
TNFSF14-Derived Molecules as a Novel Treatment for Obesity and Type 2 Diabetes

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
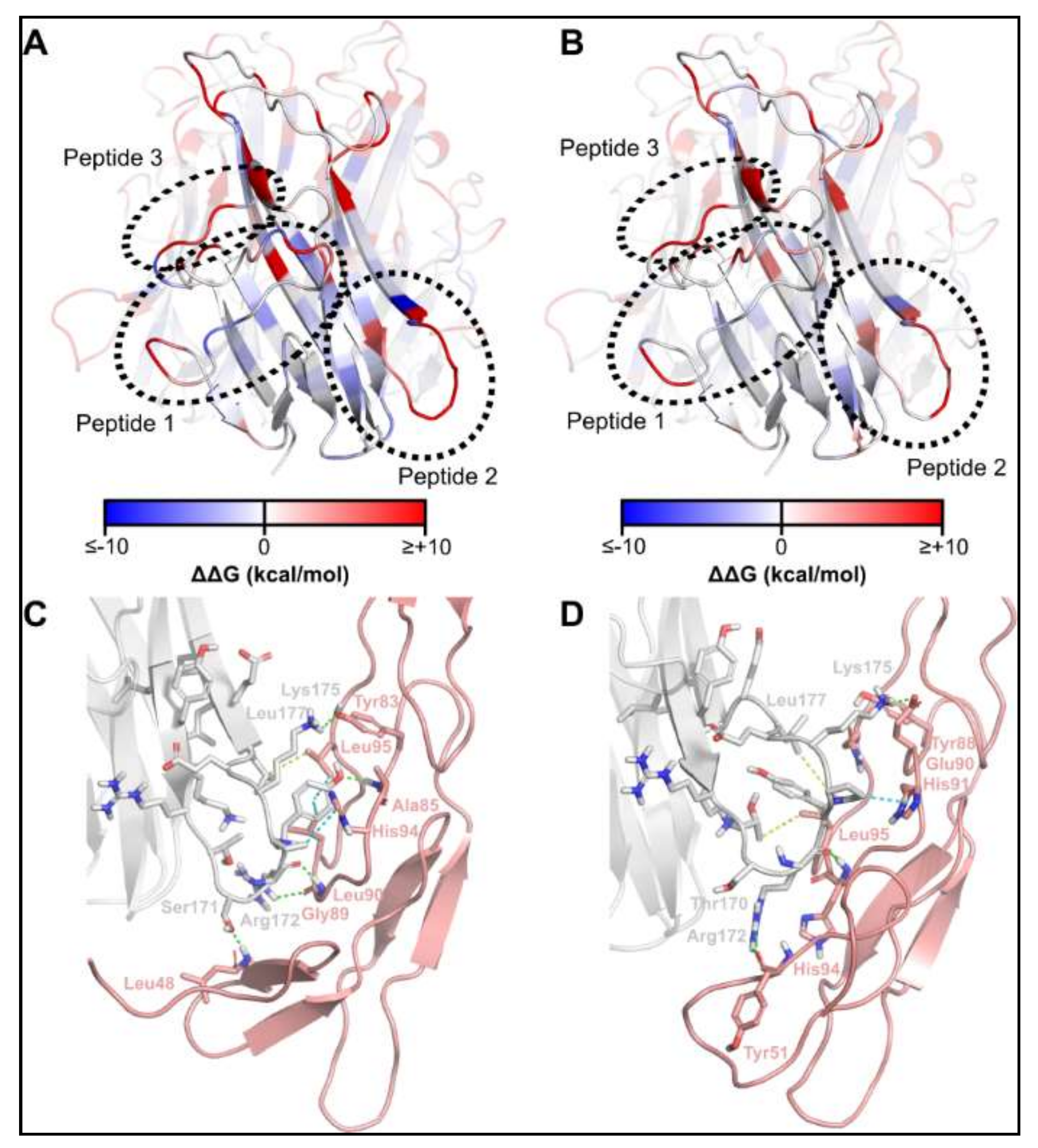
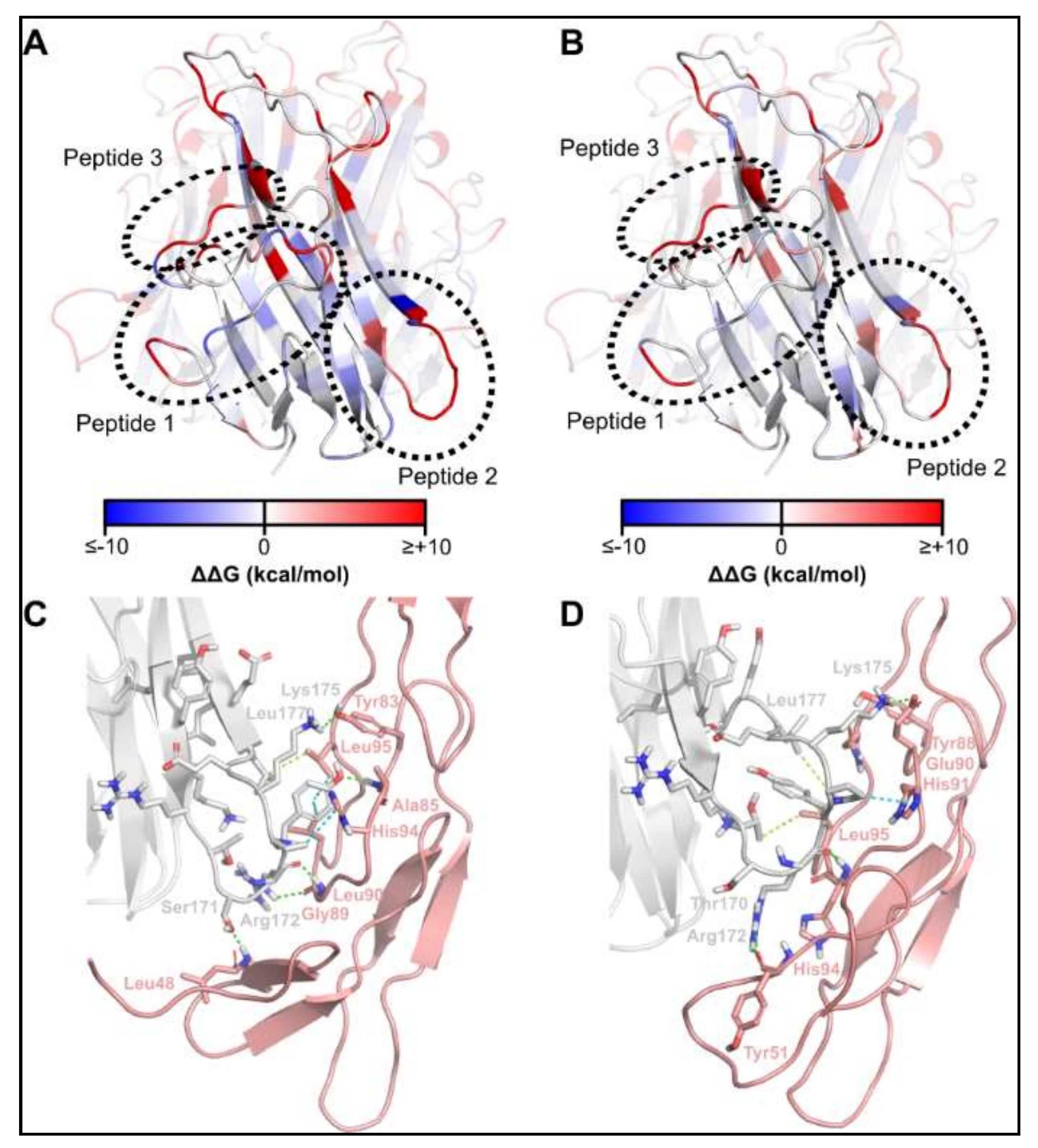
2.1. Identification of TNFSF14 Regions Contributing to Receptor Binding
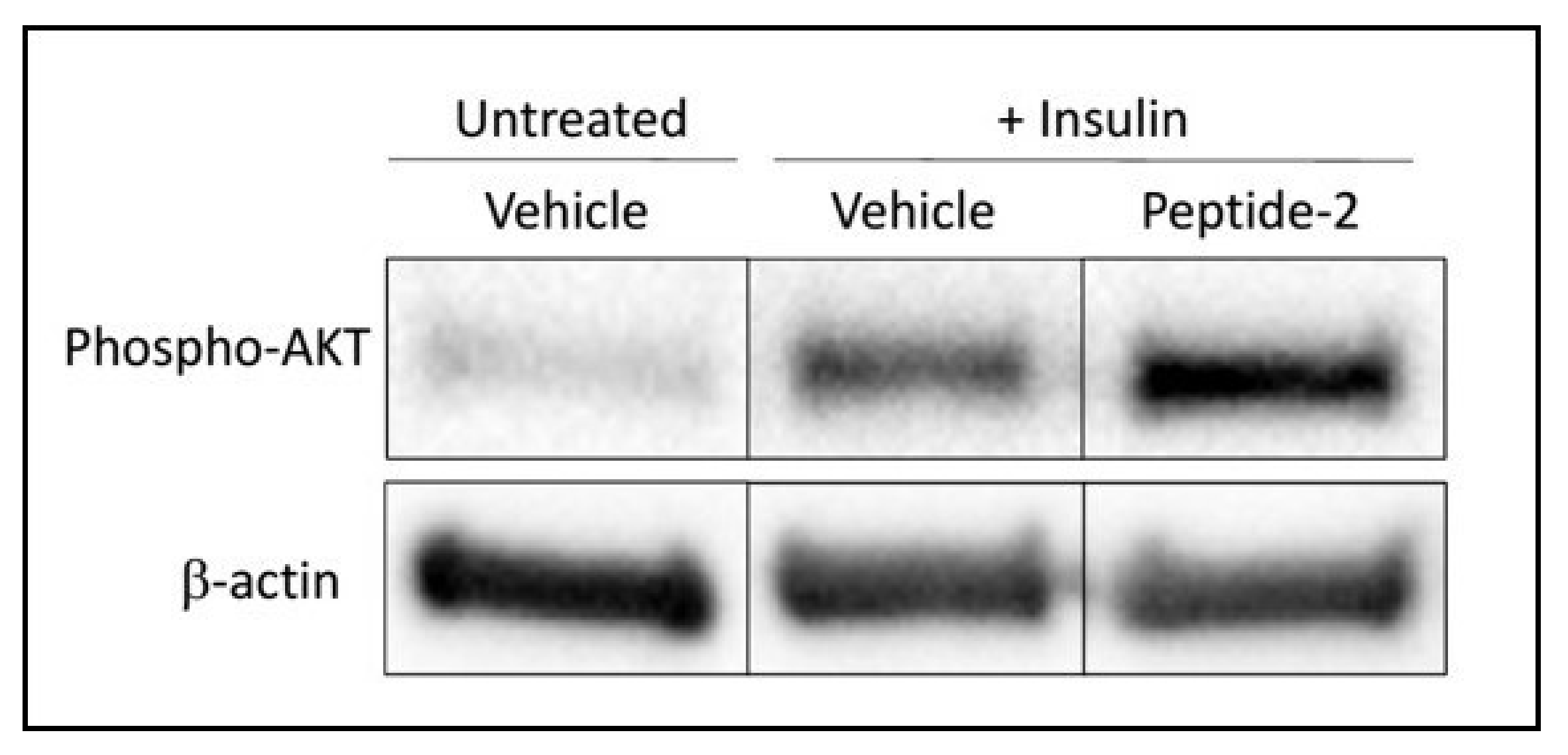
2.2. TNSFS14 Peptide-2 Promotes Insulin Sensitivity In Vitro
2.3. Optimisation of the TNFSF14 Peptide-2
2.4. Synthesis of TNFSF14 pep2 Analogues
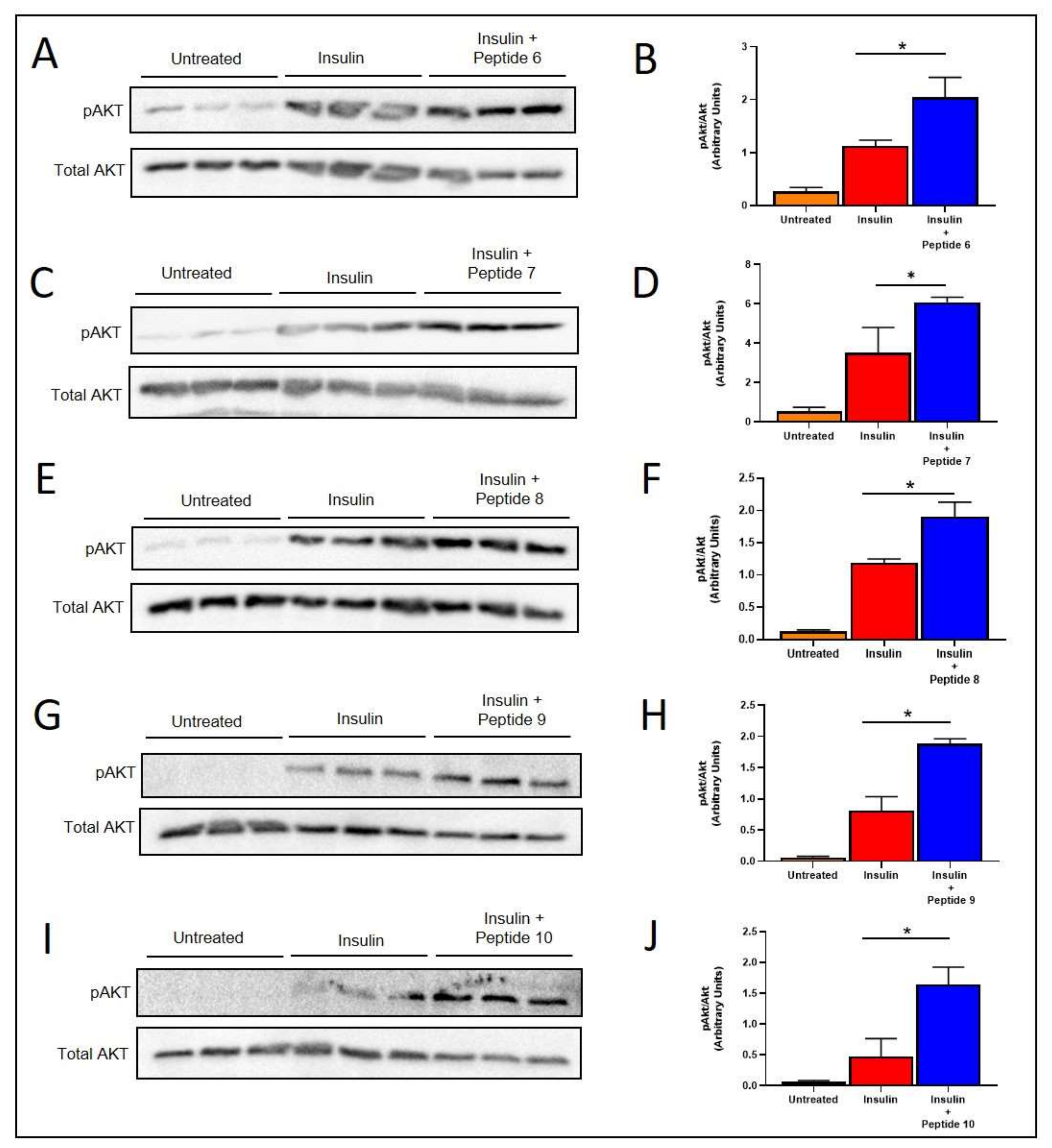
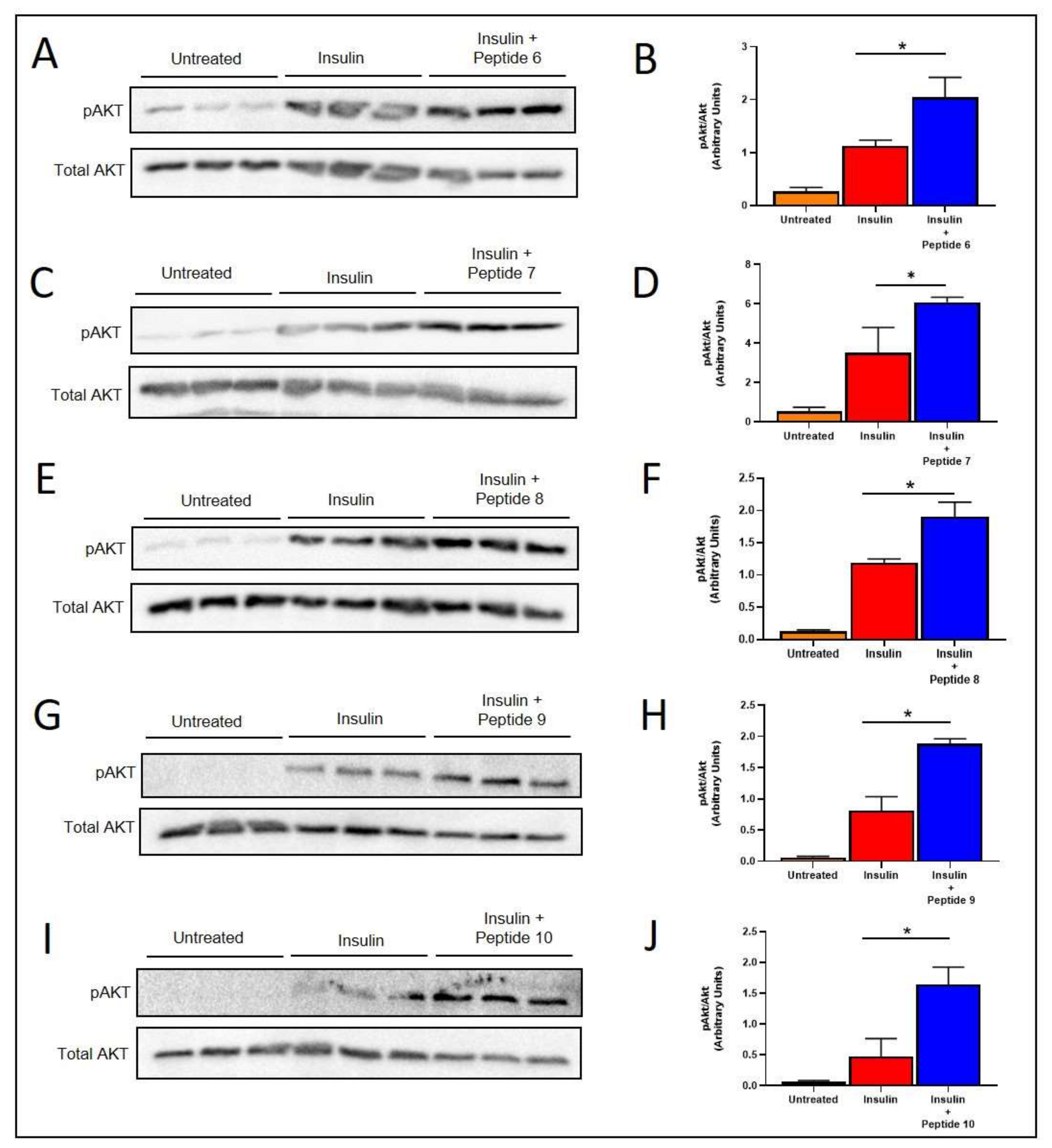
2.5. TNFSF14 Peptides Promote Insulin Signalling in L6 Skeletal Muscle Myotubes
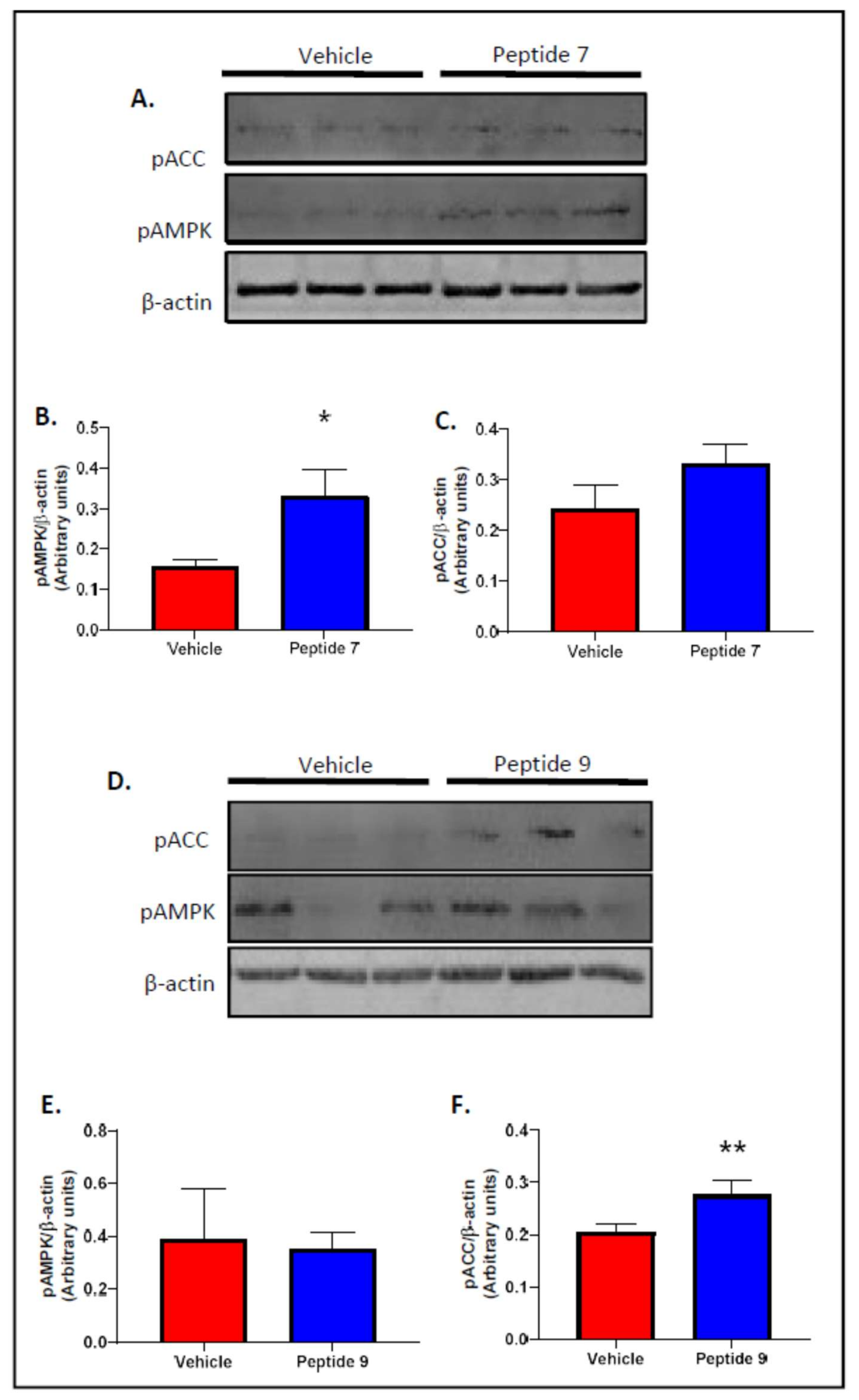
2.6. Peptides 7 and 9 Increased Fatty Acid Oxidation Signalling in Skeletal Muscle
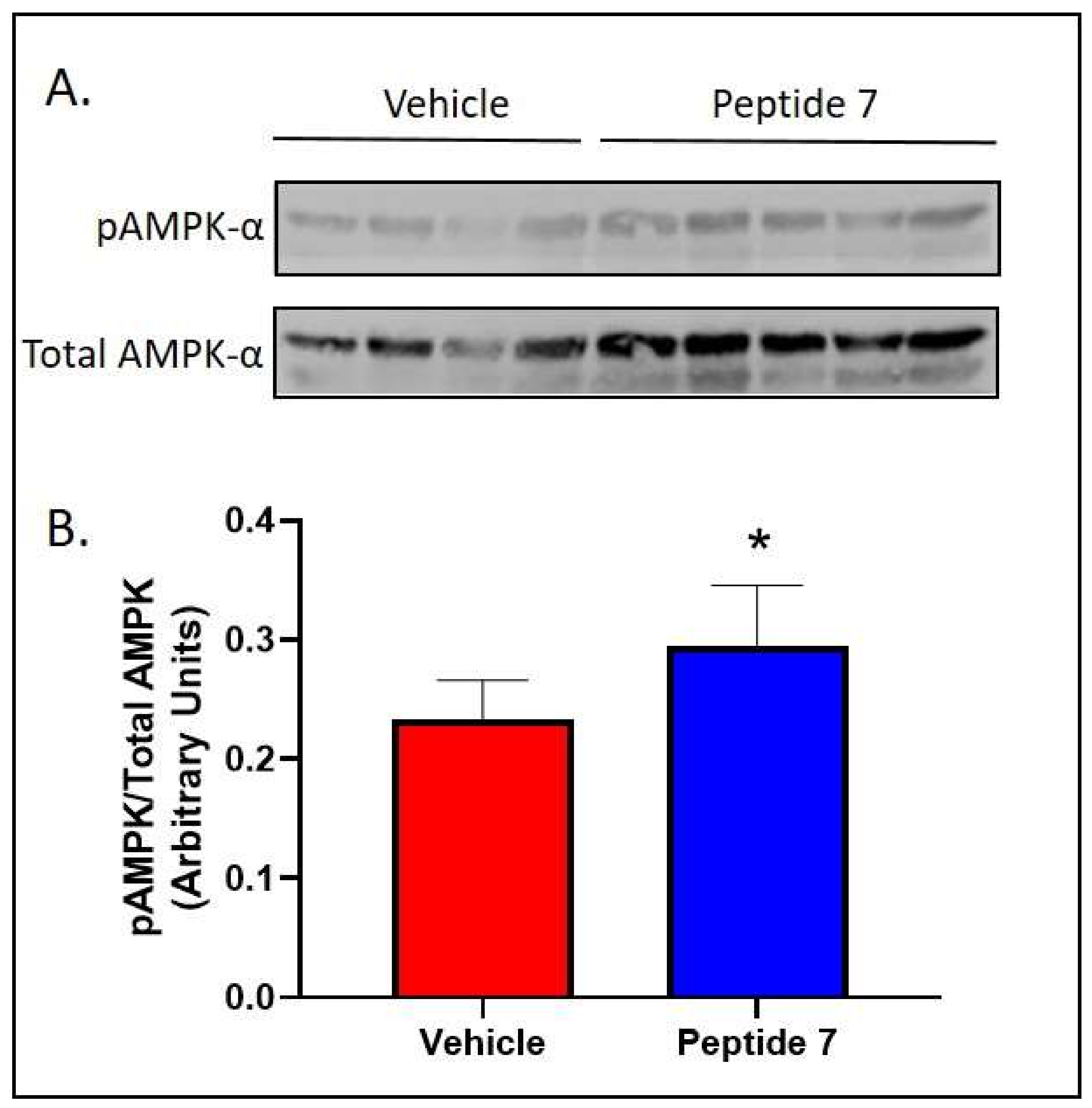
2.7. Peptide 7 Increased Fatty Acid Oxidation Signalling in the Liver
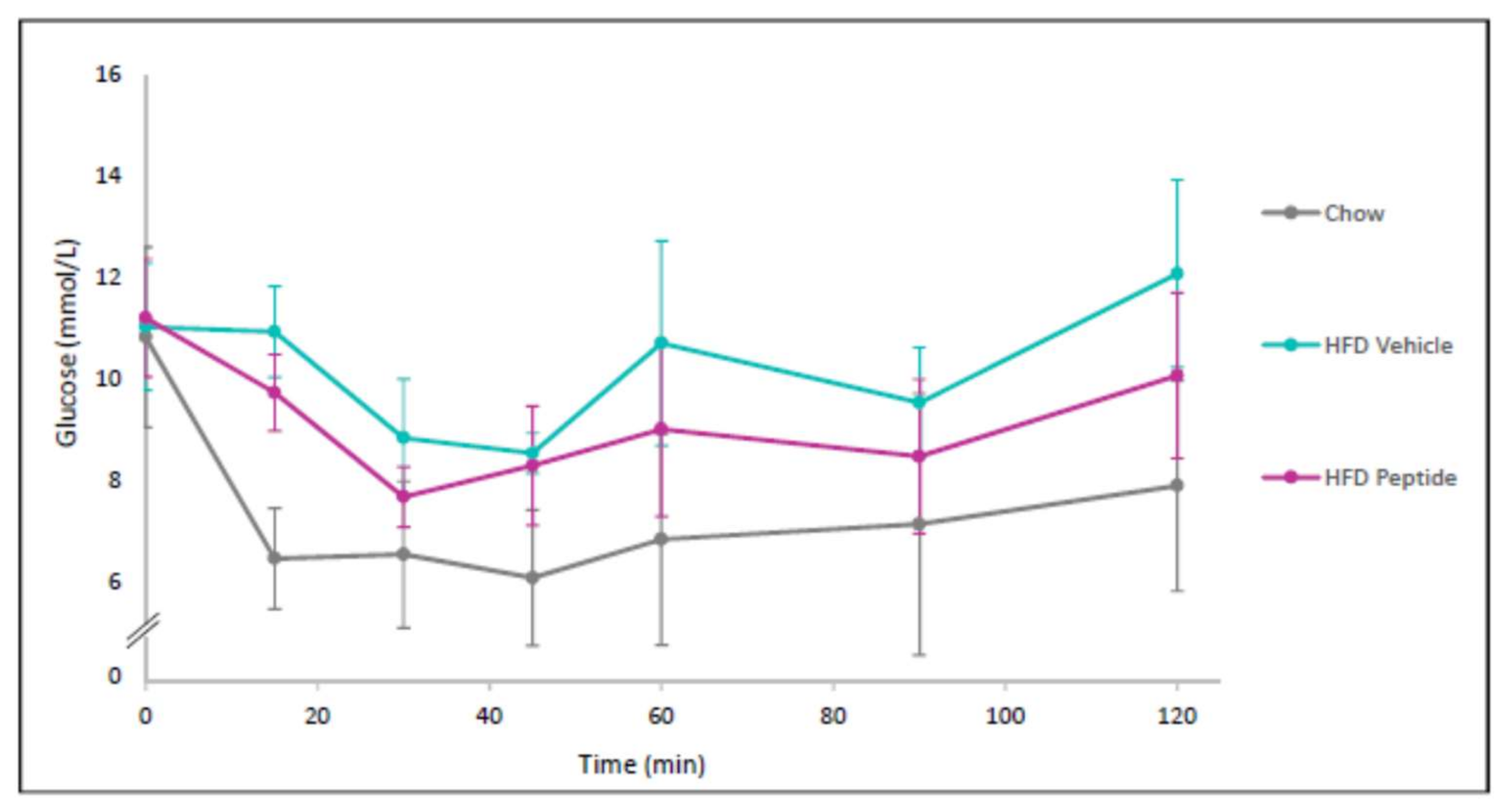
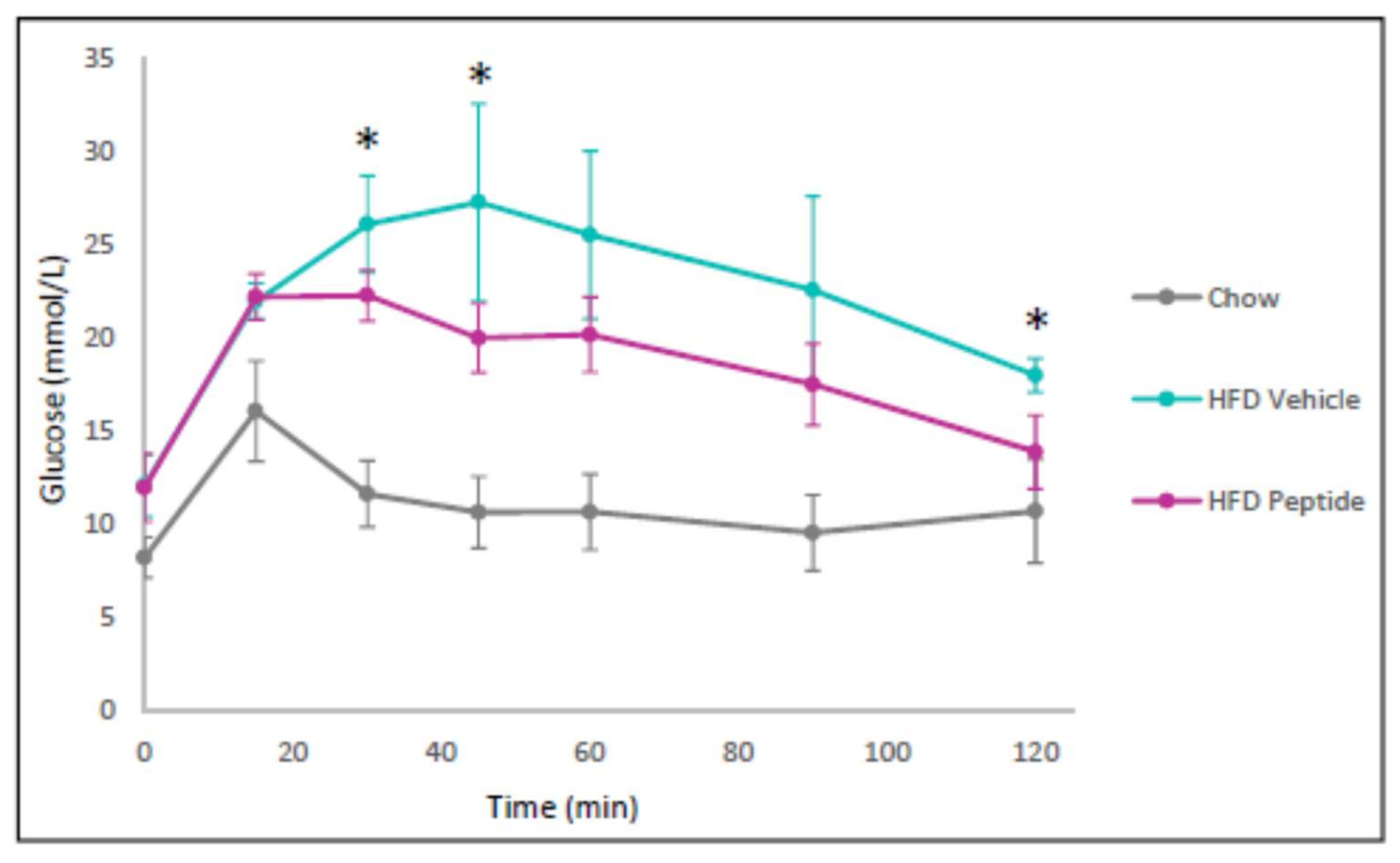
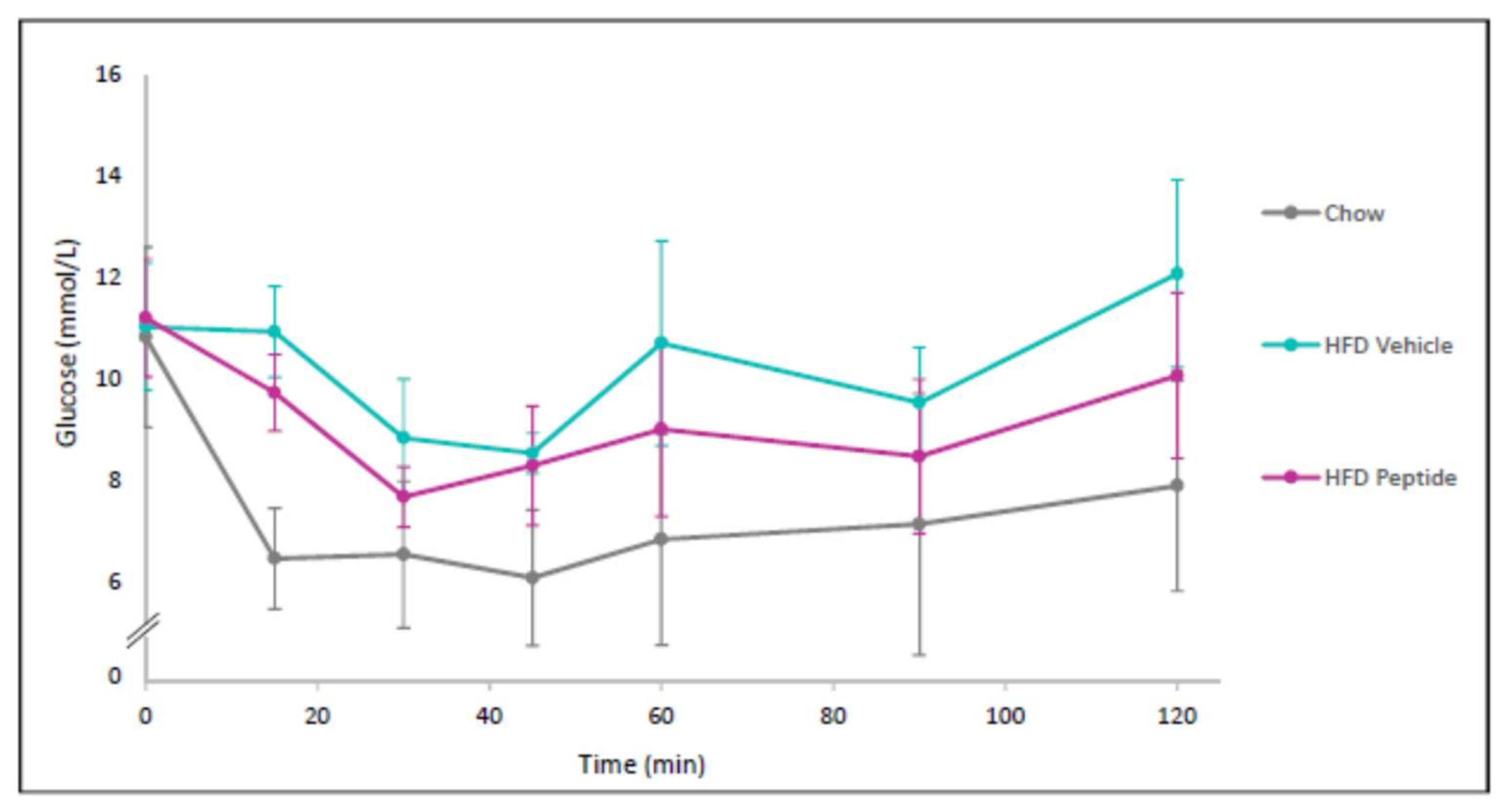
2.8. The TNFSF14 Peptide 7 Decreased Hyperglycaemia and Glucose Intolerance In Vivo
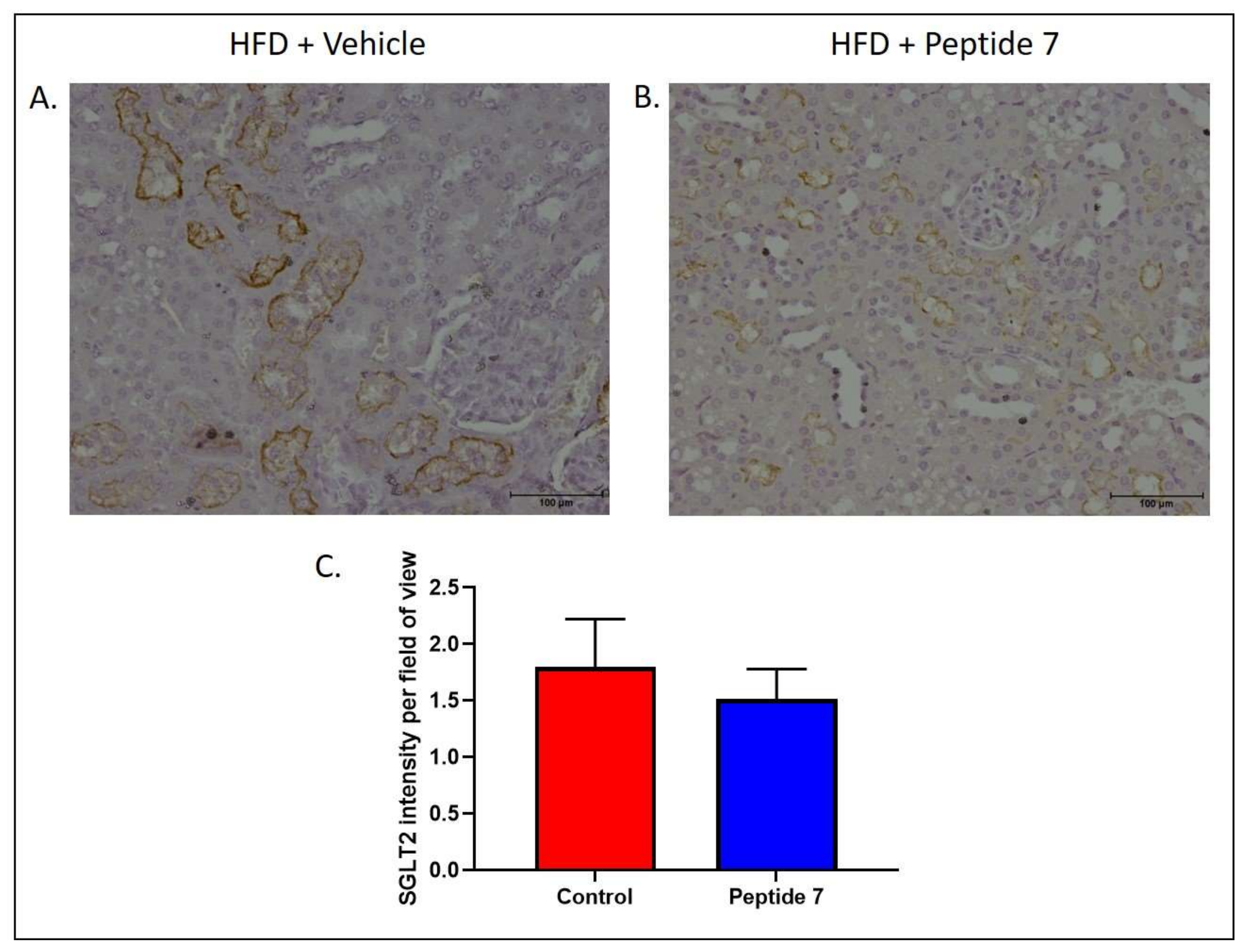
2.9. The TNFSF14 Peptide 7 Reduced SGLT2 Expression
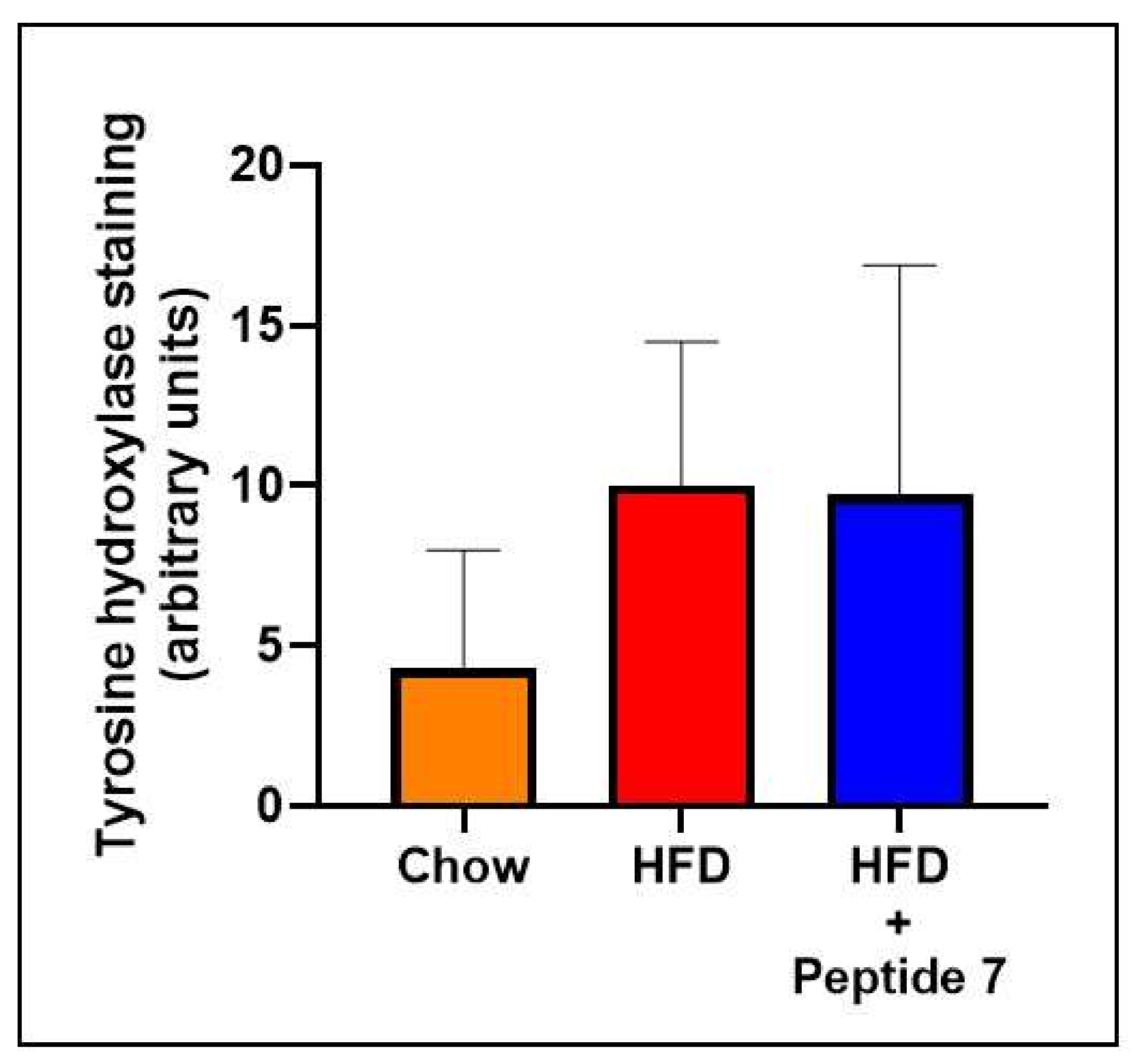
2.10. TNFSF14 Peptide 7 Was Unable to Decrease HFD-Induced Increased Sympathetic Innervation
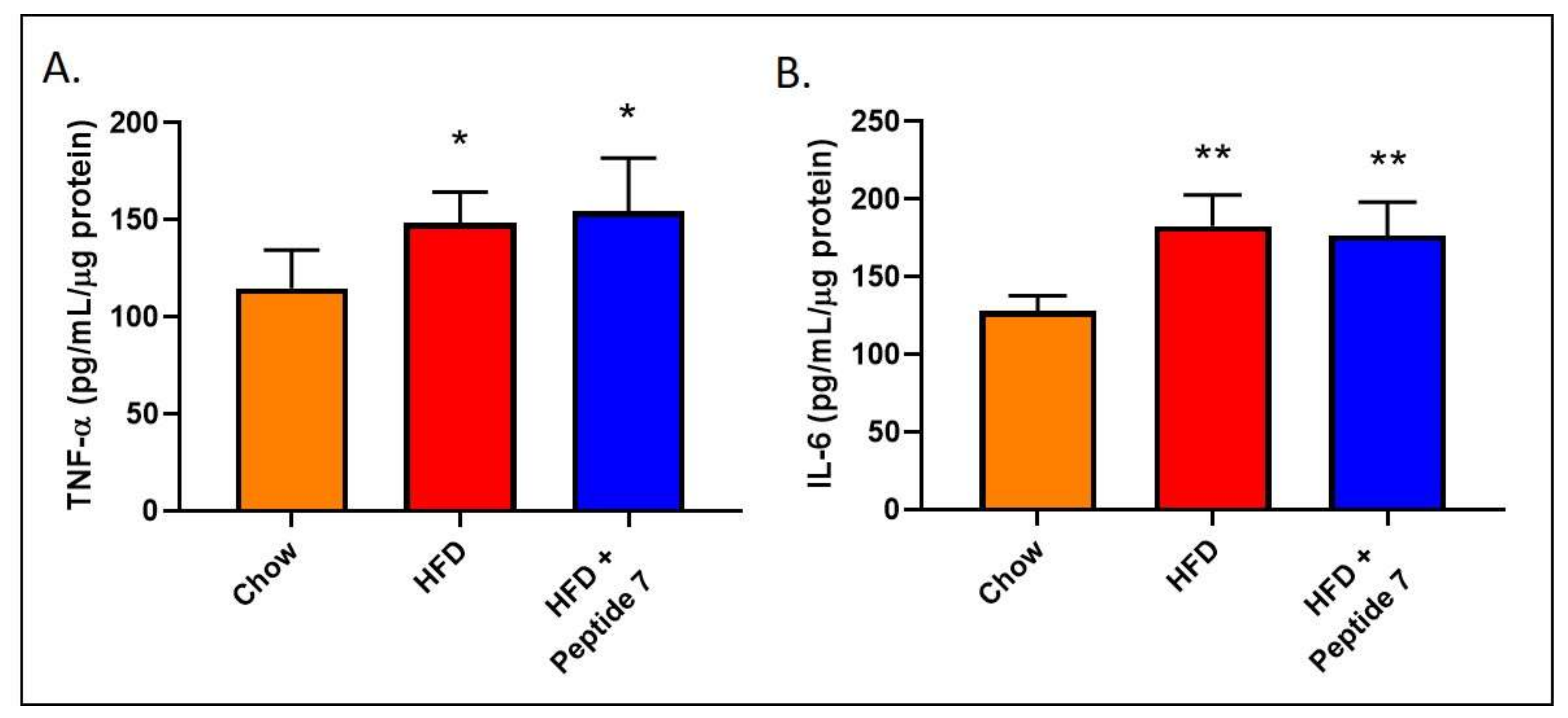
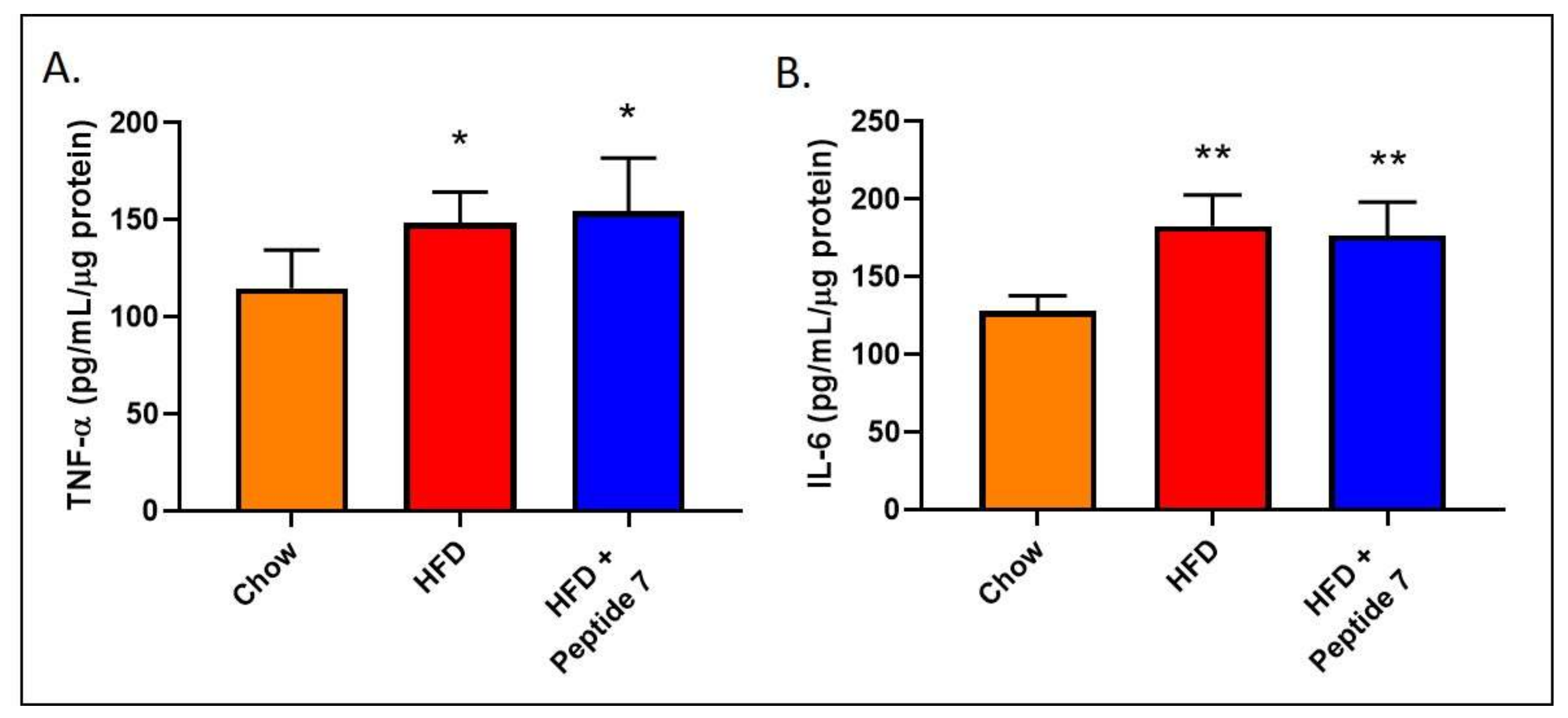
2.11. Peptide 7 Does Not Alter Pro-Inflammatory Cytokine Production
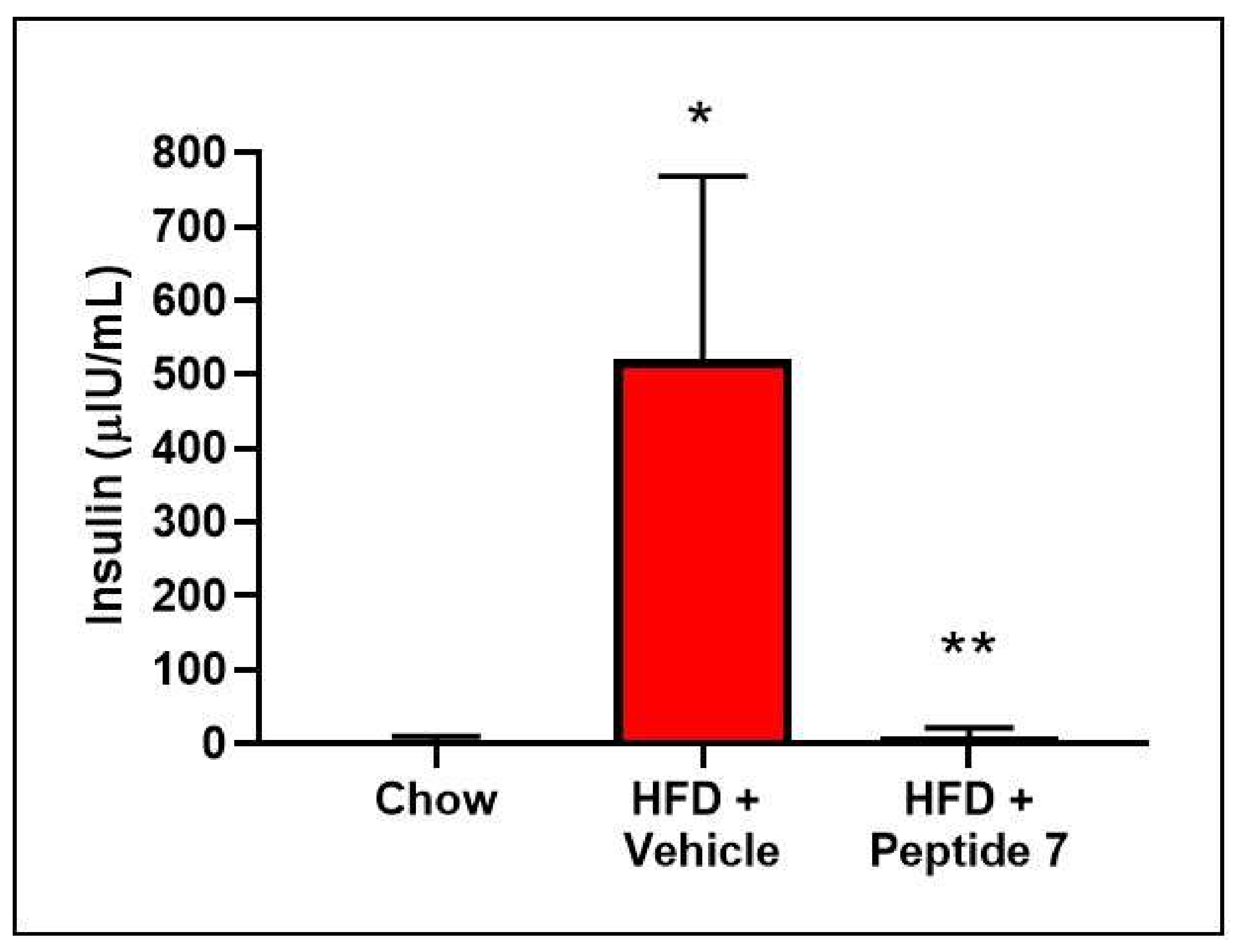
2.12. Peptide 7 Reduces HFD-Induced Insulin Resistance
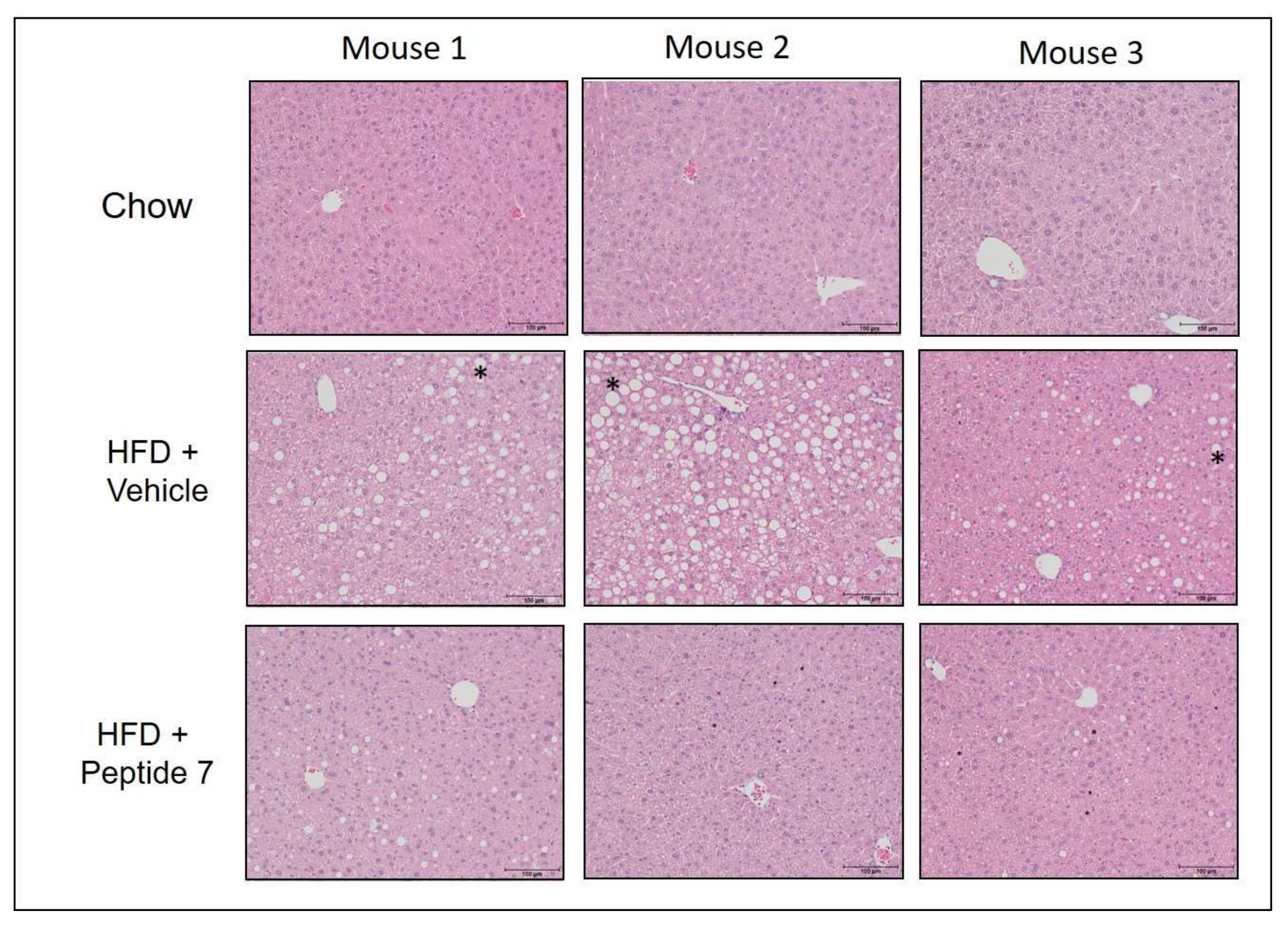
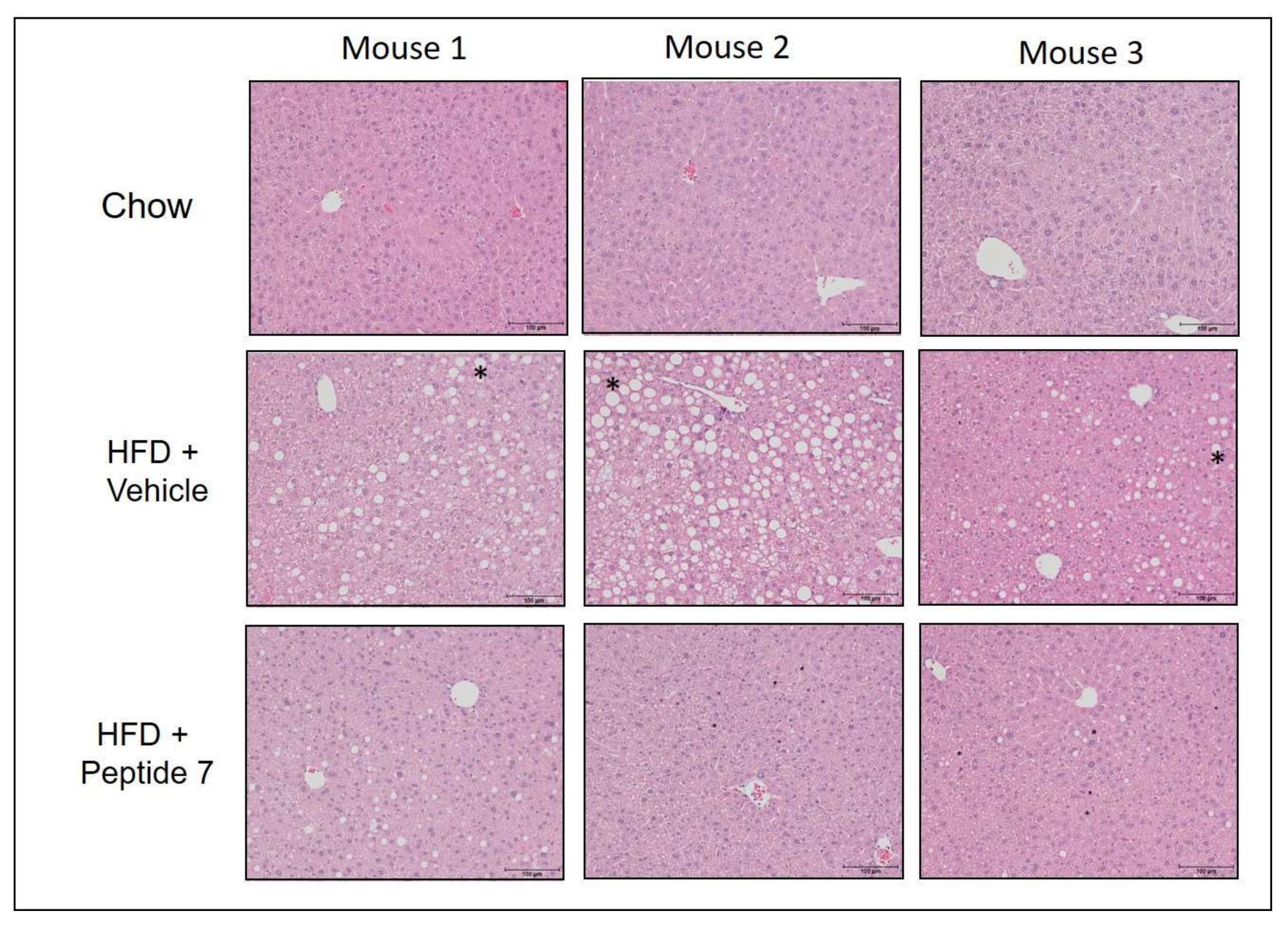
2.13. Peptide 7 Reduces HFD-Induced Liver Steatosis
3. Discussion
4. Materials and Methods
4.1. In Silico Modelling and Optimisation
