye, S.M.; Rebbeck, T.R. Evaluation of prostate cancer characteristics in four populations worldwide. Can. J. Urol. 2008, 15, 4056–4064. [Google Scholar]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Pearson, H.B.; Li, J.; Meniel, V.S.; Fennell, C.M.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; Macpherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca mutation as a genetic driver of prostate cancer that cooperates with Pten loss to accelerate progression and castration-resistant growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschos, A.; Pandya, R.; Duivenvoorden, W.C.M.; Pinthus, J.H. Oxidative stress in prostate cancer: Changing research concepts towards a novel paradigm for prevention and therapeutics. Prostate Cancer Prostatic Dis. 2013, 16, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Cazares, L.H.; Drake, R.R.; Esquela-Kirscher, A.; Lance, R.S.; Semmes, O.J.; Troyer, D.A. Molecular pathology of prostate cancer. Cancer Biomark. 2010, 9, 441–459. [Google Scholar] [CrossRef]
- Yu, S.; Khor, T.O.; Cheung, K.L.; Li, W.; Wu, T.Y.; Huang, Y.; Foster, B.A.; Kan, Y.W.; Kong, A.-N. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS ONE 2010, 5, e8579. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Suh, N.; Kong, A.N. Does vitamin E prevent or promote cancer? Cancer Prev. Res. 2012, 5, 701–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karppi, J.; Kurl, S.; Laukkanen, J.A.; Kauhanen, J. Serum β-carotene in relation to risk of prostate cancer: The Kuopio Ischaemic heart disease risk factor study. Nutr. Cancer 2012, 64, 361–367. [Google Scholar] [CrossRef]
- Khandrika, L.; Kumar, B.; Koul, S.; Maroni, P.; Koul, H.K. Oxidative stress in prostate cancer. Cancer Lett. 2009, 282, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Panov, A.; Orynbayeva, Z. Bioenergetic and antiapoptotic properties of mitochondria from cultured human prostate cancer cell lines PC-3, DU145 and LNCaP. PLoS ONE 2013, 8, e72078. [Google Scholar] [CrossRef] [Green Version]
- Mills, I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Rodrigues, D.N.; Gurel, B.; Clarke, M.; et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Kallio, H.M.L.; Hieta, R.; Latonen, L.; Brofeldt, A.; Annala, M.; Kivinummi, K.; Tammela, T.L.; Nykter, M.; Isaacs, W.B.; Lilja, H.G.; et al. Constitutively active androgen receptor splice variants AR-V3, AR-V7 and AR-V9 are co-expressed in castration-resistant prostate cancer metastases. Br. J. Cancer 2018, 119, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudard, S.; Fizazi, K.; Sengeløv, L.; Daugaard, G.; Saad, F.; Hansen, S.; Hjälm-Eriksson, M.; Jassem, J.; Thiery-Vuillemin, A.; Caffo, O.; et al. Cabazitaxel versus docetaxel as first-line therapy for patients with metastatic castration-resistant prostate cancer: A randomized phase III trial-FIRSTANA. J. Clin. Oncol. 2017, 35, 3189–3197. [Google Scholar] [CrossRef]
- Kiokias, S.; Proestos, C.; Varzakas, T.H. A review of the structure, biosynthesis, absorption of carotenoids-analysis and properties of their common natural extracts. Curr. Res. Nutr. Food Sci. 2015, 4, 25–37. [Google Scholar] [CrossRef]
- Dulińska-Litewka, J.; Sharoni, Y.; Hałubiec, P.; Łazarczyk, A.; Szafrański, O.; McCubrey, J.A.; Gąsiorkiewicz, B.; Laidler, P.; Bohn, T. Recent progress in discovering the role of carotenoids and their metabolites in prostatic physiology and pathology with a focus on prostate cancer—A review—Part I: Molecular mechanisms of carotenoid action. Antioxidants 2021, 10, 585. [Google Scholar] [CrossRef] [PubMed]
- Dulińska-Litewka, J.; Hałubiec, P.; Łazarczyk, A.; Szafrański, O.; Sharoni, Y.; McCubrey, J.A.; Gąsiorkiewicz, B.; Bohn, T. Recent progress in discovering the role of carotenoids and metabolites in prostatic physiology and pathology—A review—Part II: Carotenoids in the human studies. Antioxidants 2021, 10, 319. [Google Scholar] [CrossRef] [PubMed]
- The NHS Website. Available online: https://www.nhs.uk/conditions/vitamins-and-minerals/vitamin-a/ (accessed on 26 July 2021).
- Explore Chemistry. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 10 August 2021).
