
Zebrafish and Medaka: Important Animal Models for Human Neurodegenerative Diseases

1
State Key Laboratory of Freshwater Ecology and Biotechnology, Institute of Hydrobiology, Chinese Academy of Sciences, Donghu South Road 7#, Wuhan 430072, China
2
College of Advanced Agriculture Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(19), 10766; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910766
Submission received: 27 July 2021
/
Revised: 30 September 2021
/
Accepted: 30 September 2021
/
Published: 5 October 2021
(This article belongs to the Special Issue Molecular Pathogenesis of Neurodegeneration: From Fish to Human)
Abstract
:Animal models of human neurodegenerative disease have been investigated for several decades. In recent years, zebrafish (Danio rerio) and medaka (Oryzias latipes) have become popular in pathogenic and therapeutic studies about human neurodegenerative diseases due to their small size, the optical clarity of embryos, their fast development, and their suitability to large-scale therapeutic screening. Following the emergence of a new generation of molecular biological technologies such as reverse and forward genetics, morpholino, transgenesis, and gene knockout, many human neurodegenerative disease models, such as Parkinson’s, Huntington’s, and Alzheimer’s, were constructed in zebrafish and medaka. These studies proved that zebrafish and medaka genes are functionally conserved in relation to their human homologues, so they exhibit similar neurodegenerative phenotypes to human beings. Therefore, fish are a suitable model for the investigation of pathologic mechanisms of neurodegenerative diseases and for the large-scale screening of drugs for potential therapy. In this review, we summarize the studies in modelling human neurodegenerative diseases in zebrafish and medaka in recent years.
1. Introduction
Neurodegenerative diseases are a major threat to human health. With the increase in the elderly population, these age-dependent diseases are becoming increasingly prevalent [1]. These disorders are devastating to families, and they represent a huge burden for society. Hence, it is urgent to develop novel and more effective therapeutic strategies to remedy these diseases. Animal models were confirmed as a useful tool to investigate the complex mechanisms of neurodegenerative diseases.
Over the past several decades, animal models, such as mice, monkeys, dogs, pigs, fruit flies, and fish, have contributed greatly to our understanding of the genetic basis of the cellular and molecular mechanisms behind neurodegenerative diseases [2,3,4,5,6]. In particular, small fish such as zebrafish (Danio rerio) and medaka (Oryzias latipes) offer several advantages as model organisms for human neurodegenerative disease studies and drug discovery. Due to their relatively small size and short lifespan, they require less space and are more cost-efficient for laboratory maintenance compared with other vertebrate model organisms, such as the mouse. In addition, they have very high fecundity, and their embryos are transparent during development, which facilitates the non-invasive visualization of its development, and complex mechanisms of neurodegeneration can be analysed more rapidly than in mouse and other vertebrate animal models [7,8,9,10,11].
Finally, drugs can be administered by intraperitoneal injection or oral gavage in adult zebrafish [12] or medaka [13], whereas in larvae or embryos, they are always administered by adding them to the water and drug solution [14]. Due to their small size, they can be easily treated in the 24-well plate, 96-well plate, or 10-cm Petri dish. This facilitates subsequent analysis of phenotypes after drug treatment. Therefore, all these characteristics make them suitable for large-scale and high-throughput drug screening scans.
On the other hand, the identity of nucleotide or amino acid sequences between zebrafish and human homologues is approximately 71% [15], which is much higher than some invertebrate animal models such as roundworms (Caenorhabditis elegans) (30–60%) [16] and fruit flies (Drosophila melanogaster) (40%) [17]. Notably, zebrafish possess a vertebrate neural structural organisation, and all of the major structures are similar to the mammalian brain. Furthermore, zebrafish also possesses a functional Blood–Brain Barrier (BBB), similar to humans [18]. Many important neurotransmitters were detected in the neurotransmitter profile of zebrafish, which is very important for neuroscientific studies [19].
Although the zebrafish is the most widely used fish model globally, medaka is also used extensively, especially in Europe and Asia [20]. Compared with the zebrafish, the embryos of medaka tolerate a wider temperature range (4–35 °C until the onset of heartbeat and 18–35 °C thereafter, compared to 25–33 °C in zebrafish) [11,21]. This provides great convenience in screens for isolation of low temperature-sensitive gene mutations and the manipulation of developmental rates [11]. In addition, medaka has a long history as a genetic model system. Therefore, a lot of inbred strains from different populations with a high degree of genetic polymorphism are available. This facilitates the generation of high-resolution genetic maps and the genetic analysis of monogenic traits and quantitative trait loci [21].
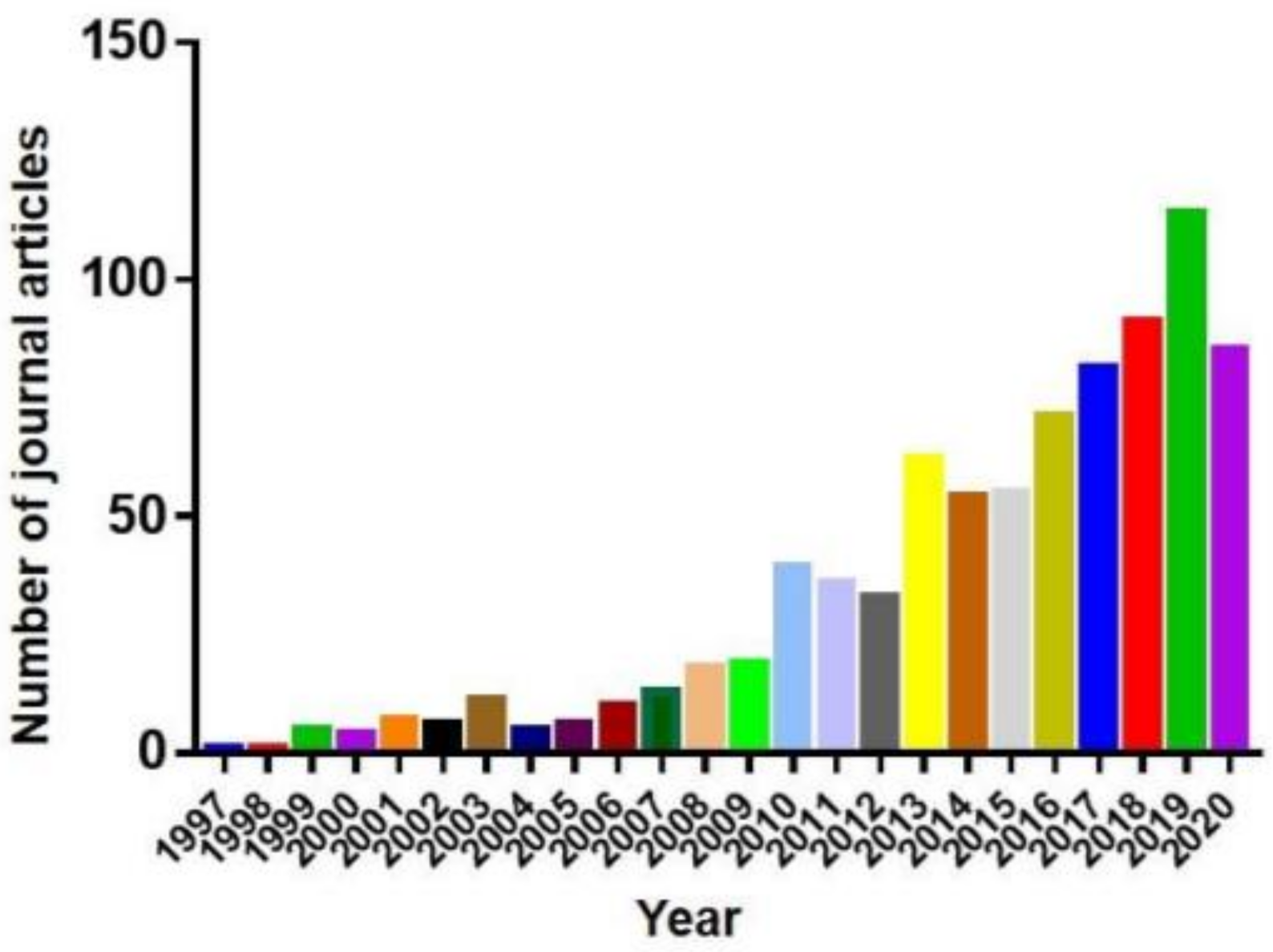
Therefore, all these factors make zebrafish and medaka of great value in studies of neurodegenerative diseases [22]. As a result, the publications in PubMed using zebrafish, the more popular model of the two, as the neurodegenerative disease model increased sharply in recent years (Figure 1). This review summarizes the use of zebrafish and medaka as models in neurodegenerative disease research.