4.2. Peptide Synthesis
4.3. Cell Culture Experiments
4.4. Insulin Signalling
4.5. Mice (Mus Musculus)
4.6. Diet-Induced Obesity Mouse Model
4.7. Western Blotting
4.8. Haematoxylin and Eosin Staining
4.9. SGLT2 and TH Immunohistochemistry
4.10. Cytokine ELISAs
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hossain, P.; Kawar, B.; El Nahas, M. Obesity and diabetes in the developing world—A growing challenge. N. Engl. J. Med. 2007, 356, 213–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deloitte Access Economics. The Economic Value of Informal Care in Australia in 2015; Deloitte Access Economics: Canberra, ACT, Australia, 2015. [Google Scholar]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Biryukov, B.S.; Abbafati, C.; Semaw Ferede Abera, M.A.; Achoki, T.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Magliano, D.; Zimmet, P. The worldwide epidemiology of type 2 diabetes mellitus-present and future prospectives. Nat. Rev. Endocrinol. 2012, 8, 228–236. [Google Scholar] [CrossRef]
- Farag, Y.; Gaballa, M. Diabesity: An overview of a rising epidemic. Nephrol. Dial. Transpl. 2011, 26, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Lazzaroni, E.; Nasr, M.B.; Loretelli, C.; Pastore, I.; Plebani, L.; Lunati, M.E.; Vallone, L.; Bolla, A.M.; Rossi, A.; Montefusco, L.; et al. Anti-diabetic drugs and weight loss in patients with type 2 diabetes. Pharmacol. Res. 2021, 171, 105782. [Google Scholar] [CrossRef]
- Carey, A.; Steinberg, G.; Macaulay, S.L.; Thomas, W.; Holmes, A.G.; Ramm, G.; Prelovsek, O.; Hohnen-Behrens, C.; Watt, M.J.; James, D.; et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 2006, 55, 2688–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.A.; Clegg, D.J.; Smith, K.B.; Tolod-Richer, E.G.; Matter, E.K.; Picard, L.S.; Seeley, R.J. GM-CSF action in the CNS decreases food intake and body weight. J. Clin. Investig. 2005, 115, 3035–3044. [Google Scholar] [CrossRef] [PubMed]