- Barrua, A.B.; Furr, H.C. Properties of retinoids. Mol. Biotech. 1998, 10, 167–182. [Google Scholar] [CrossRef]
- Napoli, J.L. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol. Ther. 2017, 173, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Dao, D.Q.; Ngo, T.C.; Thong, N.M.; Nam, P.C. Is vitamin A an antioxidant or a pro-oxidant? J. Phys. Chem. B. 2017, 12, 9348–9357. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.H.; Collings, J.C.; Whiting, A.; Przyborski, S.A.; Marder, T.B. Synthetic retinoids: Structure-activity relationships. Chem.—Eur. J. 2009, 15, 11430–11442. [Google Scholar] [CrossRef] [PubMed]
- Lund, B.W.; Knapp, A.E.; Piu, F.; Gauthier, N.K.; Begtrup, M.; Hacksell, U.; Olsson, R. Design, synthesis, and structure−activity analysis of isoform-selective retinoic acid receptor β ligands. J. Med. Chem. 2009, 52, 1540–1545. [Google Scholar] [CrossRef]
- le Maire, A.; Alvarez, S.; Shankaranarayanan, P.; Lera, A.R.; Bourguet, W.; Gronemeyer, H. Retinoid receptors and therapeutic applications of RAR/RXR modulators. Curr. Top. Med. Chem. 2012, 12, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, A.D.; Goncalves, M.B.; Corcoran, J.P.T. Recent advances in the design of RAR α and RAR β agonists as orally bioavailable drugs. A review. Bioorganic Med. Chem. 2020, 28, 115664. [Google Scholar] [CrossRef]
- Krężel, W.; Rühl, R.; de Lera, A.R. Alternative retinoid X receptor (RXR) ligands. Mol. Cell. Endocrinol. 2019, 491, 110436. [Google Scholar] [CrossRef]
- Li, B.; Cai, S.-Y.; Boyer, J.L. The role of the retinoid receptor, RAR/RXR heterodimer, in liver physiology. Biochim. Biophys. Acta Mol. Basis. Dis. 2021, 1867, 166085. [Google Scholar] [CrossRef]
- Detailed Review Paper on Retinoid Pathway Signalling. Available online: https://www.oecd.org/chemicalsafety/testing/draft-review-paper-retinoid-pathway-signalling.pdf (accessed on 26 July 2021).
- Li, M.-T.; Richter, F.; Chang, C.; Irwin, R.J.; Huang, H. Androgen and retinoic acid interaction in LNCaP cells, effects on cell proliferation and expression of retinoic acid receptors and epidermal growth factor receptor. BMC Cancer 2002, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Jung, B.-J.; Yoo, H.-S.; Shin, S.; Park, Y.-J.; Jeon, S.-M. Dysregulation of NRF2 in cancer: From molecular mechanisms to therapeutic opportunities. Biomol. Ther. 2018, 26, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Xue, D.; Zhou, C.; Shi, Y.; Lu, H.; Xu, R.; He, X. Nuclear transcription factor Nrf2 suppresses prostate cancer cells growth and migration through upregulating ferroportin. Oncotarget 2016, 7, 78804–78812. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, D.A.; McCabe, M.T.; Arnold, R.S.; Day, M.L. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 2008, 27, 4353–4362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Qin, Y.; Lei, F.; Chen, X.; Zhou, Z. Retinoic acid receptors α and γ are involved in antioxidative protection in renal tubular epithelial cells injury induced by hypoxia/reoxygenation. Free Radic. Res. 2017, 51, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Gad, A.; Hamed, S.A.; Khalifa, M.; Amin, A.; El-Sayed, A.; Swiefy, S.A.; El-Assal, S. Retinoic acid improves maturation rate and upregulates the expression of antioxidant-related genes in in vitro matured buffalo (Bubalus bubalis) oocytes. Int. J. Vet. Sci. Med. 2018, 6, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Das, B.C.; Thapa, P.; Karki, R.; Das, S.; Mahapatra, S.; Liu, T.C.; Torregroza, I.; Wallace, D.P.; Kambhampati, S.; Veldhuizen, P.V.; et al. Retinoic acid signaling pathways in development and diseases. Bioorganic Med. Chem. 2014, 22, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Hail, N., Jr.; Chen, P.; Kepa, J.J. Selective apoptosis induction by the cancer chemopreventive agent N-(4-hydroxyphenyl)retinamide is achieved by modulating mitochondrial bioenergetics in premalignant and malignant human prostate epithelial