2. Parkinson’s Disease Models
Parkinson’s disease (PD) is one of the most common neurodegenerative diseases that affects the motor system. Surveys, medical records, and death certificates demonstrate that the prevalence of PD has notably increased worldwide in recent years, possibly due to the growing elderly population worldwide [23,24,25]. The prevalence of PD was approximately 8.52 million and the incidence was 1.02 million in 2017 globally [26], whereas approximately 0.34 million people died from PD in 2017 globally [27]. It is predicted that the number of cases will reach 12 million by 2050 [28]. In spite of extensive studies that focus on the epidemiology and possible treatments of PD, its pathogenic mechanism has not been fully elucidated, and there is still no effective therapeutic strategy to cure this disease [29]. Compared with some traditional mammal models such as mice, zebrafish and medaka have comparative advantages for the pathological research of PD due to their short life cycles and high fecundity, which makes them particularly suitable for large scale drug screening [14,30,31,32]. In addition, as vertebrate species, zebrafish and medaka have higher genetic similarity to humans than invertebrate model animals such as roundworms and fruit flies [15,16,17,20]. In this review, we summarize several studies of PD in zebrafish, focusing on those published in recent years (Table 1) and several studies of PD in medaka. We discuss two main types of models: neurotoxin-induced and genetic models.
2.1. Neurotoxin-Induced PD Models in Zebrafish and Medaka
Some neurotoxins, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), 6-hydroxydopamine (6-OHDA), paraquat (1,1’-dimethyl-4,4’-bipyridinium dichloride), and rotenone are often used to selectively induce harmful effects to the dopaminergic neurons, which leads to dopaminergic neuronal loss and increases the risk of PD in model animals.
2.1.1. MPTP
MPTP is a neurotoxin that is widely used in the animal models of PD. It targets dopaminergic neurons and induces their loss in the substantia nigra, which leads to the appearance of motor features of PD [66,67,68]. It was observed that MPTP can induce PD-like symptoms in other animal species such as rodents, primates, and zebrafish [9]. A recent study demonstrated that the mechanism of action of MPTP toxicity is conserved both in zebrafish and humans [33], which suggests that it may be suitable to be used as a neurotoxin to induce the PD model in zebrafish. After the exposure to MPTP, zebrafish exhibited obvious movement impairment, including reduced locomotory activity and aberrant swimming behaviour, equivalent to the bradykinesia-like symptoms in humans. In addition, the exposure to MPTP also weakens the tactile sensitivity in zebrafish [34,35]. Another study found that the intraperitoneal injection of MPTP in adult zebrafish caused a significant reduction of locomotory activity, accompanied by the loss of dopaminergic neurons and the over-expression of synuclein in the optic tectum [36]. Furthermore, 78 proteins differentially expressed in the brains of MPTP-treated zebrafish (compared to the control) were involved in several neurological pathways [36].
Similarly, the MPTP also effectively triggered PD-like symptoms in medaka [69]. Shortly after the MPTP-exposure treatment, an obvious reduction of swimming movement was observed in the larvae of medaka. The histochemical examination of the medaka brain showed that the immunoreactivity of tyrosine hydroxylase (TH)-positive neurons in the diencephalon almost totally disappeared, suggesting a damage to the dopamine system [69,70].
2.1.2. 6-OHDA
6-OHDA is a synthetic neurotoxin which also has been used to construct the PD animal models. In zebrafish, after the injection of 6-OHDA into the ventral diencephalon, a significant reduction of dopaminergic neurons and reduced swimming speed and distance travelled were observed [37]. A recent study also reported the death of DA neurons and the reduction of dopamine levels in the lesioned brain area of the 6-OHDA administrated zebrafish [38]. Similarly, the exposure of zebrafish larvae to 6-OHDA produced a reduction in dopaminergic neurons [39]. Morphological alternations were also observed in the 6-OHDA-exposed zebrafish embryos (blood pooling, cardiac edema, delayed development) [40] and larvae (reduced total length and head length) [39].
Interestingly, there was no substantial reduction in the dopaminergic neurons in the medaka after 6-OHDA exposure [70]. The authors suspected that 6-OHDA could not permeate to the BBB in the adult fish, so they injected 6-OHDA directly into the cerebrospinal fluid (CSF) of medaka, which did cause a loss of dopamine neurons. Similar to the MPTP treatment in medaka, only the middle diencephalic cluster among the dopamine neuron clusters exhibited a robust cell loss [70].
2.1.3. Paraquat
Paraquat is a herbicide that has similar structure to the MPTP [71]. Its effects on zebrafish have been tested using embryonic, larval, and adult stages. The exposure of zebrafish embryos to paraquat triggered premature hatching and increased locomotory activity in the larvae [41]. Another study showed that the exposure of 18 hpf (hours post-fertilization) embryonic zebrafish to a low dose of paraquat (0.04 ppm) produced neurodegenerative phenotypes and motor deficits in 50% of the embryos. Furthermore, various developmental anomalies including small eyes, flat head, a pinched midbrain–hindbrain boundary, thin yolk extension, and curved-up body were detected in the hatched larvae [42]. However, in contrast to the observations in mice models, the exposure to paraquat resulted in an increased dopamine level in adult zebrafish, which indicated that the paraquat-treated zebrafish did not recapitulate the PD pathology [43]. Moreover, further studies showed that both the expression level of TH and the numbers of dopaminergic neurons were not affected in the paraquat-treated zebrafish [41,44]. Nevertheless, the paraquat-treated zebrafish still showed impaired locomotory activity [43,44,72].
2.1.4. Rotenone
Rotenone is a rotenoid extracted from the roots of plants from the Leguminosae family, commonly used as an insecticide, herbicide, and piscicide [73]. It can easily permeate the BBB and enter the central nervous system (CNS) [73,74], and its destructive effect on dopaminergic neurons in rodents makes it a popular drug of choice to trigger PD models [75,76]. In zebrafish, rotenone also reduces the dopamine population, marked by a decreased expression level of TH [45,46]. Studies also showed that after the rotenone administration, adult zebrafish exhibited reduced swimming speed and shorter travel distances compared to the control group [46,47]. As mentioned above, this aberrant swimming behaviour is reflective of bradykinesia-like symptoms of PD in humans [45]. In addition, a study found impaired olfaction in a rotenone-administrated zebrafish, which is a typical non-motor symptom of PD [48].
2.2. Genetic PD Models in Zebrafish and Medaka
Accompanied by the development of gene-editing technology, extensive studies have applied genetic PD models to study the PD development in zebrafish and medaka. These widely used methods include the MO (morpholino antisense technology) [77], TALEN (transcription activator-like effector nucleases technology) [78] and CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/Cas9 technology) [79].
2.2.1. Parkinson’s Disease Protein 2 (PARK2)
Mutations of the PARK2 gene are correlated with the early onset of PD, and they are the most prevalent cause of autosomal recessive PD [80]. PARK2 encodes the Parkin protein, a ubiquitin ligase that targets other proteins for degradation. A loss-of-function Parkin protein encoded by a mutated PARK2 gene will destroy its function, which in turn increases the risk of inducing PD [49]. The zebrafish Parkin protein consists of 458 amino acids and exhibits 62% identity to the human orthologue, with 78% identity in functionally relevant regions [50]. In addition, although neurodegenerative diseases are always late onset in human patients, we can still detect some similar pathological characteristics and phenotypes in the early developmental stages of zebrafish, therefore, the administration of MO technology could reveal some important mechanisms of neurodegenerative diseases. For instance, a previous study using the MO-mediated knockdown of PARK2 in zebrafish reported that knockdown of PAKR2 gene in zebrafish displayed a 20% loss of the DA neuron numbers in the diencephalon, but the swimming behaviour was not apparently affected [50]. However, the MO technology still has some disadvantages, such as the occurrence of false positives, cytotoxicity, and the inability to obtain stable genetic lines. The negative phenotype of Parkin-mutant medaka was also similar to Parkin deficient mice [70,81].
2.2.2. PTEN (Phosphatase/Tensin Homolog)-Induced Kinase 1 (Pink1)
The PINK1 protein is the second most common cause of autosomal recessive early onset cases in PD patients [49,82,83]. The zebrafish Pink1 gene shares 57.8% identity to the human orthologue [51]. A previous study using the antisense MOs injection into the zebrafish embryo showed a reduction of dopaminergic neurons, but the locomotory activity remained unchanged [51]. However, another study of MO of Pink1 in the zebrafish embryos did not detect a loss of dopaminergic neurons, but obviously disorganized diencephalic dopaminergic neurons were observed [52]. It is possible that these differences may have been caused by different target regions of this gene in different studies, which remains to be elucidated in the future studies.
Some studies used the Targeting Induced Local Lesions In Genomes (TILLING) library [70] to screen for loss-of-function mutations in human autosomal recessive PD homologous genes in medaka and identified a mutation in the homologous medaka Pink1 gene, the Pink1Q178X mutation [70]. RT-PCR and in situ hybridization analysis detected a relatively small number of PINK1 transcripts, demonstrating its loss-of-function at the mRNA level. A homozygous Pink1Q178X medaka showed some phenotypes similar to the human PD patients, such as late-onset reduction of spontaneous movements and a decreased level of 3,4-dihydroxyphenylacetic acid, a dopamine metabolic product [70].