- Fruebis, J.; Tsao, T.S.; Javorschi, S.; Ebbets-Reed, D.; Erickson, M.R.; Yen, F.T.; Bihain, B.E.; Lodish, H.F. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2005–2010. [Google Scholar] [CrossRef]
- Pelleymounter, M.A.; Cullen, M.J.; Baker, M.B.; Hecht, R.; Winters, D.; Boone, T.; Collins, F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995, 269, 540–543. [Google Scholar] [CrossRef]
- Bassols, J.; Moreno-Navarrete, J.; Ortega, F.; Ricart, W.; Fernandez-Real, J. Light is associated with hypertriglyceridemia in obese subjects and increased cytokine secretion from cultured human adipocytes. Int. J. Obes. 2010, 34, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, B.S.; Tan, K.B.; Ni, J.; Kwi-Ok-Oh, Z.H.; Kim, K.K.; Kim, Y.J.; Wang, S.; Gentz, R.; Yu, G.L.; Harrop, J.; et al. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J. Biol. Chem. 1997, 272, 14272–14276. [Google Scholar] [CrossRef] [Green Version]
- Tiller, G.; Laumen, H.; Fischer-Posovszky, P.; Finck, A.; Skurk, T.; Keuper, M.; Brinkmann, U.; Wabitsch, M.; Link, D.; Hauner, H. Light (TNFSF14) inhibits adipose differentiation without affecting adipocyte metabolism. Int. J. Obes. 2011, 35, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, B.M.; Rudnicka, C.; Filipovska, A.; Davies, S.; Ward, N.; Hricova, J.; Schlaich, M.P.; Matthews, V.B. Shining Light on the metabolic role of the cytokine TNFSF 14 and the implications on hepatic IL-6 production. Immunol. Cell Biol. 2018, 96, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Persaud, S.J. Islet G-protein coupled receptors: Therapeutic potential for diabetes. Curr. Opin. Pharmacol. 