cells. Apoptosis 2009, 14, 849–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hail, N., Jr.; Chen, P.; Kepa, J.J.; Bushman, L.R.; Shearn, C. Dihydroorotate dehydrogenase is required for N-(4-hydroxyphenyl)retinamide-induced reactive oxygen species production and apoptosis. Free Radic. Biol. Med. 2010, 49, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Hail, N., Jr.; Chen, P.; Wempe, M.F. The hydroxyl functional group of N-(4-hydroxyphenyl)retinamide mediates cellular uptake and cytotoxicity in premalignant and malignant human epithelial cells. Free Radic. Biol. Med. 2010, 49, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- Benelli, R.; Monteghirfo, S.; Venè, R.; Tosetti, F.I.; Ferrari, N. The chemopreventive retinoid 4HPR impairs prostate cancer cell migration and invasion by interfering with FAK/AKT/GSK3β pathway and β-catenin stability. Mol. Cancer 2010, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Gouazé-Andersson, V.; Flowers, M.; Karimi, R.; Fabriás, G.; Delgado, A.; Casas, J.; Cabot, M.C. Inhibition of acid ceramidase by a 2-substituted aminoethanol amide synergistically sensitizes prostate cancer cells to N-(4-hydroxyphenyl) retinamide. Prostate 2010, 71, 1064–1073. [Google Scholar] [CrossRef]
- Mbatia, H.W.; Ramalingam, S.; Ramamurthy, V.P.; Martin, M.S.; Kwegyir-Afful, A.K.; Njar, V.C. Novel C-4 heteroaryl 13-cis-retinamide Mnk/AR degrading agents inhibit cell proliferation and migration and induce apoptosis in human breast and prostate cancer cells and suppress growth of MDA-MB-231 human breast and CWR22Rv1 human prostate tumor xenografts in mice. J. Med. Chem. 2015, 58, 1900–1914. [Google Scholar]
- Ramamurthy, V.P.; Ramalingam, S.; Gediya, L.; Kwegyir-Afful, A.K.; Njar, V.C.O. Simultaneous targeting of androgen receptor (AR) and MAPK-interacting kinases (MNKs) by novel retinamides inhibits growth of human prostate cancer cell lines. Oncotarget 2015, 6, 3195–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamurthy, V.P.; Ramalingam, S.; Gediya, L.K.; Njar, V.C.O. The retinamide VNLG-152 inhibits f-AR/AR-V7 and MNK-eIF4E signaling pathways to suppress EMT and castration-resistant prostate cancer xenograft growth. FEBS J. 2018, 285, 1051–1063. [Google Scholar] [CrossRef]
- Shen, D.; Wang, H.; Zheng, Q.; Sheng, C.; Liwei, X.; Mingchao, W.; Li, G.H.; Xia, L.Q. Synergistic effect of a retinoid X receptor-selective ligand bexarotene and docetaxel in prostate cancer. Onco Targets Ther. 2019, 12, 7877–7886. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Sun, Y.; Luo, J.; He, X.; Ye, M.; Li, G.; Zhang, Y.; Bai, J.; Zhang, D.; Chang, C. Targeting TR4 nuclear receptor with antagonist bexarotene increases docetaxel sensitivity to better suppress the metastatic castration-resistant prostate cancer progression. Oncogene 2020, 39, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Bahmad, H.F.; Samman, H.; Monzer, A.; Hadadeh, O.; Cheaito, K.; Abdel-Samad, R.; Hayar, B.; Pisano, C.; Msheik, H.; Liu, Y.-N.; et al. The synthetic retinoid ST1926 attenuates prostate cancer growth and potentially targets prostate cancer stem-like cells. Mol. Carinog. 2019, 58, 1208–1220. [Google Scholar] [CrossRef]
- Ishigami-Yuasa, M.; Ekimoto, H.; Kagechika, H. Class IIb HDAC inhibition enhances the inhibitory effect of Am80, a synthetic retinoid, in prostate cancer. Biol. Pharm. Bull. 2019, 42, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N. Antioxidant properties of N-(4-Hydroxyphenyl)retinamide (fenretinide). Biol. Pharm. Bull. 2000, 23, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuperus, R.; Leen, R.; Tytgat, G.A.; Caron, H.N.; van Kuilenburg, A.B. Fenretinide induces mitochondrial ROS and inhibits the mitochondrial respiratory chain in neuroblastoma. Cell. Mol. Life Sci. 2010, 67, 807–816. [Google Scholar] [CrossRef] [Green Version]