2.2.3. LRRK2 (Leucine-Rich Repeat Kinase 2)
Mutations in the LRRK2 gene are the most prevalent underlying reason for the late-onset autosomal dominant PD [54]. The LRRK2 protein consists of several repeat regions followed by ROC (Ras of complex proteins)-COR (C-terminal of ROC) GTPase, kinase, and WD40 domains [53]. A previous study using the MO-mediated LRRK2 knockdown in zebrafish displayed a severe embryonic lethality, and the surviving larvae morphants showed several developmental aberrations, including slow growth, heart edema, reduced brain size, and reduced number of dopaminergic neurons [54]. Another study employing the deletion of the LRRK2 WD40 domain revealed diencephalic DA neurodegeneration and a shortened swimming distance in zebrafish [55]. Some recent studies observed that mutations of the LRRK2 gene caused a loss of neuronal cells and synuclein aggregation in zebrafish, similar to the PD phenotype in humans [56].
2.2.4. PARK7 (Parkinson’s Disease Protein 7)
The PARK7 gene encodes a protein named DJ-1, which plays an important role in the antioxidative stress reaction and the protection of neuron survival [84,85]. It was reported that mutations in the PARK7 could induce the early onset of PD [57]. The zebrafish PARK7 gene encodes a 189 amino acid protein, which share 83% identity to the DJ-1 of humans [58]. A previous study showed that a transient inactivation of PARK7 mediated by the MO-injection in zebrafish embryo inactivated the DJ-1 production, and the knockdown of PARK7 caused dopaminergic neuronal cell death without toxin exposure [59]. In a recent study, PARK7-knockout zebrafish line was constructed using the CRISPR/Cas9 method, by targeting the exon 1 of PARK7. This DJ-1 deficient zebrafish appeared to develop normally during the larval and young adult stages. With aging, however, they exhibited lower TH levels, respiratory failure in skeletal muscle, and lower body mass, particularly pronounced in male fish [60].
2.2.5. Other Genes
The SNCA gene in humans encodes the α-synuclein protein. It was reported that misfolded α-synuclein proteins form aggregates and thus cause the formation of Lewy bodies (LBs) [86], which is a hallmark of PD pathology [87]. However, the zebrafish lacks the α-synuclein; instead, it possesses three synuclein genes named SNCB, SNCG1, and SNCG2, which encode for β-, γ1-, and γ2-synuclein proteins, respectively [88]. Some previous studies constructed transgenic models that express human wild-type α-synuclein protein in zebrafish [61,89,90]. It was observed that the overexpression of α-synuclein protein in the zebrafish PD model caused a moderate cell death in larval zebrafish sensory neurons [61]. Another study showed that the overexpression of α-Synuclein in zebrafish is a critical factor of the mitochondrial dysfunction in dopaminergic neurons [90].
Mutations in the GBA gene are very important causes of PD [91]. In a recent study, a GBA−/− mutant zebrafish line was generated through the TALEN-mediated technology [62,63]. These GBA-null homozygous fish showed a decreased level and activity of the GBA protein in the brain, and reduced numbers of dopaminergic and noradrenergic neurons were detected in the 3-month-old mutants [63].
3. Alzheimer’s Disease Models
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive memory loss, cognitive impairment, behavioural changes, and loss of functional abilities [92,93,94,95]. AD is the most prevalent form of dementia. It is estimated that nowadays more than 50 million people worldwide have dementia, and this number is expected to reach over 150 million by the 2050 [93]. AD is irreversible and it causes about 70% of all dementia cases [94]. Unfortunately, it still cannot be prevented, treated, or cured. The drug discovery for AD is very challenging, so no new drugs have been approved since 2003, when Memantine was approved [92,93,94,96]. Including four drugs that are approved by the Food and Drug Administration (FDA), at present there are only five approved drugs on the market available for the treatment of AD [94,97]. In addition, a previous study demonstrated that oxidative stress may induce behavioural and cognitive impairments in the aging zebrafish, just as it does in mammals [98]. Below, we describe previous studies of the molecular mechanisms of AD in zebrafish (Table 2).
3.1. Neurotoxic Agents-Induced AD Model in Zebrafish
Although the pathological mechanisms of AD progression remain to be elucidated, two principal neuropathological hallmarks are widely approbated: the extracellular accumulation of the amyloid β (Aβ) plaques and the intracellular neurofibrillary tangles (NFT) [113,114,115]. In addition, several previous studies have reported that the elimination of Aβ through a vascular route is a pivotal mechanism and that its failure could induce the formation of vascular abnormalities, which consequently leads to AD pathogenesis [116,117]. It was reported that Aβ can be degraded in the extracellular space by diverse proteases, such as insulin-degrading enzymes and neprilysin [118]. In addition, cells in the neurovascular unit also have the capacity to endocytose Aβ and eliminate it through lysosomal degradation [119]. In addition, the transportation of Aβ across the BBB also contributes to the removal of soluble interstitial Aβ from the brain [116]. In addition, altered CSF dynamics were widely observed in many AD patients [120]. It might cause an imbalance in the production and clearance of soluble Aβ, thus causing the accumulation of interstitial Aβ, with increased likelihood of plaque formation [121]. When amyloid-β42 (Aβ42) peptides were injected into the cerebral ventricle of adult zebrafish, they aggregated in the brain, where they were mostly seen as intracellular depositions, but also around the blood vessels [99]. Based on this AD model, a subsequent study compared young and old zebrafish to investigate the effects of aging on regenerative ability after Aβ42 deposition [100]. Some previous studies already demonstrated that aging aggravates the neurodegeneration phenotypes and impedes the proliferative response of stem cells in various animal models [122,123]. A study revealed that Aβ42 toxicity causes synapses to degenerate at a higher level in the ventricular region of old zebrafish (1.5-year-old) compared with young adult zebrafish (6 months of age); the same study also found that young zebrafish seemed to activate macrophages and produce newborn neurons at a much higher rate than old zebrafish [100]. The authors of that study speculated that the activated microglia help the zebrafish brain to limit the synaptic degeneration and to promote neurogenesis after Aβ42-induced neurodegeneration [100].
Okadaic acid (OA) is an inhibitor of two main cytosolic, broad-specificity protein phosphatases, PP1 and PP2A. It has been widely used in studies of neurodegeneration in various cell types and rodent models [124]. OA induced AD pathologies in several studies. For instance, it was observed that a decrease in PP2A expression and function caused by OA directly triggered the pathophysiology of AD [101]. In zebrafish, Nada et al. observed some major morphological hallmarks such as mini hemorrhagic transformations or micro-bleeds at the later stages of AD in the OA-induced fish [102]. Another separate research utilized the OA-treated zebrafish for drug screening [103]. In this study, researchers found that LKE (lanthionine ketimine-5-ethyl ester), a derivative of a naturally occurring brain sulfur metabolite, can exhibit a neuroprotective function against OA by improving the levels of the brain-derived neurotrophic factor (BDNF), anti-apoptotic kinase pAkt (Ser473), and the transcription factor phospho-cAMP response element-binding protein (pCREB) (Ser133). When simultaneously exposed to OA and LKE, the zebrafish were able to successfully perform the learning and memory ability, whereas, when exposed to the OA only, the fish could not demonstrate learning and memorizing ability. This suggests that LKE can inhibit the cognitive impairment triggered by OA [103].
Cigarette smoke extract (CSE) consists of 4500 identified and almost 1000 unidentified molecules, many of which could lead to oxidative damage and trigger pro-inflammatory and carcinogenic reactions [125]. Several recent studies revealed that after the CSE treatment, an elevation in the acetylcholinesterase activity was detected in zebrafish, which contributed to its cognitive impairment [104,105].
3.2. Metals and AD in Zebrafish
Metals are abundant in our environment. The disturbance of the metal homeostasis is harmful for brain health and can induce dementia. Some studies used metals to trigger memory loss and neurodegeneration in zebrafish. For example, Xu et al. reported that the lead (Pb) exposure caused learning impairments that persisted for at least three generations, showing trans-generational effects of embryonic exposure to Pb [126]. Chronic Aluminum (Al) exposure is regarded as a risk factor for the AD pathogenesis [127,128]. A previous study revealed that after the aluminium chloride (AlCl3) treatment, adult zebrafish exhibited AD-like behaviour in locomotory activity tests and activity-avoidance conditioning paradigms, suggesting that locomotory activity and learning and memory abilities were damaged in the Al-exposed zebrafish [106]. Acosta et al. exposed larvae and adult zebrafish to another metal, copper (Cu) [107]. The exposure to the 60 μg/L concentration of Cu produced decreased body length of larvae, spatial memory of adults, and the glutathione S-transferase (GST) activity in the gill [107]. In a recent study, Altenhofen et al. investigated the effects of manganese (Mn) exposure on the cognition and exploratory behavior in larval and adult zebrafish [108]. It caused an impairment of the aversive long-term memory in the MnCl2-treated adult zebrafish and reduced distance traveled and movement time in both larvae and adult fish [108].
