
Utilization of Pharmacological Ascorbate to Enhance Hydrogen Peroxide-Mediated Radiosensitivity in Cancer Therapy

 ,
,  , ,
, ,  , and
, and 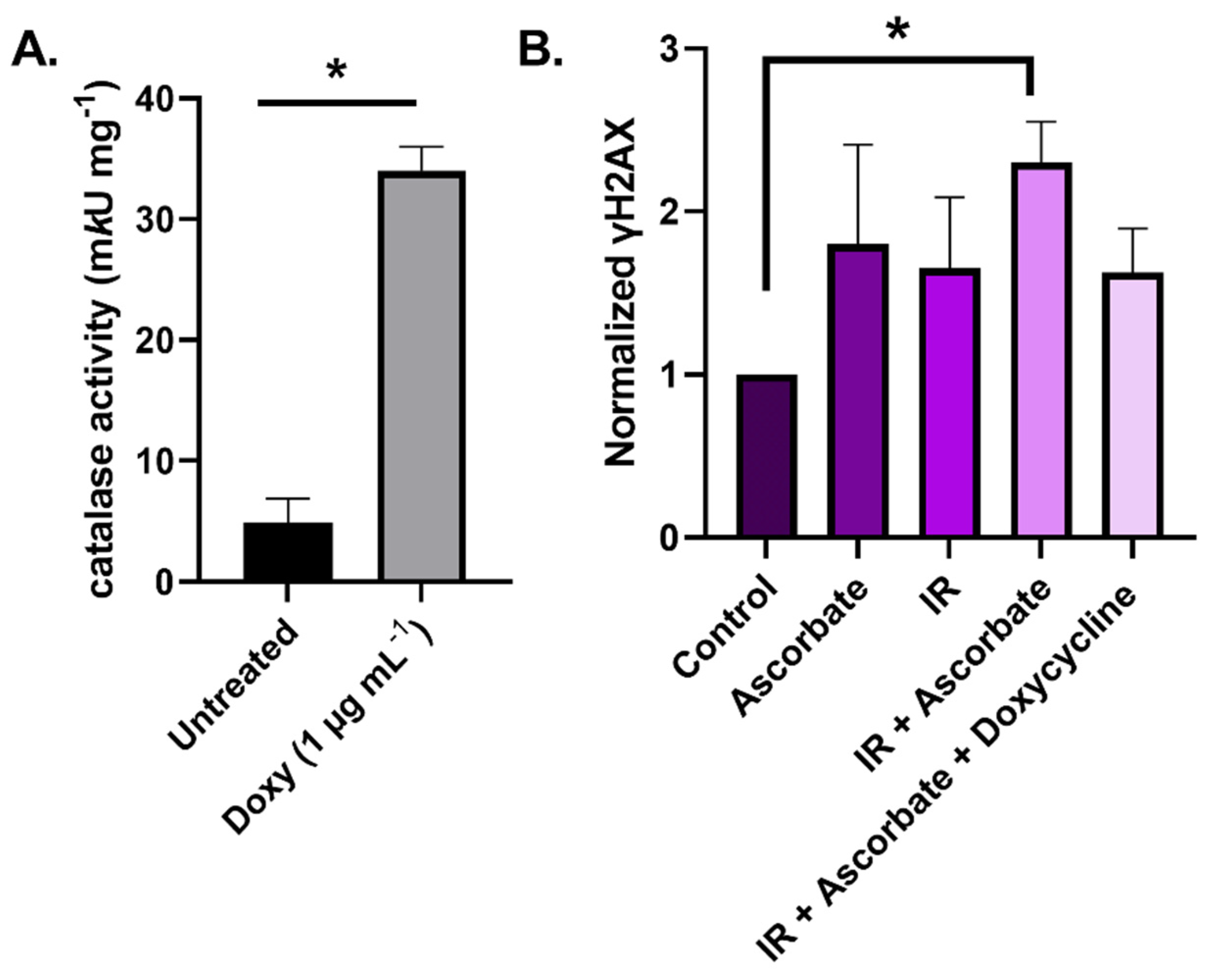{kind=link}
{kind=link}
Abstract
:1. Introduction
2. History of the Use of P-AscH− as a Cancer Drug
3. Radiation-Induced Injury in Cancer and the Role of Hydrogen Peroxide
4. P-AscH− and the Flux of Hydrogen Peroxide
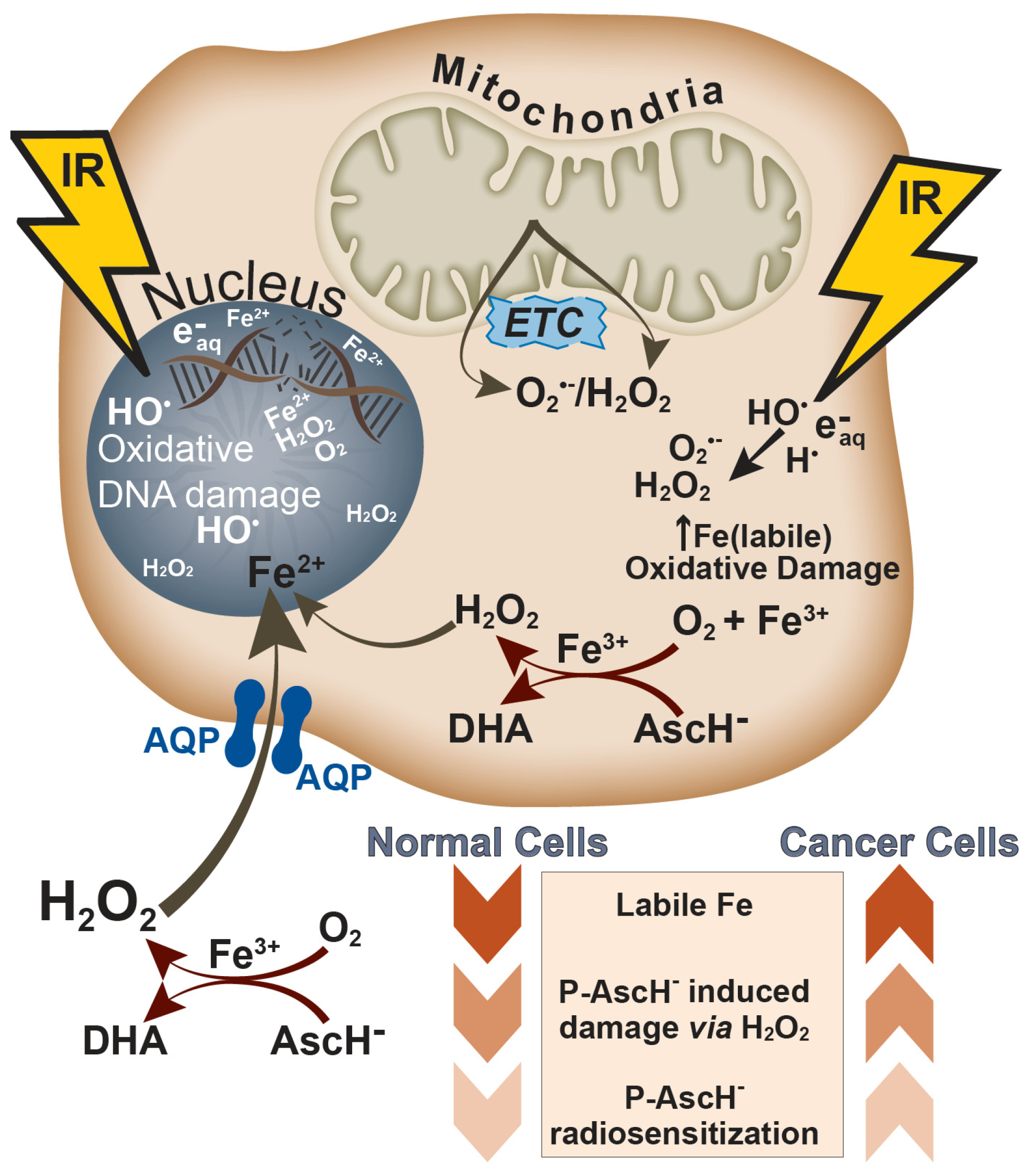
5. Targeting Intracellular Iron to Enhance Radiation-induced Oxidative Damage
5.1. Disruption of Intracellular Iron Metabolism by P-AscH−
5.2. Interactions of Iron and Ionizing Radiation
6. P-AscH− as a Radioprotector in Normal Tissue
7. Conclusions
8. Materials and Methods
8.1. Catalase Activity Assay
8.2. Lentiviral Transduction of U87 Cells with Catalase
8.3. γH2AX Staining and Flow Cytometry
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manna, B.; Cooper, J.S. Radiation Therapy Induced Skin Ulcer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Borek, C. Antioxidants and radiation therapy. J. Nutr. 2004, 134, 3207S–3209S. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Cieslak, J.A., 3rd; Welsh, J.L.; Sibenaller, Z.A.; Allen, B.G.; Wagner, B.A.; Kalen, A.L.; Doskey, C.M.; Strother, R.K.; Button, A.M.; et al. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer Res. 2015, 75, 3314–3326. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2•− and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 32, 268. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [Green Version]
- Cameron, E.; Pauling, L. Ascorbic acid and the glycosaminoglycans. An orthomolecular approach to cancer and other diseases. Oncology 1973, 27, 181–192. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Dorzi, H.M.; Alruwaita, A.A.; Marae, B.O.; Alraddadi, B.S.; Tamim, H.M.; Ferayan, A.; Arabi, Y.M. Incidence, risk factors and outcomes of seizures occurring after craniotomy for primary brain tumor resection. Neurosciences 2017, 22, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.H.; Horton, J.; Schoenfeld, D.; Salazer, O.; Perez-Tamayo, R.; Kramer, S.; Weinstein, A.; Nelson, J.S.; Tsukada, Y. Comparison of postoperative radiotherapy and combined postoperative radiotherapy and chemotherapy in the multidisciplinary management of malignant gliomas. A joint Radiation Therapy Oncology Group and Eastern Cooperative Oncology Group study. Cancer 1983, 52, 997–1007. [Google Scholar] [CrossRef]