2017, 37, 24–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, R.; Avogaro, A.; Formoso, G.; Piro, S.; Purrello, F.; Targher, G.; Consoli, A. Beneficial Effects of Glucagon-Like Peptide 1 Receptor Agonists on Glucose Control, Cardiovascular Risk Profile, and Non-Alcoholic Fatty Liver Disease. An Expert Opinion of the Italian Diabetes Society. Nutr. Metab. Cardiovasc. Diseases 2021, in press. [Google Scholar] [CrossRef]
- Bucheit, J.D.; Pamulapati, L.G.; Carter, N.; Malloy, K.; Dixon, D.L.; Sisson, E.M. Oral semaglutide: A review of the first oral glucagon-like peptide 1 receptor agonist. Diabetes Technol. Ther. 2020, 22, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, B.; Chen, J.; You, H.; Sheng, C.; Yang, P.; Qu, S. Glucagon-like Peptide-1 Improves Fatty Liver and Enhances Thermogenesis in Brown Adipose Tissue via Inhibiting BMP4-Related Signaling Pathway in High-Fat-Diet-Induced Obese Mice. Int. J. Endocrinol. 2021, 26, 2021. [Google Scholar] [CrossRef]
- Tarantino, G.; Citro, V.; Capone, D. Nonalcoholic fatty liver disease: A challenge from mechanisms to therapy. J. Clin. Med. 2020, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Kou, Y.; Liu, Q.; Liu, W.; Sun, H.; Liang, M.; Kong, F.; Zhang, B.; Wei, Y.; Liu, Z.; Wang, Y. LIGHT/TNFSF14 signaling attenuates beige fat biogenesis. FASEB J. 2019, 33, 1595–1604. [Google Scholar] [CrossRef]
- Herrero-Cervera, A.; Vinue, A.; Burks, D.J.; Gonzalez-Navarro, H. Genetic inactivation of the LIGHT (TNFSF14) cytokine in mice restores glucose homeostasis and diminishes hepatic steatosis. Diabetologia 2019, 62, 2143–2157. [Google Scholar] [CrossRef]