3.3. Genetic Technology-Mediated AD Model in Zebrafish
Tau is an important microtubule-associated protein for tubule formation which is widely expressed in neurons. Hyperphosphorylation of the tau protein causes the intracellular neurofibrillary tangle (NFT) formation, which is one of the neuropathological hallmarks related to AD [109,110,129]. In a previous study, a transgenic zebrafish line was generated through the Gal4-UAS system. A mutant human gene, 4R/2N-TauP301L, was expressed under the regulation of a novel bidirectional UAS promoter, which allows the co-expression of a red fluorescent reporter gene in Tau-expressing cells [110]. It was observed that a high level of the mutant P301L Tau protein triggered a transient motor phenotype during the embryogenesis, likely caused by the peripheral motor axonal developmental abnormalities. This result is very important because phenotypic abnormalities at the larval stages make it suitable for high-throughput screening [110].
4. Huntington’s Disease Models
Huntington’s disease (HD) is an autosomal dominant, incurable, and fatal neurodegenerative disorder. Initially, HD patients display excessive movements of the limbs and face, and then gradually progress to exaggerated body movements described as chorea. Patients exhibit progressive symptoms, such as psychiatric, cognitive, and motor dysfunction, and this disease is usually lethal 10–20 years after the onset [130,131,132]. HD is caused by an expansion of the polyglutamine-coding region in the N-terminus of the huntingtin protein (HTT) [133]. HTT is a 350 kDa protein that is ubiquitously expressed, evolutionarily conserved, and likely to be involved in many cellular processes [134,135,136]. However, the precise mechanisms underlying the functions of the HTT gene remain incompletely understood.
The zebrafish HTT protein consists of 3121 amino acids and shares 70% identity with the human HTT orthologue [137]. Compared with the HTT-null mutation mice [135], HTT-null mutation zebrafish are viable, so the zebrafish is believed to be a suitable model to study the mechanisms of HD. To investigate the roles of HTT, several previous zebrafish HD models used MO to observe the effects of HTT deficiency in the early zebrafish development [138,139,140]. One study revealed that HTT-deficient zebrafish had hypochromic blood because of the decrease in hemoglobin production, despite the presence of iron within blood cells, and speculated that the disturbance of HTT’s normal function in the iron pathway leads to HD pathology and especially to its neuronal specificity [138]. By use of the same HTT-deficient model, Henshall et al. reported the effects of the loss-of-function of HTT on the developing nervous system and found obvious defects in the morphology of olfactory placode, neuromasts, and branchial arches, which led them to postulate that HTT may have a specific function that enables the formation of telencephalic progenitor cells and preplacodal cells in the forebrain [139]. Another study of the morpholino-based HTT loss-of-function zebrafish observed massive apoptosis of neuronal cells, accompanied by impaired neuronal development, small eyes and heads, and the enlargement of brain ventricles. Interestingly, it was observed that the expression of brain-derived neurotrophic factor (BDNF) was reduced. Notably, treatment of HTT-MO zebrafish embryos with exogenous BDNF rescued these defects, which suggests that increasing the BDNF expression might be a useful strategy for HD treatment [140].
In addition, some scientists established HD zebrafish models through the transgenic technology [141,142,143]. Schiffer et al. transiently expressed 102 polyglutamine repeats in the N-terminal fragment of the HTT protein fused with GFP (Q102-GFP) in zebrafish and found an accumulation of this mutant protein in large SDS-insoluble inclusions in the zebrafish embryos, thus reproducing an important feature of the HD pathology. The expression of the mutant HTT protein resulted in an increase in abnormal morphology and the occurrence of apoptosis in zebrafish embryos. A further study found that soluble mutant HTT protein forms are responsible for toxicity and aberrant polyglutamine aggregates in zebrafish [141]. The same study also found that its toxicity can be suppressed by the heat-shock proteins Hsp40 and Hsp70. Importantly, by the use of this HD transgenic model, two inhibitors of the Q102-GFP aggregation in vivo were identified, both of which are compounds of the N′-benzylidene-benzohydrazide class (293G02 and 306H03). In another study, a stable transgenic zebrafish line, which expressed a Q71-GFP fusion protein under the control of the rhodopsin promoter, was constructed to screen FDA-approved drugs to identify novel autophagy-inducing pathways. Three drugs (L-type Ca2+ channel antagonists, the K+ATP channel opener minoxidil, and the Gi signalling activator clonidine), which participate in a cyclical mTOR-independent pathway that regulates autophagy, were detected. This pathway has lots of candidate points to induce autophagy and reduce aggregates [142].
Cre-loxP system was also sometimes used to generate conditionally inducible transgenic zebrafish to study HD. For example, Veldman et al. created an inducible zebrafish HD model, in which the N-terminal 17 amino acids (N17) in the context of the exon 1 fragment of HTT were deleted, coupled with 97Q expansion (mHTT-ΔN17-exon1). That study found that, compared with the mHTT-ΔN17-exon1 line, fish with intact N17 and 97Q expansion (mHTT-exon1) had more delayed-onset movement deficits with slower progression. This model confirmed that the deletion of N17 terminal amino acids of the HTT will lead to an accelerated HD-like phenotype in zebrafish [143]. Recently, a separate study treated a transgenic HD zebrafish model with a phosphodiesterase 5 (PDE5) inhibitor and found an obvious decrease in the mutant HTT protein levels, cell death, and morphological abnormalities [144].
5. Other Neurodegenerative Disease Models
In addition to the above studies, zebrafish and medaka were also used in the investigation of some other rare neurodegenerative disorders. Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterised by the motor neuron loss, and thus progressive muscle weakness and eventual death, primarily due to respiratory failure. The most prevalent genetic cause of ALS and frontotemporal dementia (FTD) is a hexanucleotide repeat expansion (HRE) within the first intron of the C9orf72 gene [145,146]. Shaw et al. generated two zebrafish lines to express C9orf72 HREs. This model recapitulates the motor deficits, cognitive impairment, muscle atrophy, motor neuron loss, and mortality in early adulthood that was observed in human C9orf72-ALS/FTD patients. Moreover, this stable transgenic model represents a powerful potential for the screening of therapeutic compounds [147]. In another study, several transgenic C9orf72-associated repeat zebrafish lines were generated by TOL2-mediated transposition. These models confirm the poly-GA toxicity in zebrafish. The reduction of poly-GA protein rescues toxicity, indicating its potential therapeutic value to treat C9orf72 repeat expansion carriers [148]. Conversely, Yeh et al. constructed two transient loss-of-function zebrafish larvae (C9orf72u-DENN, C9orf72c-DENN) using a morpholino injection. These models facilitate advances in the understanding of the functions of C9orf72 and provide potential mechanisms to elucidate the pathogenesis of ALS-FTD [149]. Mutations in the superoxide dismutase 1 (SOD1) gene were identified as another cause of ALS. In a previous study, by outcrossing the G93Ros10-SH1 line with the wildtype AB zebrafish strain, a mutant SOD1 zebrafish model was generated and used for high throughput screening to identify neuroprotective compounds [150].
Spinocerebellar ataxias (SCAs) are global neurodegenerative diseases leading to motor discoordination, which is always caused by the affected cerebellar Purkinje cells (PCs). A previous study generated a transgenic SCA type 13 (SCA13) model, which mimics a human pathological SCA13R420H mutation. This model exhibited neuropathological and behavioural changes similar to those manifested by SCA-affected patients [151]. Based on the same model, Namikawa et al. reported an SCA13-triggered cell-autonomous PC degeneration, which results in eye movement deficits [152]. In a previous study in our lab, we constructed an NPC1 knock-out zebrafish model using the CRISPR/Cas9-mediated technology [153]. This model developed symptoms similar to those observed in human patients of Niemann-Pick type C disease (NPC). We observed the loss of Purkinje cells in the cerebella of the NPC1−/− homozygous fish [153] and the aberrant motor behaviour, i.e., ataxias, a typical pathological character of human NPC1 patients (unpublished data), indicating its potential value for investigating the molecular mechanisms of NPC1.
In addition, a previous study generated a Gaucher disease (GD) model in medaka by the use of a high-resolution melting assay in the TILLING library for the glucocerebrosidase (GBA) gene [154]. In this study, it was observed that the GBAW337X/W337X (GBA−/−) medaka displayed complete deficiency in GCase activity, and it showed similar pathological phenotypes with human neuronopathic GD. Importantly, compared with the perinatal death in humans and mice lacking the GCase activity, the GBA−/− medaka survived for months, enabling the investigation of the pathological progression [154].
6. Conclusions
Various zebrafish and medaka models have been generated for neurodegenerative disorder studies in recent years. Compared with some other vertebrate models, such as rodents, these small fish are particularly well-suited for forward and reverse genetic and high-throughput screens for chemical compounds and optical analysis in vivo. These advantages should be considered thoroughly when designing a study aimed at neurodegenerative disorders.