- Kong, D.S.; Lee, J.I.; Park, K.; Kim, J.H.; Lim, D.H.; Nam, D.H. Efficacy of stereotactic radiosurgery as a salvage treatment for recurrent malignant gliomas. Cancer 2008, 112, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Alexander, M.S.; Wilkes, J.G.; Schroeder, S.R.; Buettner, G.R.; Wagner, B.A.; Du, J.; Gibson-Corley, K.; O’Leary, B.R.; Spitz, D.R.; Buatti, J.M.; et al. Pharmacologic Ascorbate Reduces Radiation-Induced Normal Tissue Toxicity and Enhances Tumor Radiosensitization in Pancreatic Cancer. Cancer Res. 2018, 78, 6838–6851. [Google Scholar] [CrossRef] [Green Version]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Cameron, E.; Pauling, L. The orthomolecular treatment of cancer. I. The role of ascorbic acid in host resistance. Chem. Biol. Interact. 1974, 9, 273–283. [Google Scholar] [CrossRef]
- Cameron, E.; Campbell, A. The orthomolecular treatment of cancer. II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem. Biol. Interact. 1974, 9, 285–315. [Google Scholar] [CrossRef]
- Cameron, E.; Campbell, A.; Jack, T. The orthomolecular treatment of cancer. III. Reticulum cell sarcoma: Double complete regression induced by high-dose ascorbic acid therapy. Chem. Biol. Interact. 1975, 11, 387–393. [Google Scholar] [CrossRef]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689. [Google Scholar] [CrossRef] [Green Version]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1978, 75, 4538–4542. [Google Scholar] [CrossRef] [Green Version]
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N. Engl. J. Med. 1979, 301, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Wittes, R.E. Vitamin C and cancer. N. Engl. J. Med. 1985, 312, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Graumlich, J.F.; Ludden, T.M.; Conry-Cantilena, C.; Cantilena, L.R., Jr.; Wang, Y.; Levine, M. Pharmacokinetic model of ascorbic acid in healthy male volunteers during depletion and repletion. Pharm. Res. 1997, 14, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Padayatty, S.J.; Espey, M.G. Vitamin C: A concentration-function approach yields pharmacology and therapeutic discoveries. Adv. Nutr. 2011, 2, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riordan, H.D.; Riordan, N.H.; Jackson, J.A.; Casciari, J.J.; Hunninghake, R.; Gonzalez, M.J.; Mora, E.M.; Miranda-Massari, J.R.; Rosario, N.; Rivera, A. Intravenous vitamin C as a chemotherapy agent: A report on clinical cases. Health Sci. J. 2004, 23, 115–118. [Google Scholar]