- Dhall, S.; Karim, Z.; Khasawneh, F.; Martins-Green, M. Platelet hyperactivity in TNFSF14/LIGHT knockout mouse model of impaired healing. Adv. Wound Care 2016, 5, 421–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, S.; Noh, E.; Gwon, G.; Kim, J.Y.; Jo, J.C.; Choi, Y.; Koh, S.; Baek, J.H.; Min, Y.J.; Kim, H. LIGHT (TNFSF14) Increases the Survival and Proliferation of Human Bone Marrow-Derived Mesenchymal Stem Cells. PLoS ONE 2016, 11, e0166589. [Google Scholar] [CrossRef]
- Waldemer-Streyer, R.; Chen, J. Myocyte-derived Tnfsf14 is a survival factor necessary for myoblast differentiation and skeletal muscle regeneration. Cell Death Dis. 2015, 6, e2026. [Google Scholar] [CrossRef] [Green Version]
- Malmestrom, C.; Gillet, A.; Jernas, M.; Khademi, M.; Axelsson, M.; Kockum, I.; Mattsson, N.; Zetterberg, H.; Blennow, K.; Alfredsson, L.; et al. Serum levels of LIGHT in MS. Mult. Scler. J. 2013, 19, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Mana, P.; Linares, D.; Silva, D.; Fordham, S.; Scheu, S.; Pfeffer, K.; Staykova, M.; Bertram, E.M. LIGHT (TNFSF14/CD258) is a decisive factor for recovery from experimental autoimmune encephalomyelitis. J. Immunol. 2013, 191, 154–163. [Google Scholar] [CrossRef]
- Krause, P.; Zahner, S.; Kim, G.; Shaikh, R.; Steinberg, M.; Kronenberg, M. The tumor necrosis factory family member TNFSF14 (LIGHT) is required for resolution of intestinal inflammation in mice. Gastroenterology 2014, 146, 1752–1762. [Google Scholar] [CrossRef] [Green Version]
- Qiao, G.; Qin, J.; Kunda, N.; Calata, J.F.; Mahmud, D.L.; Gann, P.; Fu, Y.