Author Contributions
Conceptualization, J.W. and H.C.; Investigation, J.W. and H.C.; Writing—original draft preparation, J.W. and H.C.; Writing—review and editing, H.C.; Funding acquisition, H.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key R & D Program of China: 2018YFD0900804.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank the National Key R&D Program of China (2018YFD0900804) for their support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Heemels, M.T. Neurodegenerative diseases. Nature 2016, 539, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Models Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Fernaández-Trapero, M.; Espejo-Porras, F.; Rodrıguez-Cueto, C.; Coates, J.R.; Peérez-Díaz, C.; De Lago, E.; Fernaández-Ruiz, J. Upregulation of CB2 receptors in reactive astrocytes in canine degenerative myelopathy, a disease model of amyotrophic lateral sclerosis. Dis. Model Mech. 2017, 10, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Snyder, B.R.; Chan, A.W.S. Progress in Developing Transgenic Monkey Model for Huntington’s Disease. J. Neural Transm. 2018, 125, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Story, B.D.; Miller, M.E.; Bradbury, A.M.; Million, E.D.; Duan, D.; Taghian, T.; Faissler, D.; Fernau, D.; Beecy, S.J.; Gray-Edwards, H.L. Canine Models of Inherited Musculoskeletal and Neurodegenerative Diseases. Front. Vet. Sci. 2020, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Bolus, H.; Crocker, K.; Boekhoff-Falk, G.; Chtarbanova, S. Modeling Neurodegenerative Disorders in Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 3055. [Google Scholar] [CrossRef] [PubMed]
- Horzmann, K.A.; Freeman, J.L. Making waves: New developments in toxicology with the zebrafish. Toxicol. Sci. 2018, 163, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Paone, C.; Diofano, F.; Park, D.D.; Rottbauer, W.; Just, S. Genetics of cardiovascular disease: Fishing for causality. Front. Cardiovasc. Med. 2018, 5, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz, R.L.; Outeiro, T.F.; Ferreira, J.J. Zebrafish as an animal model for drug discovery in parkinson’s disease and other movement disorders: A systematic review. Front. Neurol. 2018, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Saleem, S.; Kannan, R.R. Zebrafish: An emerging real-time model system to study Alzheimer’s disease and neurospecific drug discovery. Cell Death Discov. 2018, 4, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furutani-Seiki, M.; Wittbrodt, J. Medaka and zebrafish, an evolutionary twin study. Mech. Dev. 2004, 121, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Henderson, R.E.; Garraway, L.A.; Zon, L.I. Long-term drug administration in the adult zebrafish using oral gavage for cancer preclinical studies. Dis. Model Mech. 2016, 9, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, Y.; Hosokai, H.; Mizuguchi, Y.; Fukamachi, S.; Shimizu, A.; Saya, H. Establishment of HRAS(G12V) transgenic medaka as a stable tumor model for in vivo screening of anticancer drugs. PLoS ONE 2013, 8, e54424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fior, R.; Póvoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, M.G. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Apfeld, J.; Alper, S. What Can We Learn About Human Disease from the Nematode C. elegans? Methods Mol. Biol. 2018, 1706, 53–75. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Yoshida, H. Drosophila as a Model Organism. Adv. Exp. Med. Biol. 2018, 1076, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Im, S.H.; Yang, J.Y.; Lee, Y.R.; Kim, G.R.; Chae, J.S.; Shin, D.S.; Song, J.S.; Ahn, S.; Lee, B.H.; et al. Zebrafish as a Screening Model for Testing the Permeability of Blood-Brain Barrier to Small Molecules. Zebrafish 2017, 14, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Panula, P.; Chen, Y.C.; Priyadarshini, M.; Kudo, H.; Semenova, S.; Sundvik, M.; Sallinen, V. The comparative neuroanatomy and neurochemistry of zebrafish CNS systems of relevance to human neuropsychiatric diseases. Neurobiol. Dis. 2010, 40, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Uemura, N.; Yamakado, H.; Takeda, S.; Takahashi, R. Exploring the pathogenetic mechanisms underlying Parkinson’s disease in medaka fish. J. Parkinsons Dis. 2014, 4, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Wittbrodt, J.; Shima, A.; Schartl, M. Medaka—a model organism from the Far East. Nat. Rev. Genet. 2002, 3, 53–64. [Google Scholar]
- Stewart, A.M.; Braubach, O.; Spitsbergen, J.; Gerlai, R.; Kalueff, A.V. Zebrafish models for translational neuroscience research: From tank to bedside. Trends Neurosci. 2014, 37, 264–278. [Google Scholar] [CrossRef] [Green Version]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Ahlskog, J.E.; Boeve, B.F.; Graff-Radford, J.; Rocca, W.A.; Mielke, M.M. Survival and Causes of Death among People with Clinically Diagnosed Synucleinopathies with Parkinsonism: A Population-Based Study. JAMA Neurol. 2017, 74, 839–846. [Google Scholar] [CrossRef]
- Darweesh, S.K.L.; Raphael, K.G.; Brundin, P.; Matthews, H.; Wyse, R.K.; Chen, H.; Bloem, B.R. Parkinson Matters. J. Parkinsons Dis. 2018, 8, 495–498. [Google Scholar] [CrossRef] [Green Version]
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, Regional, and National Age-Sex-Specific Mortality for 282 Causes of Death in 195 Countries and Territories, 1980–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Rocca, W.A. The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol. 2018, 17, 928–929. [Google Scholar] [CrossRef] [Green Version]
- Pirtošek, Z.; Bajenaru, O.; Kovács, N.; Milanov, I.; Relja, M.; Skorvanek, M. Update on the Management of Parkinson’s Disease for General Neurologists. Parkinson’s Dis. 2020, 2020, 9131474. [Google Scholar] [CrossRef]
- Hoo, J.Y.; Kumari, Y.; Shaikh, M.F.; Hue, S.M.; Goh, B.H. Zebrafish: A Versatile Animal Model for Fertility Research. BioMed Res. Int. 2016, 2016, 9732780. [Google Scholar] [CrossRef] [Green Version]
- Rahman Khan, F.; Sulaiman Alhewairini, S. Zebrafish (Danio rerio) as a Model Organism. Curr. Trends Cancer Manag. 2019, 2019, 81517. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Takahashi, R. Parkinson’s disease pathogenesis from the viewpoint of small fish models. J. Neural Transm. 2018, 125, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Barnhill, L.M.; Murata, H.; Bronstein, J.M. Studying the pathophysiology of Parkinson’s disease using zebrafish. Biomedicines 2020, 8, 8070197. [Google Scholar] [CrossRef]
- Lam, C.S.; Korzh, V.; Strahle, U. Zebrafish embryos are susceptible to the dopaminergic neurotoxin MPTP. Eur. J. Neurosci. 2005, 21, 1758–1762. [Google Scholar] [CrossRef]
- Wasel, O.; Freeman, J.L. Chemical and genetic zebrafish models to define mechanisms of and treatments for dopaminergic neurodegeneration. Int. J. Mol. Sci. 2020, 21, 5981. [Google Scholar] [CrossRef]
- Sarath, B.N.; Murthy, C.H.L.; Kakara, S.; Sharma, R.; Brahmendra, S.C.V.; Idris, M.M. 1-Methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine induced Parkinson’s disease in zebrafish. Proteomics 2016, 16, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Vijayanathan, Y.; Lim, F.T.; Lim, S.M.; Long, C.M.; Tan, M.P.; Majeed, A.B.A.; Ramasamy, K. 6-OHDA-Lesioned Adult Zebrafish as a Useful Parkinson’s Disease Model for Dopaminergic Neuroregeneration. Neurotox. Res. 2017, 32, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Vaz, R.L.; Sousa, S.; Chapela, D.; van der Linde, H.C.; Willemsen, R.; Correia, A.D.; Outeiro, T.F.; Afonso, N.D. Identification of antiparkinsonian drugs in the 6-hydroxydopamine zebrafish model. Pharmacol. Biochem. Behav. 2020, 189, 172828. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, F.; Xu, T.; Song, H.; Lu, B. Acteoside protects against 6-OHDA-induced dopaminergic neuron damage via Nrf2-ARE signaling pathway. Food Chem. Toxicol. 2018, 119, 6–13. [Google Scholar] [CrossRef]
- Cronin, A.; Grealy, M. Neuroprotective and neuro-restorative effects of minocycline and rasagiline in a zebrafish 6-hydroxydopamine model of Parkinson’s disease. Neuroscience 2017, 367, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Souders, C.L.; Zhao, Y.H.; Martyniuk, C.J. Paraquat affects mitochondrial bioenergetics, dopamine system expression, and locomotor activity in zebrafish (Danio rerio). Chemosphere 2018, 191, 106–117. [Google Scholar] [CrossRef]