- Monti, D.A.; Mitchell, E.; Bazzan, A.J.; Littman, S.; Zabrecky, G.; Yeo, C.J.; Pillai, M.V.; Newberg, A.B.; Deshmukh, S.; Levine, M. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS ONE 2012, 7, e29794. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J., 2nd; et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.G.; Bodeker, K.L.; Smith, M.C.; Monga, V.; Sandhu, S.; Hohl, R.; Carlisle, T.; Brown, H.; Hollenbeck, N.; Vollstedt, S.; et al. First-in-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined with Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2019, 25, 6590–6597. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 8th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2019; 597p. [Google Scholar]
- Ohashi, Y.; Sakai, K.; Hase, H.; Joki, N. Dry weight targeting: The art and science of conventional hemodialysis. Semin. Dial. 2018, 31, 551–556. [Google Scholar] [CrossRef]
- Spitz, D.R.; Azzam, E.I.; Li, J.J.; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer Metastasis Rev. 2004, 23, 311–322. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Oberley, L.W.; Buettner, G.R. Role of superoxide dismutase in cancer: A review. Cancer Res. 1979, 39, 1141–1149. [Google Scholar] [PubMed]
- Ahmad, I.M.; Aykin-Burns, N.; Sim, J.E.; Walsh, S.A.; Higashikubo, R.; Buettner, G.R.; Venkataraman, S.; Mackey, M.A.; Flanagan, S.W.; Oberley, L.W.; et al. Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J. Biol. Chem. 2005, 280, 4254–4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babior, B.M. NADPH oxidase: An update. Blood 1999, 93, 1464–1476. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [Green Version]
- Afifi, A.K.; Jacoby, C.G.; Bell, W.E.; Menezes, A.H. Aqueductal stenosis and neurofibromatosis: A rare association. J. Child. Neurol. 1988, 3, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anticancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuranno, L.; Ient, J.; De Ruysscher, D.; Vooijs, M.A. Radiation-Induced Lung Injury (RILI). Front. Oncol. 2019, 9, 877. [Google Scholar] [CrossRef] [Green Version]
- Brandes, A.A.; Tosoni, A.; Spagnolli, F.; Frezza, G.; Leonardi, M.; Calbucci, F.; Franceschi, E. Disease progression or pseudoprogression after concomitant radiochemotherapy treatment: Pitfalls in neurooncology. Neuro Oncol. 2008, 10, 361–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gondi, V.; Pugh, S.L.; Tome, W.A.; Caine, C.; Corn, B.; Kanner, A.; Rowley, H.; Kundapur, V.; DeNittis, A.; Greenspoon, J.N.; et al. Preservation of memory with conformal avoidance of the hippocampal neural stem-cell compartment during whole-brain radiotherapy for brain metastases (RTOG 0933): A phase II multi-institutional trial. J. Clin. Oncol. 2014, 32, 3810–3816. [Google Scholar] [CrossRef]