-X.; Rosetnberg, S.A.; Prabhakar, B.S.; Maker, A.V.; et al. LIGHT elevation enhances immune eradication of colon cancer metastases. Cancer Res. 2017, 77, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, X.; Li, D.; Cui, L.; Li, X.; Ye, X. DcR3 promotes hepatoma cell migration by downregulating E-cadherin expression. Oncol. Rep. 2017, 38, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, C.H.; Holloway, M.K. Thermodynamics of ligand binding and efficiency. ACS Med. Chem. Lett. 2011, 2, 433–437. [Google Scholar] [CrossRef] [Green Version]
- Kohut, G.; Liwo, A.; Bősze, S.; Beke-Somfai, T.; Samsonov, S.A. The molecular mechanism of structural changes in the antimicrobial peptide CM15 upon complex formation with drug molecule suramin: A computational analysis. J. Phys. Chem. B 2018, 122, 7821–7827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyansky, A.A.; Zubac, R.; Zagrovic, B. Estimation of conformational entropy in protein–ligand interactions: A computational perspective. In Computational Drug Discovery and Design; Baron, R., Ed.; Springer: New York, NY, USA, 2012; pp. 327–353. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Hemming, I.A.; Clément, O.; Gladwyn-Ng, I.E.; Cullen, H.D.; Ng, H.L.; See, H.B.; Ngo, L.; Ulgiati, D.; Pfleger, K.D.G.; Agostino, M.; et al. Disease-associated missense variants in ZBTB18 disrupt DNA binding and impair the development of neurons within the embryonic cerebral cortex. Hum. Mutat. 2019, 40, 1841–1855. [Google Scholar] [CrossRef]
- Dagdeviren, S.; Jung, D.Y.; Lee, E.; Friedline, R.H.; Noh, H.L.; Kim, J.H.; Patel, P.R.; Tsitsilianos, N.; Tsitsilianos, A.V.; Tran, D.A.; et al. Altered interleukin-10 signaling in skeletal muscle regulates obesity-mediated inflammation and insulin resistance. Mol. Cell. Biol. 2016, 36, 2956–2966. [Google Scholar] [CrossRef] [Green Version]