- Nellore, J.P.N. Paraquat exposure induces behavioral deficits in larval zebrafish during the window of dopamine neurogenesis. Toxicol. Rep. 2015, 2, 950–956. [Google Scholar] [CrossRef] [Green Version]
- Bortolotto, J.W.; Cognato, G.P.; Christoff, R.R.; Roesler, L.N.; Leite, C.E.; Kist, L.W.; Bogo, M.R.; Vianna, M.R.; Bonan, C.D. Long-term exposure to paraquat alters behavioral parameters and dopamine levels in adult zebrafish (Danio rerio). Zebrafish 2014, 11, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.E.; Nunes, M.E.; Menezes, C.C.; Marins, A.T.; Leitemperger, J.; Gressler, A.C.L.; Carvalho, F.B.; de Freitas, C.M.; Quadros, V.A.; Fachinetto, R.; et al. Sodium Selenite Prevents Paraquat-Induced Neurotoxicity in Zebrafish. Mol. Neurobiol. 2018, 55, 1928–1941. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.J.; Li, L.X.; Chen, J.; Wei, S.Z.; Wang, F.; Hu, H.; Xie, A.M.; Liu, C.F. Sleep deprivation caused a memory defects and emotional changes in a rotenone-based zebrafish model of Parkinson’s disease. Behav. Brain Res. 2019, 372, 112031. [Google Scholar] [CrossRef] [PubMed]
- Ramli, M.D.B.C.; Hashim, N.H.B.; Uzid, M.B.M.; Weinheimeri, A.Z. Zebrafish parkinson’s model: The effects of tocotrienol rich fraction towards rotenone induced zebrafish. Int. J. Med. Toxicol. Legal Med. 2020, 23, 1–7. [Google Scholar] [CrossRef]
- Ünal, İ.; Üstündağ, Ü.V.; Ateş, P.S.; Eğilmezer, G.; Alturfan, A.A.; Yiğitbaşı, T.; Emekli-Alturfan, E. Rotenone impairs oxidant/antioxidant balance both in brain and intestines in zebrafish. Int. J. Neurosci. 2019, 129, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Trans. Neurodegener. 2015, 4, 40–42. [Google Scholar] [CrossRef] [Green Version]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mole Neurodegener. 2020, 15, 7. [Google Scholar] [CrossRef] [Green Version]
- Flinn, L.; Mortiboys, H.; Volkmann, K.; Kster, R.W.; Ingham, P.W.; Bandmann, O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 2009, 132, 1613–1623. [Google Scholar] [CrossRef] [Green Version]
- Sallinen, V.; Kolehmainen, J.; Priyadarshini, M.; Toleikyte, G.; Chen, Y.C.; Panula, P. Dopaminergic cell damage and vulnerability to MPTP in PINK1 knockdown zebrafish. Neurobiol. Dis. 2010, 40, 93–101. [Google Scholar] [CrossRef]
- Xi, Y.; Ryan, J.; Noble, S.; Yu, M.; Yilbas, A.E.; Ekker, M. Impaired dopaminergic neuron development and locomotor function in zebrafish with loss of pink1 function. Eur. J. Neurosci. 2010, 31, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Sloan, M.; Alegre-Abarrategui, J.; Wade-Martins, R. Insights into LRRK2 function and dysfunction from transgenic and knockout rodent models. Biochem. Soc. Trans. 2012, 40, 1080–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhudesai, S.; Bensabeur, F.Z.; Abdullah, R.; Basak, I.; Baez, S.; Alves, G.; Holtzman, N.G.; Larsen, J.P.; Møller, S.G. LRRK2 knockdown in zebrafish causes developmental defects, neuronal loss, and synuclein aggregation. J. Neurosci. Res. 2016, 94, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Sheng, D.; Qu, D.; Kwok, K.H.; Ng, S.S.; Lim, A.Y.; Aw, S.S.; Lee, C.W.; Sung, W.K.; Tan, E.K.; Lufkin, T.; et al. Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS Genet. 2010, 6, e1000914. [Google Scholar] [CrossRef] [Green Version]
- Seegobin, S.P.; Heaton, G.R.; Liang, D.; Choi, I.; Blanca Ramirez, M.; Tang, B.; Yue, Z. Progress in LRRK2-Associated Parkinson’s Disease Animal Models. Front. Neurosci. 2020, 14, 674. [Google Scholar] [CrossRef]
- Hughes, G.L.; Lones, M.A.; Bedder, M.; Currie, P.D.; Smith, S.L.; Pownall, M.E. Machine learning discriminates a movement disorder in a zebrafish model of Parkinson’s disease. Dis. Model Mech. 2020, 13, dmm045815. [Google Scholar] [CrossRef]
- Bai, Q.; Mullett, S.J.; Garver, J.A.; Hinkle, D.A.; Burton, E.A. Zebrafish DJ-1 is evolutionarily conserved and expressed in dopaminergic neurons. Brain Res. 2006, 1113, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Bretaud, S.; Allen, C.; Ingham, P.W.; Bandmann, O. p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson’s disease. J. Neurochem. 2007, 100, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Edson, A.J.; Hushagen, H.A.; Frøyset, A.K.; Elda, I.; Khan, E.A.; Di Stefano, A.; Fladmark, K.E. Dysregulation in the Brain Protein Profile of Zebrafish Lacking the Parkinson’s Disease-Related Protein DJ-1. Mol. Neurobiol. 2019, 56, 8306–8322. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.C.; Lulla, A.; Stahl, M.C.; Wheat, N.D.; Bronstein, J.M.; Sagasti, A. Axon degeneration and PGC-1a-mediated protection in a zebrafish model of a-synuclein toxicity. Dis. Models Mech. 2014, 7, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Keatinge, M.; Bui, H.; Menke, A.; Chen, Y.C.; Sokol, A.M.; Bai, Q.; Ellett, F.; Da Costa, M.; Burke, D.; Gegg, M.; et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015, 24, 6640–6652. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Ito, J.; Matsui, N.; Uechi, T.; Onodera, O.; Kakita, A. Cytosolic dsDNA of mitochondrial origin induces cytotoxicity and neurodegeneration in cellular and zebrafish models of Parkinson’s disease. Nat. Commun. 2021, 12, 3101. [Google Scholar] [CrossRef]
- Noble, S.; Ismail, A.; Godoy, R.; Xi, Y.; Ekker, M. Zebrafish Parla- and Parlb-deficiency affects dopaminergic neuron patterning and embryonic survival. J. Neurochem. 2012, 122, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Merhi, R.; Kalyn, M.; Zhu-Pawlowsky, A.; Ekker, M. Loss of parla Function Results in Inactivity, Olfactory Impairment, and Dopamine Neuron Loss in Zebrafish. Biomedicines 2021, 9, 205. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.R.; Dadgar, J.; Trevor, A.; Singer, T.P. Energy-driven uptake of N-methyl-4-phenylpyridine by brain mitochondria mediates the neurotoxicity of MPTP. Life Sci. 1986, 39, 581–588. [Google Scholar] [CrossRef]
- Colpo, G.D.; Ribeiro, F.M.; Rocha, N.P.; Teixeira, A.L. Study of Human Disease. Animal Models for the Study of Human Disease; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1109–1129. [Google Scholar]
- Mastrangelo, L. The genetics of Parkinson disease. Adv. Genet. 2017, 98, 43–62. [Google Scholar]
- Matsui, H.; Gavinio, R.; Takahashi, R. Medaka fish Parkinson’s disease model. Exp. Neurobiol. 2012, 21, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Vaccari, C.; El Dib, R.; Gomaa, H.; Lopes, L.C.; de Camargo, J.L. Paraquat and Parkinson’s disease: A systematic review and meta-analysis of observational studies. J. Toxicol. Environ. Health Part B Crit. Rev. 2019, 22, 172–202. [Google Scholar] [CrossRef]
- Nunes, M.E.; Müller, T.E.; Braga, M.M.; Fontana, B.D.; Quadros, V.A.; Marins, A.; Rodrigues, C.; Menezes, C.; Rosemberg, D.B.; Loro, V.L. Chronic Treatment with Paraquat Induces Brain Injury, Changes in Antioxidant Defenses System, and Modulates Behavioral Functions in Zebrafish. Mol. Neurobiol. 2017, 54, 3925–3934. [Google Scholar] [CrossRef] [PubMed]
- Bastías-Candia, S.; Zolezzi, J.M.; Inestrosa, N.C. Revisiting the Paraquat-Induced Sporadic Parkinson’s Disease-Like Model. Mole Neurob. 2019, 56, 1044–1055. [Google Scholar] [CrossRef]
- Robea, M.A.; Strungaru, A.; Lenzi, C.; Nicoară, M.; Ciobică, A. The Importance of Rotenone in Generating Neurological and Psychiatric Features in Zebrafish-Relevance for a Parkinson’s Disease Model. Acad. Roman Sci. 2018, 7, 55–67. [Google Scholar]
- Jiang, X.; Tang, P.C.; Chen, Q.; Zhang, X.; Fan, Y.Y.; Yu, B.C.; Gu, X.X.; Sun, Y.; Ge, X.Q.; Zhang, X.L. Cordycepin Exerts Neuroprotective Effects via an Anti-Apoptotic Mechanism based on the Mitochondrial Pathway in a Rotenone-Induced Parkinsonism Rat Model. CNS Neurol. Disord. Drug Targets 2019, 18, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Askar, M.H.; Hussein, A.M.; Al-Basiony, S.F.; Meseha, R.K.; Metias, E.F.; Salama, M.M.; Antar, A.; El-Sayed, A. Effects of Exercise and Ferulic Acid on Alpha Synuclein and Neuroprotective Heat Shock Protein 70 in An Experimental Model of Parkinsonism Disease. CNS Neurol. Disord. Drug Targets 2019, 18, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- Hisano, Y.; Ota, S.; Kawahara, A. Genome editing using artificial site-specific nucleases in zebrafish. Dev. Growth Differ. 2014, 56, 26–33. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Kaini, P.; Sander, J.D.; Joung, J.K.; Peterson, R.T.; Yeh, J.R. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS ONE 2013, 8, e68708. [Google Scholar] [CrossRef]
- Selvaraj, S.; Piramanayagam, S. Impact of gene mutation in the development of Parkinson’s disease. Genes Dis. 2019, 6, 120–128. [Google Scholar] [CrossRef]