- Hursting, S.D.; Shen, J.C.; Sun, X.Y.; Wang, T.T.; Phang, J.M.; Perkins, S.N. Modulation of cyclophilin gene expression by N-4-(hydroxyphenyl)retinamide: Association with reactive oxygen species generation and apoptosis. Mol. Carcinog. 2002, 33, 16–24. [Google Scholar] [CrossRef]
- Stattin, P.; Bylund, A.; Rinaldi, S.; Biessy, C.; Dechaud, H.; Stenman, U.H.; Egevad, L.; Riboli, E.; Hallmans, G.; Kaaks, R. Plasma insulin-like growth factor-I, insulinlike growth factor-binding proteins, and prostate cancer risk: Aprospective study. J. Natl. Cancer Inst. 2000, 92, 1910–1917. [Google Scholar] [CrossRef] [Green Version]
- Munier-Lehmann, H.; Vidalain, P.O.; Tangy, F.; Janin, Y.L. On dihydroorotate dehydrogenases and their inhibitors and uses. J. Med. Chem. 2013, 56, 3148–3167. [Google Scholar] [CrossRef]
- Christian, S.; Merz, C.; Evans, L.; Gradl, S.; Seidel, H.; Friberg, A.; Eheim, A.; Lejeune, P.; Brzezinka, K.; Zimmerman, K.; et al. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia 2019, 33, 2403–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardy, G.W.; Brewster, S.F. Wnt signalling and prostate cancer. Prostate Cancer Prostatic Dis. 2005, 8, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, M.M.; Stockler, M.; Lim, R.; Mok, T.S.; Millward, M.; Boyer, M.J. A phase II study of fenretinide in patients with hormone refractory prostate cancer: A trial of the Cancer Therapeutics Research Group. Cancer Chemother. Pharmacol. 2010, 66, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Meyskens, F.L., Jr. Another negative chemoprevention trial: What can we learn? Clin. Cancer Res. 2008; 14, 2–3. [Google Scholar]
- Cheung, E.; Pinski, J.; Dorff, T.; Groshen, S.; Quinn, D.I.; Reynolds, C.P.; Maurer, B.J.; Lara, P.N., Jr.; Tsao-Wei, D.D.; Twardowski, P.; et al. Oral fenretinide in biochemically recurrent prostate cancer: A California cancer consortium phase II trial. Clin. Genitourin. Cancer 2009, 7, 43–50. [Google Scholar] [CrossRef]
- Andriole, G.L.; Crawford, E.D.; Grubb, R.L., III; Buys, S.S.; Chia, D.; Church, T.R.; Fouad, M.N.; Gelmann, E.P.; Kvale, P.A.; Reding, D.J.; et al. Mortality results from a randomized prostate-cancer screening trial. N. Engl. J. Med. 2009, 360, 1310–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, F.H.; Hugosson, J.; Roobol, M.J.; Tammela, T.L.; Ciatto, S.; Nelen, V.; Kwiatkowski, M.; Lujan, M.; Lilja, H.; Zappa, M.; et al. Screening and prostate-cancer mortality in a randomized European study. N. Engl. J. Med. 2009, 360, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Srivastava, J.K.; Shankar, E.; Kanwal, R.; Nawab, A.; Sharma, H.; Bhaskaran, N.; Ponsky, L.E.; Fu, P.; MacLennan, G.T.; et al. Oxidative stress and antioxidant status in high-risk prostate cancer subjects. Diagnostics 2020, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Hariton, E.; Locascio, J.J. Randomised controlled trials—The gold standard for effectiveness research. BJOG 2018, 125, 1716. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.P.; Reynolds, C.P.; Cho, H.; Kang, M.H. Clinical development of fenretinide as an antineoplastic drug: Pharmacology perspectives. Exp. Biol. Med. 2017, 242, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Martínez, A.; Sesé, M.; Losa, J.H.; Robichaud, N.; Sonenberg, N.; Aasen, T.; y Cajal, S.R. Phosphorylation of eIF4E confers resistance to cellular stress and DNA-damaging agents through an interaction with 4E-T: A rationale for novel therapeutic approaches. PLoS ONE 2015, 10, e0123352. [Google Scholar]
- Sandeman, L.Y.; Kang, W.X.; Wang, X.; Jensen, K.B.; Wong, D.; Bo, T.; Gao, L.; Zhao, J.; Byrne, C.D.; Page, A.J.; et al. Disabling MNK protein kinases promotes oxidative metabolism and protects against diet-induced obesity. Mol. Metab. 2020, 42, 101054. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Pallichankandy, S.; Thayyullathil, F.; Galadari, S. Critical role of H2O2 in mediating sanguinarine-induced apoptosis in prostate cancer cells via facilitating ceramide generation, ERK1/2 phosphorylation, and Par-4 cleavage. Free Radic. Biol. Med. 2019, 134, 527–544. [Google Scholar] [CrossRef]