- Stanke-Labesque, F.; Gautier-Veyret, E.; Chhun, S.; Guilhaumou, R.; French Society of Pharmacology and Therapeutics. Inflammation is a major regulator of drug metabolizing enzymes and transporters: Consequences for the personalization of drug treatment. Pharmacol. Ther. 2020, 215, 107627. [Google Scholar] [CrossRef]
- Thévenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tuffery, P. PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Sudhamsu, J.; Yin, J.; Chiang, E.Y.; Starovasnik, M.A.; Grogan, J.L.; Hymowitz, S.G. Dimerization of LTβR by LTα1β2 is necessary and sufficient for signal transduction. Proc. Natl. Acad. Sci. USA 2012, 110, 19896–19901. [Google Scholar] [CrossRef] [Green Version]
- Salam, N.K.; Adzhigirey, M.; Sherman, W.; Pearlman, D.A. Structure-based approach to the prediction of disulfide bonds in proteins. Protein Eng. Des. Sel. 2014, 27, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef] [PubMed]
- Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals 8th Edition. In National Research Council of the National Academies, 8th ed.; Institute for Laboratory Animal Research: Washington, DC, USA, 2011. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Residue Numbers in TNFSF14 | Sequence |
|---|---|---|
| 1 | 98–117 | GANASLIGIGGPLLWETRLG |
| 2 | 166–180 | LYKRTSRYPKELELL |
| 3 | 219–228 | PGNRLVRPRD |
| Peptide | Site 1 a | Site 2 a | Δ Affinity for TNFRSF14 at Site 1 b | Δ Affinity for TNFRSF14 at Site 2 b | Δ Affinity for LTβR at Site 1 b | Δ Affinity for LTβR at Site 2 b |
|---|---|---|---|---|---|---|
| 2.1 | Leu1 | Leu14 | −1.32 | −0.70 | −0.24 | −0.25 |
| 2.2 | Leu1 | Leu15 | −1.32 | −1.75 | −0.24 | −0.82 |
| 2.3 | Tyr2 | Glu11 | −2.37 | −6.38 | −1.06 | −3.18 |
| 2.4 | Lys3 | Leu12 | +6.01 | +24.51 | +3.77 | +5.97 |
| Peptide | Sequence | Δ Affinity for TNFRSF14 a | Δ Affinity for LTBR a | AGGSCORE |