- Blandini, F.; Armentero, M.T. Animal models of Parkinson’s disease. FEBS J. 2012, 279, 1156–1166. [Google Scholar] [CrossRef] [Green Version]
- Maynard, M.E.; Redell, J.B.; Kobori, N.; Underwood, E.L.; Fischer, T.D.; Hood, K.N.; LaRoche, V.; Waxham, M.N.; Moore, A.N.; Dash, P.K. Loss of PTEN-induced kinase 1 (Pink1) reduces hippocampal tyrosine hydroxylase and impairs learning and memory. Exp. Neurol. 2020, 323, 113081. [Google Scholar] [CrossRef]
- Kelm-Nelson, C.A.; Brauer, A.F.L.; Barth, K.J.; Lake, J.M.; Sinnen, M.L.K.; Stehula, F.J.; Muslu, C.; Marongiu, R.; Kaplitt, M.G.; Ciucci, M.R. Characterization of early-onset motor deficits in the Pink1−/− mouse model of Parkinson disease. Brain Res. 2018, 1680, 1–12. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Giangrasso, D.M.; Furlong, T.M.; Keefe, K.A. Characterization of striatum-mediated behavior and neurochemistry in the DJ-1 knock-out rat model of Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104673. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [Green Version]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. Parkinson’s Dis. 2018, 1, 3–26. [Google Scholar] [CrossRef]
- Toni, M.; Cioni, C. Fish Synucleins: An Update. Mar. Drugs 2015, 13, 6665–6686. [Google Scholar] [CrossRef]
- Prabhudesai, S.; Sinha, S.; Attar, A.; Kotagiri, A.; Fitzmaurice, A.G.; Lakshmanan, R.; Ivanova, M.I.; Loo, J.A.; Klärner, F.G.; Schrader, T.; et al. A novel “molecular tweezer” inhibitor of α-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics 2012, 9, 464–476. [Google Scholar] [CrossRef] [Green Version]
- Van Laar, V.S.; Chen, J.; Zharikov, A.D.; Bai, Q.; Di Maio, R.; Dukes, A.A.; Hastings, T.G.; Watkins, S.C.; Greenamyre, J.T.; St Croix, C.M.; et al. α-Synuclein amplifies cytoplasmic peroxide flux and oxidative stress provoked by mitochondrial inhibitors in CNS dopaminergic neurons in vivo. Redox Biol. 2020, 37, 101695. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International (ADI): London, UK, 2019. [Google Scholar]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Ruhl, T.; Jonas, A.; Seidel, N.I.; Prinz, N.; Albayram, O.; Bilkei-Gorzo, A.; von der Emde, G. Oxidation and Cognitive Impairment in the Aging Zebrafish. Gerontology 2015, 62, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, P.; Thomas, A.K.; Cosacak, M.I.; Papadimitriou, C.; Mashkaryan, V.; Zhang, Y.; Kizil, C. Modeling Amyloid-β42 Toxicity and Neurodegeneration in Adult Zebrafish Brain. J. Vis. Exp. 2017, 128, 56014. [Google Scholar] [CrossRef]
- Bhattarai, P.; Thomas, A.K.; Zhang, Y.; Kizil, C. The effects of aging on Amyloid-β42-induced neurodegeneration and regeneration in adult zebrafish brain. Neurogenesis 2017, 4, e1322666. [Google Scholar] [CrossRef]
- Rudrabhatla, P.; Pant, H.C. Role of protein phosphatase 2A in Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Nada, S.E.; Williams, F.E.; Shah, Z.A. Development of a Novel and Robust Pharmacological Model of Okadaic Acid-induced Alzheimer’s Disease in Zebrafish. CNS Neurol. Disord. Drug Targets 2016, 15, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Koehler, D.; Williams, F.E. Utilizing zebrafish and okadaic acid to study Alzheimer’s disease. Neural Regen. Res. 2018, 13, 1538–1541. [Google Scholar] [CrossRef] [PubMed]
- Muthuraman, A.; Thilagavathi, L.; Jabeen, S.; Ravishankar, S.B.; Ahmed, S.S.; George, T.; Rishitha, N.; Paramakrishnan, N. Curcumin prevents cigarette smoke extract induced cognitive impairment. Front. Biosci. 2019, 11, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Muthuraman, A.; Nafisa, K.; Sowmya, M.S.; Arpitha, B.M.; Choedon, N.; Sandy, C.D.; Rishitha, N.; Johurul, I. Role of ambrisentan (selective endothelin-A receptor antagonist) on cigarette smoke exposure induced cognitive impairment in Danio rerio. Life Sci. 2019, 222, 133–139. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhong, Z.M.; Che, Y. Locomotor activity and learning and memory abilities in Alzheimer’s disease induced by aluminum in an acid environment in zebrafish. Dongwuxue Yanjiu 2012, 33, 231–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, D.D.S.; Danielle, N.M.; Altenhofen, S.; Luzardo, M.D.; Costa, P.G.; Bianchini, A.; Bonan, C.D.; da Silva, R.S.; Dafre, A.L. Copper at low levels impairs memory of adult zebrafish (Danio rerio) and affects swimming performance of larvae. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2016, 185–186, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Altenhofen, S.; Wiprich, M.T.; Nery, L.R.; Leite, C.E.; Vianna, M.R.M.R.; Bonan, C.D. Manganese(II) chloride alters behavioral and neurochemical parameters in larvae and adult zebrafish. Aquat. Toxicol. 2017, 182, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Garver, J.A.; Hukriede, N.A.; Burton, E.A. Generation of a transgenic zebrafish model of Tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res. 2007, 35, 6501–6516. [Google Scholar] [CrossRef] [Green Version]
- Paquet, D.; Bhat, R.; Sydow, A.; Mandelkow, E.M.; Berg, S.; Hellberg, S.; Fälting, J.; Distel, M.; Köster, R.W.; Schmid, B.; et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Investig. 2009, 119, 1382–1395. [Google Scholar] [CrossRef] [Green Version]
- Sundvik, M.; Chen, Y.C.; Panula, P. Presenilin1 regulates histamine neuron development and behavior in zebrafish, danio rerio. J. Neurosci. 2013, 33, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- van Bebber, F.; Hruscha, A.; Willem, M.; Schmid, B.; Haass, C. Loss of Bace2 in zebrafish affects melanocyte migration and is distinct from Bace1 knock out phenotypes. J. Neurochem. 2013, 127, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Bejanin, A.; Schonhaut, D.R.; La Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M.; et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef] [Green Version]
- Mazza, M.; Marano, G.; Traversi, G.; Bria, P.; Mazza, S. Primary cerebral blood flow deficiency and Alzheimer’s disease: Shadows and lights. J. Alzheimers Dis. 2011, 23, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, J.J.; Veronese, M.; Marchitelli, L.; Bodini, B.; Tonietto, M.; Stankoff, B.; Brooks, D.J.; Bertoldo, A.; Edison, P.; Turkheimer, F.E. Dynamic 11C-PiB PET Shows Cerebrospinal Fluid Flow Alterations in Alzheimer Disease and Multiple Sclerosis. J. Nucl. Med. 2019, 60, 1452–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef]
- Jiang, T.; Tan, L.; Zhu, X.C.; Zhou, J.S.; Cao, L.; Tan, M.S.; Wang, H.F.; Chen, Q.; Zhang, Y.D.; Yu, J.T. Silencing of TREM2 exacerbates tau pathology, neurodegenerative changes, and spatial learning deficits in P301S tau transgenic mice. Neurobiol. Aging 2015, 36, 3176–3186. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Avila, J.; Villanueva, N. Use of okadaic acid to identify relevant phosphoepitopes in pathology: A focus on neurodegeneration. Mar. Drugs 2013, 11, 1168–1656. [Google Scholar] [CrossRef] [Green Version]
- Ryu, A.R.; Kim, D.H.; Kim, E.; Lee, M.Y. The Potential Roles of Extracellular Vesicles in Cigarette Smoke-Associated Diseases. Oxid. Med. Cell Longev. 2018, 4692081. [Google Scholar] [CrossRef]
- Xu, X.; Weber, D.; Burge, R.; VanAmberg, K. Neurobehavioral impairments produced by developmental lead exposure persisted for generations in zebrafish (Danio rerio). Neurotoxicology 2016, 52, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Bondy, S.C. Low levels of aluminum can lead to behavioral and morphological changes associated with Alzheimer’s disease and age-related neurodegeneration. Neurotoxicology 2016, 52, 222–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wei, X.; Yang, J.; Suo, J.; Chen, J.; Liu, X.; Zhao, X. Chronic exposure to aluminum and risk of Alzheimer’s disease: A meta-analysis. Neurosci. Lett. 2016, 610, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Duan, Y.; Hu, Y.; Zhao, Z. Advances in the pathogenesis of Alzheimer’s disease: A re-evaluation of amyloid cascade hypothesis. Transl. Neurodegener. 2012, 1, 18. [Google Scholar] [CrossRef] [Green Version]