- Yen, W.C.; Lamph, W.W. A selective retinoid X receptor agonist bexarotene (LGD1069, Targretin) prevents and overcomes multidrug resistance in advanced prostate cancer. Prostate 2006, 66, 305–316. [Google Scholar] [CrossRef]
- Wu, J.; Wang, H.; Tang, X. Rexinoid inhibits Nrf2-mediated transcription through retinoid X receptor alpha. Biochem. Biophys. Res. Commun. 2014, 452, 554–559. [Google Scholar] [CrossRef]
- Hacioglu, C.; Kar, F.; Kanbak, G. Ex vivo investigation of bexarotene and nicotinamide function as a protective agent on rat synaptosomes treated with Aβ(1–42). Neurochem. Res. 2021, 46, 804–818. [Google Scholar] [CrossRef]
- Zuo, Y.; Huang, L.; Enkhjargal, B.; Xu, W.; Umut, O.; Travis, Z.D.; Zhang, G.; Tang, J.; Liu, F.; Zhang, J.H. Activation of retinoid X receptor by bexarotene attenuates neuroinflammation via PPARγ/SIRT6/FoxO3a pathway after subarachnoid hemorrhage in rats. J. Neuroinflammation 2019, 16, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The role of sirtuins in antioxidant and redox signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.E.; Suino-Powell, K.M.; Xu, Y.; Chan, C.W.; Tanabe, O.; Kruse, S.W.; Reynolds, R.; Engel, J.D.; Xu, H.E. The orphan nuclear receptor TR4 is a vitamin A activated nuclear receptor. J. Biol. Chem. 2011, 286, 2877–2885. [Google Scholar] [CrossRef] [Green Version]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef] [PubMed]
- Lacher, S.E.; Levings, D.C.; Freeman, S.; Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018, 19, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Aouad, P.; Saikali, M.; Abdel-Samad, R.; Fostok, S.; El-Houjeiri, L.; Pisano, C.; Talhouk, R.; Darwiche, N. Antitumor activities of the synthetic retinoid ST1926 in two-dimensional and three-dimensional human breast cancer models. Anti Cancer Drugs 2017, 28, 757–770. [Google Scholar] [CrossRef]
- Menzel, A.; Samouda, H.; Dohet, F.; Loap, S.; Ellulu, M.S.; Bohn, T. Common and novel markers for measuring inflammation and oxidative stress ex vivo in research and clinical practice—Which to use regarding disease outcomes? Antioxidants 2021, 10, 414. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, A.; Asou, N.; Atsuta, Y.; Sakura, T.; Ueda, Y.; Sawa, M.; Dobashi, N.; Taniguchi, Y.; Suzuki, R.; Nakagawa, M.; et al. Tamibarotene maintenance improved relapse-free survival of acute promyelocytic leukemia: A final result of prospective, randomized, JALSG-APL204 study. Leukemia 2019, 33, 358–370. [Google Scholar] [CrossRef]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding failure and improving treatment using HDAC inhibitors for prostate cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Compound Name | Experimental Model | Concentration Range or Dose Used | Investigated Feature | Results | Comment | Reference |
|---|---|---|---|---|---|---|
| fenretinide (4HPR) | (1) DU145 (2) DU145 lacking mtDNA (ρ0 clones) | 5.0 µM for 24 h (for some experiments the time of culturing with fenretinide was 1–2 h or up to 48 h) | Apoptosis ROS generation Immunoblot | (1) 80% hypoploid cells vs. 20% in control ACO2↓50%, Bcl-xL↓40%, COII↓50%, EGFR↓55%, MnSOD↑40% (β-actin n/c) ROS generation rate was 6.5-fold higher than in control (2) hypoploidy was similar to control (10–15%) ROS generation rate was similar to the control | The main results of the study suggest that the anti-cancer effects of 4HPR in PC depend strongly on the ROS generation during oxidative phosphorylation. ROS generation was 3-fold greater after addition of 5.0 µM HQNO (compared to 4HPR alone). The generation of ROS and mitochondrial disruption was reduced by NAC. | Hail Jr. et al., 2009 [39] |
| fenretinide (4HPR) | (1) LNCaP (2) PC3 (3) DU145 | 5.0 µM for 150 min | ROS generation rate Apoptosis | 4HPR induced ROS production in DU145 6-fold greater in compared to the control inhibition of DHODH by TFN caused reduction of 90% ROS generation, to similar level to the control | Probably the main mechanism of 4HPR is generation of ROS mediated by DHODH. | Hail Jr. et al., 2010 [40] |