|---|---|---|---|---|
| 2.4 b | LYCRTSRYPKECELL | - | - | 58.14 |
| 5 | LRCRWNRYPRECELR | +0.455 | −13.966 | 1.492 |
| 6 | LRCRWSRYPRECELR | −7.626 | −7.649 | 2.115 |
| 7 | LRCRISRYPMECRLL | −12.161 | −10.728 | 39.79 |
| 8 | LYCRTSRLPRECELR | +7.286 | −10.077 | 44.35 |
| 9 | LYCRTTRYPRICELK | −7.479 | −8.686 | 7.35 |
| 10 | LRCRISRYRYECRLL | −24.000 | −20.632 | 24.59 |
| Peptide | Sequence | [M + 2H]2+ a | Retention Time (mins) b |
|---|---|---|---|
| 5 | LRCRWNRYPRECELR | 1024.3 m/z | 6.1 |
| 6 | LRCRWSRYPRECELR | 1011.1 m/z | 6.0 |
| 7 | LRCRISRYPMECRLL | 953.5 m/z | 7.0 |
| 8 | LYCRTSRLPRECELR | 946.9 m/z | 5.9 |
| 9 | LYCRTTRYPRICELK | 956.4 m/z | 6.4 |
| 10 | LRCRISRYRYECRLL | 999.2 m/z | 6.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agostino, M.; Rooney, J.; Herat, L.; Matthews, J.; Simonds, A.; Northfield, S.E.; Hopper, D.; Schlaich, M.P.; Matthews, V.B. TNFSF14-Derived Molecules as a Novel Treatment for Obesity and Type 2 Diabetes. Int. J. Mol. Sci. 2021, 22, 10647. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910647
Agostino M, Rooney J, Herat L, Matthews J, Simonds A, Northfield SE, Hopper D, Schlaich MP, Matthews VB. TNFSF14-Derived Molecules as a Novel Treatment for Obesity and Type 2 Diabetes. International Journal of Molecular Sciences. 2021; 22(19):10647. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910647
Chicago/Turabian StyleAgostino, Mark, Jennifer Rooney, Lakshini Herat, Jennifer Matthews, Allyson Simonds, Susan E. Northfield, Denham Hopper, Markus P. Schlaich, and Vance B. Matthews. 2021. "TNFSF14-Derived Molecules as a Novel Treatment for Obesity and Type 2 Diabetes" International Journal of Molecular Sciences 22, no. 19: 10647. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910647