- Turnquist, C.; Harris, B.T.; Harris, C.C. Radiation-induced brain injury: Current concepts and therapeutic strategies targeting neuroinflammation. Neurooncol. Adv. 2020, 2, vdaa057. [Google Scholar] [CrossRef] [PubMed]
- Vigliani, M.C.; Duyckaerts, C.; Hauw, J.J.; Poisson, M.; Magdelenat, H.; Delattre, J.Y. Dementia following treatment of brain tumors with radiotherapy administered alone or in combination with nitrosourea-based chemotherapy: A clinical and pathological study. J. Neurooncol. 1999, 41, 137–149. [Google Scholar] [CrossRef]
- Petronek, M.S.; Stolwijk, J.M.; Murray, S.D.; Steinbach, E.J.; Zakharia, Y.; Buettner, G.R.; Spitz, D.R.; Allen, B.G. Utilization of redox modulating small molecules that selectively act as pro-oxidants in cancer cells to open a therapeutic window for improving cancer therapy. Redox Biol. 2021, 42, 101864. [Google Scholar] [CrossRef]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef]
- Corpe, C.P.; Lee, J.H.; Kwon, O.; Eck, P.; Narayanan, J.; Kirk, K.L.; Levine, M. 6-Bromo-6-deoxy-L-ascorbic acid: An ascorbate analog specific for Na+-dependent vitamin C transporter but not glucose transporter pathways. J. Biol. Chem. 2005, 280, 5211–5220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, M.S.; O’Leary, B.R.; Wilkes, J.G.; Gibson, A.R.; Wagner, B.A.; Du, J.; Sarsour, E.; Hwang, R.F.; Buettner, G.R.; Cullen, J.J. Enhanced Pharmacological Ascorbate Oxidation Radiosensitizes Pancreatic Cancer. Radiat. Res. 2019, 191, 43–51. [Google Scholar] [CrossRef]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [Green Version]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic metals, ascorbate and free radicals: Combinations to avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Erudaitius, D.; Huang, A.; Kazmi, S.; Buettner, G.R.; Rodgers, V.G. Peroxiporin Expression Is an Important Factor for Cancer Cell Susceptibility to Therapeutic H2O2: Implications for Pharmacological Ascorbate Therapy. PLoS ONE 2017, 12, e0170442. [Google Scholar] [CrossRef]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra218. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Petronek, M.S.; Spitz, D.R.; Buettner, G.R.; Allen, B.G. Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism. Cancers 2019, 11, 1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonberg, D.L.; Miller, T.E.; Wu, Q.; Flavahan, W.A.; Das, N.K.; Hale, J.S.; Hubert, C.G.; Mack, S.C.; Jarrar, A.M.; Karl, R.T.; et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell 2015, 28, 441–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Hu, L.; Lu, K.; Wang, Y.; Xie, Y.; Gao, L.; Yang, G.; Xie, B.; He, W.; Chen, G.; et al. Ferroportin downregulation promotes cell proliferation by modulating the Nrf2-miR-17-5p axis in multiple myeloma. Cell Death Dis. 2019, 10, 624. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P.; Candeias, L.P. Fenton chemistry: An introduction. Radiat. Res. 1996, 145, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.Y.; Buettner, G.R. Iron and dioxygen chemistry is an important route to initiation of biological free radical oxidations: An electron paramagnetic resonance spin trapping study. Free Radic. Biol. Med. 1999, 26, 1447–1456. [Google Scholar] [CrossRef]
- Keyer, K.; Imlay, J.A. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 1996, 93, 13635–13640. [Google Scholar] [CrossRef] [Green Version]
- Keyer, K.; Gort, A.S.; Imlay, J.A. Superoxide and the production of oxidative DNA damage. J. Bacteriol. 1995, 177, 6782–6790. [Google Scholar] [CrossRef] [Green Version]
- McCormick, M.L.; Buettner, G.R.; Britigan, B.E. Endogenous superoxide dismutase levels regulate iron-dependent hydroxyl radical formation in Escherichia coli exposed to hydrogen peroxide. J. Bacteriol. 1998, 180, 622–625. [Google Scholar] [CrossRef] [Green Version]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J. Biol. Chem. 2000, 275, 14064–14069. [Google Scholar] [CrossRef] [Green Version]
- Gardner, P.R. Superoxide-driven aconitase FE-S center cycling. Biosci. Rep. 1997, 17, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Cantu, D.; Schaack, J.; Patel, M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE 2009, 4, e7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, W.H.; Rouault, T.A. Metabolic regulation of citrate and iron by aconitases: Role of iron-sulfur cluster biogenesis. Biometals 2007, 20, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Pain, J.; Ghosh, A.K.; Dancis, A.; Pain, D. Fe-S cluster biogenesis in isolated mammalian mitochondria: Coordinated use of persulfide sulfur and iron and requirements for GTP, NADH, and ATP. J. Biol. Chem. 2015, 290, 640–657. [Google Scholar] [CrossRef] [Green Version]
- Boveris, A.; Cadenas, E. Cellular sources and steady-state levels of reactive oxygen species. In Oxygen, Gene Expression and Cellular Function; Clerch, L.B., Massaro, D.J., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1997; pp. 1–25. [Google Scholar]
- Flint, D.H.; Tuminello, J.F.; Emptage, M.H. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 1993, 268, 22369–22376. [Google Scholar] [CrossRef]
- Fricke, H.; Morse, S. The chemical action of roentgen rays on dilute ferrosulphate solutions as a measure of dose. Am. J. Roentgenol. Radium Ther. 1927, 18, 430–432. [Google Scholar]