| fenretinide (4HPR), fenretinide metabolite 4MPR, fenretinide derivative 4TPR | LNCaP | 10.0 µM for 24 h (the time of culturing for DiOC6 retention assessment was 6 h) | Apoptosis ROS generation | 60% preapoptotic cells after 4HPR; to 5% of apoptotic cells in control and for 4MPR or 4TPR 80% hypoploid cells vs. 20% in control 4HPR induced ROS generation rate was 9.0× higher the in control; 4MRP and 4TPR were similar to control | The study design incorporated two (natural and synthetic) analogs of 4HPR that were devoid of free hydroxyl function group. As these substances were ineffective in induction of both ROS generation and apoptosis, it seems likely that hydroxyl group is vital for pharmacological activity of 4HPR. | Hail Jr. et al., 2010 [41] |
| fenretinide (4HPR) | (1) DU145 (2) PC3 | 2.5–10.0 µM for 4–96 h | Cell growth Migration Invasion Immunoblot | (1) cell growth after 24 h similar to control for all concentrations; after 96 h reduced to about 50% of control for 10.0 µM 4HPR migration after 96 h of exposure reduced by 2.5/5.0/10.0 µM 4HPR to 70/70/50%, respectively invasion after 96 h of exposure reduced by 2.5/5.0/10.0 µM 4HPR to 80/50/50%, respectively p-Fak↓, p-Akt↓, β-catenin↓, p-β-catenin↓, p-GSK-3β↓, CyD1↓, survivin↓after 4 h VEGF secretion↓after 16 h (2) cell growth after 24 h similar to control for all concentrations; after 96 h reduced to about 40% of control for 10.0 µM 4HPR migration after 96 h of exposure reduced by 2.5/5.0/10.0 µM 4HPR to 80/50/50%, respectively (effects independent of ROS production) invasion after 96 h of exposure reduced by 2.5/5.0/10.0 µM 4HPR to 60/40/30%, respectively p-Fak↓, p-Akt↓after 4 h VEGF secretion↓after 16 h | ROS scavengers such as 10.0 mM NAC or 1.0 µM DPI did not reduce the migration inhibiting effect of 4HPR in PC3 cells although 4HPR-dependent ROS production is significantly reduced. NAC do not change 4HPR’s ability to modulate β-catenin signaling. The specific PI3K/Akt inhibitors (200.0 nM wortmannin and 10.0 µM LY294002) act synergistically with 4HPR in reduction of cell migration. In DU145 4HPR reduces effect of stimulation with 100 ng/mL IGF-1 on migration and p-Akt level. Cells transfected with constitutionally active Myr.Akt are not susceptible to these effects. | Benelli et al., 2010 [42] |
| fenretinide (4HPR) | (1) PC3 (2) DU145 | 0.0–50.0 µM for 72 h (in the most of experiments 4HPR was used together with DM102) | Cell viability Combination index CASPs activity cPARP Ceramide level ROS generation | (1) cell viability was reduced with concentrations higher than 30.0 µM of 4HPR CIs for 2.5/5.0/10.0 µM 4HPR and 10 µM DM102 were 0.061/0.021/0.008, respectively CASP3/7 activity n/c; if 4HPR was given together with DM102 CASP3/7 activity↑200–300% cPARP↑ ceramide↑6× after 24 h exposure to 10.0 µM 4HPR ROS level↑30× after 24 h exposure to 10.0 µM 4HPR (2) 25.0 µM 4HPR reduced cell viability to 78%; combined with 15.0 µM reduced cell viability to 15% (CI 0.2) | 4HPR and DM102 synergistically reduce the viability of PC3 cells. Synergy of 4HPR and NOE was observed only for high concentrations of NOE (50 µM). CASPs activity, ceramide level, and ROS production are higher after simultaneous exposure to 4HPR and DM102. Myriocin failed to rescue PC3 cells from the cytotoxicity induced by the combination of these compounds. 250.0 µM of vitamin E reduced the cytotoxic effect of 4HPR on PC3 cells. | Gouaze’-Andersson et al., 2011 [43] |