- Shannon, K.M. Huntington’s disease—Clinical signs, symptoms, presymptomatic diagnosis, and diagnosis. Handb. Clin. Neurol. 2011, 100, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef] [PubMed]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Duyao, M.P.; Auerbach, A.B.; Ryan, A.; Persichetti, F.; Barnes, G.T.; McNeil, S.M.; Ge, P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 1995, 269, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, S.; Liu, J.P.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlovich, C.A.; John, R.M.; Ramirez, L.; Stainier, D.Y.; Myers, R.M. Characterization of the Huntington’s disease (HD) gene homologue in the zebrafish Danio rerio. Gene 1998, 217, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.L.; Henshall, T.L.; Dayan, S.; Lardelli, M.T.; Richards, R.I. Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Hum. Mol. Genet. 2007, 16, 1905–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henshall, T.L.; Tucker, B.; Lumsden, A.L.; Nornes, S.; Lardelli, M.T.; Richards, R.I. Selective neuronal requirement for huntingtin in the developing zebrafish. Hum. Mol. Genet. 2009, 18, 4830–4842. [Google Scholar] [CrossRef] [Green Version]
- Diekmann, H.; Anichtchik, O.; Fleming, A.; Futter, M.; Goldsmith, P.; Roach, A.; Rubinsztein, D.C. Decreased BDNF levels are a major contributor to the embryonic phenotype of huntingtin knockdown zebrafish. J. Neurosci. 2009, 29, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, N.W.; Broadley, S.A.; Hirschberger, T.; Tavan, P.; Kretzschmar, H.A.; Giese, A.; Haass, C.; Hartl, F.U.; Schmid, B. Identification of anti-prion compounds as efficient inhibitors of polyglutamine protein aggregation in a zebrafish model. J. Biol. Chem. 2007, 282, 9195–9203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Veldman, M.B.; Rios-Galdamez, Y.; Lu, X.H.; Gu, X.; Qin, W.; Li, S.; Yang, X.W.; Lin, S. The N17 domain mitigates nuclear toxicity in a novel zebrafish Huntington’s disease model. Mol. Neurodegener. 2015, 10, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VerPlank, J.J.S.; Tyrkalska, S.D.; Fleming, A.; Rubinsztein, D.C.; Goldberg, A.L. cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 14220–14230. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, P.M.; Zhou, X.; Robins, A.M.; Paushter, D.H.; Kim, D.; Smolka, M.B.; Hu, F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 2016, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.P.; Higginbottom, A.; McGown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Ohki, Y.; Wenninger-Weinzierl, A.; Hruscha, A.; Asakawa, K.; Kawakami, K.; Haass, C.; Edbauer, D.; Schmid, B. Glycine-alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration. Mol. Neurodegener. 2017, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Yeh, T.H.; Liu, H.F.; Li, Y.W.; Lu, C.S.; Shih, H.Y.; Chiu, C.C.; Lin, S.J.; Huang, Y.C.; Cheng, Y.C. C9orf72 is essential for neurodevelopment and motility mediated by Cyclin G1. Exp. Neurol. 2018, 304, 114–124. [Google Scholar] [CrossRef]
- McGown, A.; Shaw, D.P.J.; Ramesh, T. ZNStress: A high-throughput drug screening protocol for identification of compounds modulating neuronal stress in the transgenic mutant sod1G93R zebrafish model of amyotrophic lateral sclerosis. Mol. Neurodegener. 2016, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namikawa, K.; Dorigo, A.; Zagrebelsky, M.; Russo, G.; Kirmann, T.; Fahr, W.; Dübel, S.; Korte, M.; Köster, R.W. Modeling Neurodegenerative Spinocerebellar Ataxia Type 13 in Zebrafish Using a Purkinje Neuron Specific Tunable Coexpression System. J. Neurosci. 2019, 39, 3948–3969. [Google Scholar] [CrossRef] [Green Version]
- Namikawa, K.; Dorigo, A.; Köster, R.W. Neurological Disease Modelling for Spinocerebellar Ataxia Using Zebrafish. J. Exp. Neurosci. 2019, 13, 1179069519880515. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Cai, X.; Wang, G.; Ouyang, G.; Cao, H. Model construction of Niemann-Pick type C disease in zebrafish. Biol. Chem. 2018, 399, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Koike, M.; Ansai, S.; Kinoshita, M.; Ishikawa-Fujiwara, T.; Matsui, H.; Naruse, K.; Sakamoto, N.; Uchiyama, Y.; Todo, T.; et al. Viable neuronopathic Gaucher disease model in Medaka (Oryzias latipes) displays axonal accumulation of alpha-synuclein. PLoS Genet. 2015, 11, e1005065. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Absolute number of articles for zebrafish neurodegenerative diseases modelper year of publication extracted from the PubMed database.
Figure 1.
Absolute number of articles for zebrafish neurodegenerative diseases modelper year of publication extracted from the PubMed database.


{kind=link}
Table 1.
Zebrafish models of Parkinson’s disease.
| Method | Phenotype | Results | Reference |
|---|---|---|---|
| MPTP induced | Motor impairments and weakened touch sensory | Reduction of locomotor activity and dopaminergic neuron, over-expression of synuclein in the optic tectum | [33,34,35,36] |
| 6-OHDA induced | Motor impairments and anxiety | Reduction of dopaminergic neurons and morphological alternations | [37,38,39,40] |
| Paraquat induced | Motor impairments, various developmental anomalies | The paraquat-treated zebrafish did not recapitulate PD pathology | [41,42,43,44] |
| Rotenone induced | Motor impairments, anxiety, and olfactory dysfunction | In addition to motor impairments, they also show Olfactory dysfunction, which is a typical non-motor symptom of PD | [45,46,47,48] |
| PARK2 Morpholino | No abnormalities in swimming behavior | Loss of the DA neuron numbers in the diencephalon, whereas no abnormalities in swimming behavior | [49,50] |
| PINK1 Morpholino; Transgenes | Motor impairment and oxidative stress | Reduction of dopaminergic neurons, dis-organized diencephalic dopaminergic neurons, and the pink1 gene are sensitive markers of oxidative stress in zebrafish | [51,52] |
| LRRK2 Morpholino | Motor impairment | Loss of neuronal cells and synuclein aggregation, similar to the phenotype of PD in humans | [53,54,55,56] |
| PARK7 Morpholino; CRISPR/Cas9 | Motor impairment | With aging, exhibit lower TH levels, respiratory failure in skeletal muscle, and lower body mass, particularly in the male fish | [57,58,59,60] |
| Synuclein Transgenes | Motor impairment | Led to cell death in larval zebrafish sensory neurons | [61] |
| GBA TALEN | Motor impairment | Reduction of the GBA protein, dopaminergic, and noradrenergic neurons | [62,63] |
| PARL Morpholino; CRISPR/Cas9 | Motor impairment and olfactory dysfunction | Reduced DA neuronal population and dysregulation of the PINK1/Parkin mitophagy pathway | [64,65] |
Table 2.
Zebrafish models of Alzheimer’s disease.
| Method | Phenotype | Results | Reference |
|---|---|---|---|
| Amyloid-β42 induced | Intracellular depositions | Link between aging, neurogenesis, regenerative, neuroinflammation, and neural stem cell plasticity | [99,100] |
| Okadaic acid induced | Cognitive and memory impairments, neuroinflammation cholinergic dysfunction, glutamate excitotoxicity, and mitochondrial dysfunction | Provide all the major molecular hallmarks of AD | [101,102,103] |
| Cigarette smoke extract induced | Neurocognitive dysfunction | Enhancement of the acetylcholinesterase activity | [104,105] |
| Aluminum chloride induced | Neurocognitive dysfunction, memory impairment | Impaired locomotor activity, learning, and memory abilities | [106] |
| Copper induced | Memory impairment | Reduction of the glutathione S-transferase (GST) activity in the gill | [107] |
| MnCl2 induced | Cognition and exploratory behavior | Impairment of aversive long-term memory and distance traveled movement time | [108] |
| MAPT Transgenes | Motor impairment | The phenotypic abnormalities at larval stages make it suitable for high-throughput screening | [109,110] |
| PSEN1 ENU-mutagenized | Motor impairment | Regulation of histaminergic neuron development | [111] |
| BACE1/2 zinc finger nuclease; ENU-mutagenized | Hypomyelination, supernumery neuromasts, and abnormal pigmentation | Bace1 and Bace2 are proteases with different physiological functions | [112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, J.; Cao, H. Zebrafish and Medaka: Important Animal Models for Human Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 10766. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910766
AMA Style
Wang J, Cao H. Zebrafish and Medaka: Important Animal Models for Human Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(19):10766. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910766
Chicago/Turabian StyleWang, Jing, and Hong Cao. 2021. "Zebrafish and Medaka: Important Animal Models for Human Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 19: 10766. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910766
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.