- Schreiner, L.J. Review of Fricke Gel Dosimeters. J. Phys. Conf. Ser. 2004, 3, 9–21. [Google Scholar] [CrossRef]
- Zakaria, A.M.; Lertnaisat, P.; Islam, M.M.; Meesungnoen, J.; Katsumura, Y.; Jay-Gerin, J.-P. Yield of the Fricke dosimeter irradiated with the recoil α and Li ions of the 10B(n,α)7Li nuclear reaction: Effects of multiple ionization and temperature. Can. J. Chem. 2021, 99, 425–435. [Google Scholar] [CrossRef]
- Ambroz, H.B.; Bradshaw, T.K.; Kemp, T.J.; Kornacka, E.M.; Przybytniak, G.K. Role of Iron Ions in Damage to DNA: Influence of Ionising Radiation, UV Light and H2O2. J. Photochem. Photobiol. A Chem. 2001, 142, 9–18. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.U.; Xiao, J.; Ni, H.T.; Cho, K.H.; Spellman, S.R.; Low, W.C.; Hall, W.A. Changes in expression of transferrin, insulin-like growth factor 1, and interleukin 4 receptors after irradiation of cells of primary malignant brain tumor cell lines. Radiat. Res. 2003, 160, 224–231. [Google Scholar] [CrossRef]
- Cairo, G.; Ronchi, R.; Recalcati, S.; Campanella, A.; Minotti, G. Nitric oxide and peroxynitrite activate the iron regulatory protein-1 of J774A.1 macrophages by direct disassembly of the Fe-S cluster of cytoplasmic aconitase. Biochemistry 2002, 41, 7435–7442. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Hentze, M.W. Activation of iron regulatory protein-1 by oxidative stress in vitro. Proc. Natl. Acad. Sci. USA 1998, 95, 10559–10563. [Google Scholar] [CrossRef] [Green Version]
- Rafiei, J.; Yavari, K.; Moosavi-Movahedi, A.A. Preferential role of iron in heme degradation of hemoglobin upon gamma irradiation. Int. J. Biol. Macromol. 2017, 103, 1087–1095. [Google Scholar] [CrossRef]
- Ye, L.F.; Chaudhary, K.R.; Zandkarimi, F.; Harken, A.D.; Kinslow, C.J.; Upadhyayula, P.S.; Dovas, A.; Higgins, D.M.; Tan, H.; Zhang, Y.; et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem. Biol. 2020, 15, 469–484. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Alexander, M.S.; Waldron, T.J.; Sibenaller, Z.A.; Spitz, D.R.; Buettner, G.R.; Allen, B.G.; Cullen, J.J. Pharmacological Ascorbate as a Means of Sensitizing Cancer Cells to Radio-Chemotherapy While Protecting Normal Tissue. Semin. Radiat. Oncol. 2019, 29, 25–32. [Google Scholar] [CrossRef]
- Sarsour, E.H.; Kalen, A.L.; Xiao, Z.; Veenstra, T.D.; Chaudhuri, L.; Venkataraman, S.; Reigan, P.; Buettner, G.R.; Goswami, P.C. Manganese superoxide dismutase regulates a metabolic switch during the mammalian cell cycle. Cancer Res. 2012, 72, 3807–3816. [Google Scholar] [CrossRef] [Green Version]