| lead compounds: (A) VNHM-1-81 (B) VNHM-1-731 | (1) LNCaP (2) CWR22Rv1 (3) PC3 (4) castrated mice bearing CWR22Rv1 xenografts | 0.6–20.0 µM for 24 h | Cell growth Immunoblot Migration | (A) IC50 (µM) in LNCaP/CWR22Rv1/PC3: 2.69 ± 0.14/2.04 ± 0.01/5.62 ± 0.03 in LNCaP fAR↓(to 0% at 20.0 µM), Mnk1↓, Mnk2 n/c, p-eIF4E↓, CyD1↓, cPARP↑(β-actin n/c) cells migration reduced DHT-dependent AR signaling was 7-fold reduced in LNCaP after 18h treatment with 10.0 µM VNHM-1–81 (B) IC50 (µM) in LNCaP/CWR22Rv1/PC3: 1.69 ± 0.07/1.86 ± 0.06/3.54 ± 0.02 in LNCaP fAR↓(to 0% at 15 µM), Mnk1↓ (to 0% at 10.0 µM), Mnk2↓, p-eIF4E↓(to 0% at 5.0 µM), CyD1↓, cPARP↑(from 10.0 µM) (β-actin n/c) cells migration reduced DHT-dependent AR signaling was 2-fold reduced in LNCaP after 18h treatment with 10.0 µM VNHM-1–73 in mice model treatment with 20 mg/kg of VNHM-1-73 for 5 days per week for one month resulted in %T/C equal to 38.8% (p = 0.0001); immunoblot of cancer tissue showed: AR↓, AR-V7↓, Mnk1/2↓, eIF4E n/c, p-eIF4E↓, CyD1↓, Bcl-2↓, Bad↑(GAPDH n/c) | LNCaP cells transfected with si-AR and/or si-Mnk1 did not show an effect of treatment with VNHM-1-81. Supposing that VNHM-1-81 acts on them at the post-transcriptional stage. | Mbatia et al., 2015 [44] |
| lead compound VNLG-1522 | (1) LNCaP (2) CWR22Rv1 (3) C4-2B | 0.6–20.0 µM for 24 h | Apoptosis Cell growth Colony formation Immunoblot Cell cycle | after exposure to 5.0 µM VNLG-152 apoptosis in LNCaP cells was 2.5× of observed in control colony formation reduced to 15–25% of control (in LNCaP, CWR22Rv1, C4-2B) by 5.0 µM VNLG-152 after exposure to 10.0 µM VNLG-152 (results compared to control for LNCaP/CWR22Rv1/C4-2B, respectively): Mnk1↓(7/7/9%), AR↓(12/22/51%), eIF4E↓(5/9/8%), p-eIF4Eser209↓(9/5/8%), PSA↓(24/11/17%) (β-actin n/c) after exposure to 5.0 µM VNLG-152 (results compared to control for LNCaP/CWR22Rv1/C4-2B, respectively): CyD1↓(18/53/10%), CyB↓(3/53/18%), Bax↑(350/390/350%), cPARP↑(450/660/510%) | AR and Mnk1 are the most important targets of VNLG-152. They are reduced through the posttranslational mechanism. | Ramamurthy et al., 2015 [45] |
| VNLG-152 | CRPC tumor xenograft model (CWR22Rv1 cells in castrated mice) | 10 or 20 mg/kg, twice daily, for 5 days | TGI Immunoblot | VNLG-152 vs. vehicle: TGI 63.4% (at dose 10 mg/kg) or 76.3% (at dose 20 mg/kg) at dose 10 mg/kg: AR-V7↓70%, Mnk1/2↓90%, p-eIF4E↓60%, PSA↓60%, CyD1↓50%, Bcl-2↓75%, Bax↑500%, CASP3↑1500%, cPARP↑500%, E-cad↑250%, N-cad↓10%, β-catenin↓50%, claudin n/c, Snail↓5%, Slug↓20%, Twist↓50%, vimentin n/c, MMP2/9↓90–95% at dose 20 mg/kg: AR-V7↓95%, Mnk1/2↓90%, p-eIF4E↓80%, PSA↓90%, CyD1↓55%, Bcl-2↓90%, Bax↑1250%, CASP3↑1500%, cPARP↑500%, E-cad↑300%, N-cad↓80%, β-catenin↓99%, claudin↓90%, Snail↓50%, Slug↓50%, Twist↓80%, vimentin↓10%, MMP2/9↓90–95% | VNLG-152 blocks the pathways responsible for EMT. | Ramamurthy et al., 2018 [46] |
| bexarotene | (1) PC3 (2) DU145 | 20.0–40.0 µM for 24–48 h (combined with 5.0–10.0 nM docetaxel) | Cell cycle Apoptosis Immunoblot | bexarotene caused cell cycle arrest in G1 phase at 40.0 µM in both cell lines IC50: in PC3 40.6 ± 0.5 μM, in DU145 50.2 ± 4.1 μM CyB1↓, CDK1↓ in both cell lines (1) percentages of apoptotic cells: DMSO (control): 1.66%, bexarotene at 20.0 µM: 3.28%, bexarotene at 40.0 µM: 3.11%, bexarotene at 20.0 µM + docetaxel at 10.0 µM: 3.3%, bexarotene at 40.0 µM + docetaxel at 10.0 µM: 4.14% (2) percentages of apoptotic cells: DMSO (control): 4.67%, bexarotene at 20.0 µM: 9.9%%, bexarotene at 40.0 µM: 10.2%, bexarotene at 20.0 µM + docetaxel at 5.0 µM: 18.2%, bexarotene at 40.0 µM + docetaxel at 5.0 µM: 17.71% | Bexarotene acts synergistically with docetaxel in lines representing CRPC. The mechanism involves inhibition of CyB1 and CDK1. | Shen et al., 2019 [47] |
| bexarotene | (1) PC3 (2) DU145 (3) C4-2B (4) CWR22Rv1 (5) clinical samples of PC tissues | 0.0–24.0 µM for 24 h (combined with 0.0–400.0 nM docetaxel) | TR4 antagonism | TR4↑ after docetaxel chemotherapy chemoresistance was reduced after suppressing TR4 with bexarotene bexarotene at doses <8.0 μM did not influence cell proliferation in PC3 and DU145 bexarotene at 8.0 μM increased chemosensitivity of PC3 and DU145 cells with overexpression of TR4 and decreased proliferation of chemoresistant CWR22Rv1 and C4-2B cells | TR4 might be elevated in docetaxel-resistant PC. Targeting TR4/lincRNA-p21/ HIF-1α/VEGF signaling with bexarotene may increase the prostate cancer cells’ chemo-sensitivity to docetaxel. | Hu et al., 2020 [48] |
| adarotene (ST1926) | (1) DU145 (2) PC3 mouse PC cell lines: (3) PLum-AD (4) PLum-AI (5) PC xenografts in mice | 0.5–10.0 µM for 48 h | Cell growth Invasion Migration Cell cycle Apoptosis Sphere formation | cell growth reduced (in DU145/PC3/Plum-AD/Plum-AI, respectively) by 1.0 µM ST1926 to: 60/65/10/30% and by 10.0 µM ST1926 to: 50/50/5/25% number of migrating cells reduced by 1.0 µM ST1926 to 1000 (vs. 270 in control) in DU145 and to 500 (vs. 2500) in PC3 the percentage of sub-G1 increased by 1.0 µM ST1926 to 40% (vs. 10% in control) in DU145 and to 30% (vs. 10% in control) in PC3; arrest in S phase induced in 30% (vs. 15% in control) of DU145 and in 25% (vs. 15% in control) of PC3 the percentage of sphere forming colonies after 11 days of treatment with 0.01 µM ST1926 (1st sphere generation): 9% (vs. 12% in control) in DU145–mean sphere diameter 45 µm, 8% (vs. 12% in control) in PC3–mean sphere diameter 80 µm | ST1926 attenuates ATRA-resistant prostate cancer cells’ growth and potentially targets prostate cancer stem-like cells. | Bahmad et al., 2019 [49] |
| tamibarotene (Am80) | (1) LNCaP (2) PC3 | (1) 0.1–100.0 µM for 72 h to IC50 assessment (2) 25.0 µM for synergy studies (with 1.6–2.6 µM SAHA and 9.5–13.0 µM 5-AzadCyD) for 24–72h | Growth inhibition Apoptosis Immunoblot PSA in cell culture supernatant HDAC activity | IC50 in LNCaP: 36.0 µM, in PC3: 52.0 µM synergy with SAHA or 5-AzadCyD against LNCaP (with ↓PSA), but not against PC3 Am80 + SAHA increased apoptosis from 35.5% (Am80 alone) to 38.1% RARα↑70% (for Am80 + SAHA:↓20%) | The synergy between tamibarotene and SAHA results from inhibition of class IIB HDAC. Tamibarotene alone or SAHA alone increase the expression of RARα, however, combined they decrease it. | Ishigami-Yuasa et al. 2019 [50] |
| Number of PC Cases | Patients | Dose of Fenretinide | Duration of the Study | Study Endpoints | Results | Reference |
|---|---|---|---|---|---|---|
| 23 | Patients with PSA ≥2 ng/mL after radical prostatectomy and/or radical radiotherapy (with metastases excluded). Ethnicity: Americans Age: 69 years (median) | 900 mg/m2 of body surface area twice daily for 1 week every 3 weeks | 1 year, follow-up: 17.7 months (median) | PSA decline ≥50% or ≥5 ng/mL PSA-stable disease Time to PSA progression Probability of no PSA-progression in 6 months | 0% 30% (95%CI: 14–52%) 4.6 months (95%CI: 3.2–8.2) 0.37 ± 0.10 | Cheung et al., 2009 [60] |
| 27 | Patients after castration with rising PSA >10 ng/mL. Ethnicity: Australians, Asians Age: 74 years (median) | 900 mg/m2 of body surface area twice daily for 1 week every 3 weeks | 1 year | PSA decline >50% for at least 3 weeks PSA-stable disease for 6 weeks Time to treatment failure (PSA-based assessment) | 4% (maximum of 39 days) 52% 54 days | Moore et al. 2010 [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hałubiec, P.; Łazarczyk, A.; Szafrański, O.; Bohn, T.; Dulińska-Litewka, J. Synthetic Retinoids as Potential Therapeutics in Prostate Cancer—An Update of the Last Decade of Research: A Review. Int. J. Mol. Sci. 2021, 22, 10537. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910537
Hałubiec P, Łazarczyk A, Szafrański O, Bohn T, Dulińska-Litewka J. Synthetic Retinoids as Potential Therapeutics in Prostate Cancer—An Update of the Last Decade of Research: A Review. International Journal of Molecular Sciences. 2021; 22(19):10537. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910537
Chicago/Turabian StyleHałubiec, Przemysław, Agnieszka Łazarczyk, Oskar Szafrański, Torsten Bohn, and Joanna Dulińska-Litewka. 2021. "Synthetic Retinoids as Potential Therapeutics in Prostate Cancer—An Update of the Last Decade of Research: A Review" International Journal of Molecular Sciences 22, no. 19: 10537. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910537