- Vollbracht, C.; Schneider, B.; Leendert, V.; Weiss, G.; Auerbach, L.; Beuth, J. Intravenous vitamin C administration improves quality of life in breast cancer patients during chemo-/radiotherapy and aftercare: Results of a retrospective, multicentre, epidemiological cohort study in Germany. In Vivo 2011, 25, 983–990. [Google Scholar]
- Kanter, M.; Akpolat, M. Vitamin C protects against ionizing radiation damage to goblet cells of the ileum in rats. Acta Histochem. 2008, 110, 481–490. [Google Scholar] [CrossRef]
- Ito, Y.; Kinoshita, M.; Yamamoto, T.; Sato, T.; Obara, T.; Saitoh, D.; Seki, S.; Takahashi, Y. A combination of pre- and post-exposure ascorbic acid rescues mice from radiation-induced lethal gastrointestinal damage. Int. J. Mol. Sci. 2013, 14, 19618–19635. [Google Scholar] [CrossRef] [Green Version]
- Beers, R.F., Jr.; Sizer, I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef]
- Nelson, D.P.; Kiesow, L.A. Enthalpy of decomposition of hydrogen peroxide by catalase at 25 °C (with molar extinction coefficients of H2O2 solutions in the UV). Anal. Biochem. 1972, 49, 474–478. [Google Scholar] [CrossRef]
- Brandt, K.E.; Falls, K.C.; Schoenfeld, J.D.; Rodman, S.N.; Gu, Z.; Zhan, F.; Cullen, J.J.; Wagner, B.A.; Buettner, G.R.; Allen, B.G.; et al. Augmentation of intracellular iron using iron sucrose enhances the toxicity of pharmacological ascorbate in colon cancer cells. Redox Biol. 2018, 14, 82–87. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehdi, Z.; Petronek, M.S.; Stolwijk, J.M.; Mapuskar, K.A.; Kalen, A.L.; Buettner, G.R.; Cullen, J.J.; Spitz, D.R.; Buatti, J.M.; Allen, B.G. Utilization of Pharmacological Ascorbate to Enhance Hydrogen Peroxide-Mediated Radiosensitivity in Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 10880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910880
Mehdi Z, Petronek MS, Stolwijk JM, Mapuskar KA, Kalen AL, Buettner GR, Cullen JJ, Spitz DR, Buatti JM, Allen BG. Utilization of Pharmacological Ascorbate to Enhance Hydrogen Peroxide-Mediated Radiosensitivity in Cancer Therapy. International Journal of Molecular Sciences. 2021; 22(19):10880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910880
Chicago/Turabian StyleMehdi, Zain, Michael S. Petronek, Jeffrey M. Stolwijk, Kranti A. Mapuskar, Amanda L. Kalen, Garry R. Buettner, Joseph J. Cullen, Douglas R. Spitz, John M. Buatti, and Bryan G. Allen. 2021. "Utilization of Pharmacological Ascorbate to Enhance Hydrogen Peroxide-Mediated Radiosensitivity in Cancer Therapy" International Journal of Molecular Sciences 22, no. 19: 10880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910880