
Cheonggukjang-Specific Component 1,3-Diphenyl-2-Propanone as a Novel PPARα/γ Dual Agonist: An In Vitro and In Silico Study



Abstract
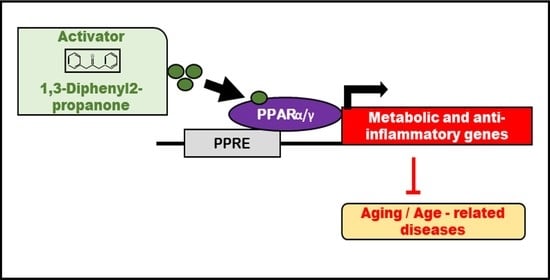
:
1. Introduction
2. Results
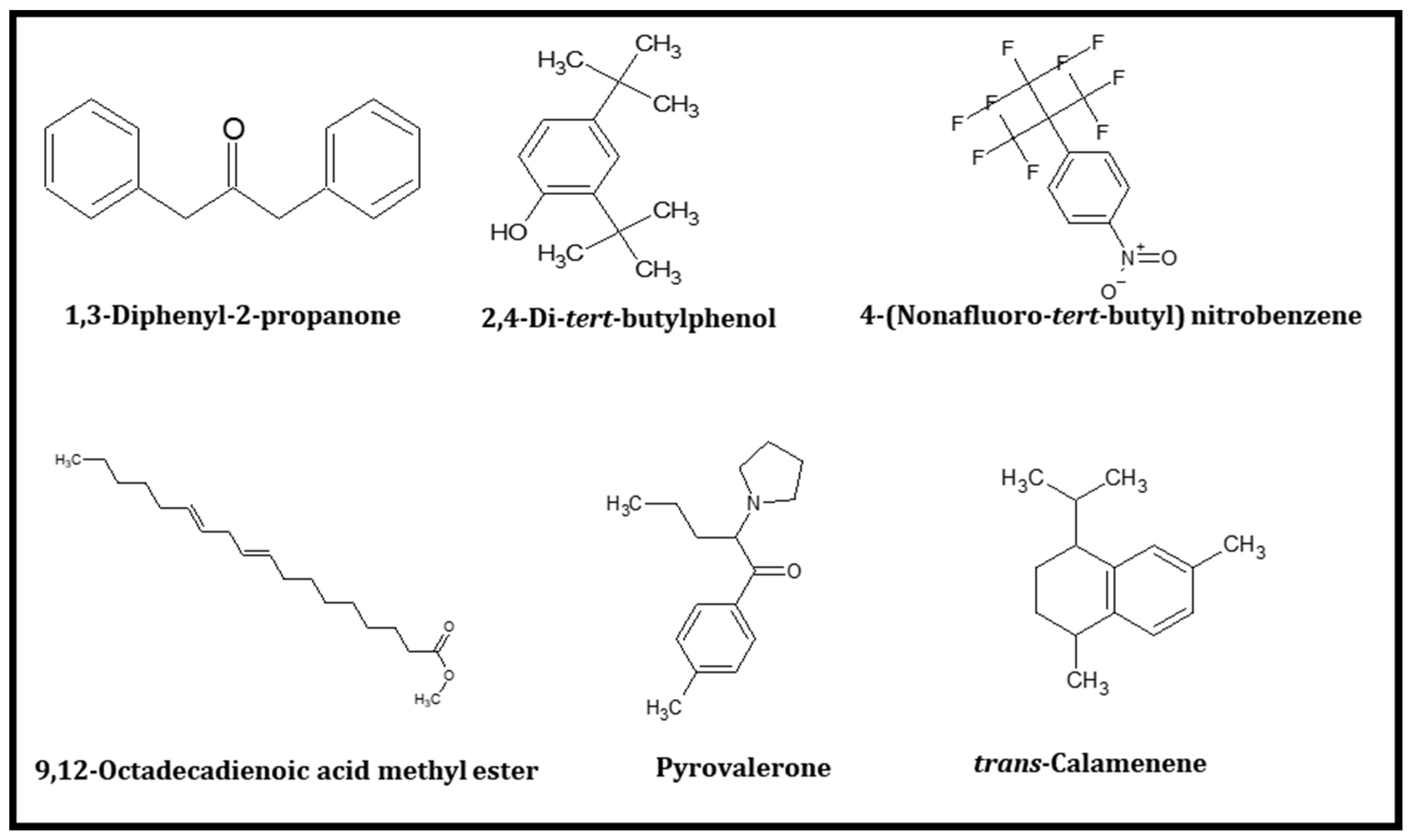
2.1. In Silico Screening of Volatile Compounds from Cheonggukjang by Culturing with B. subtilis
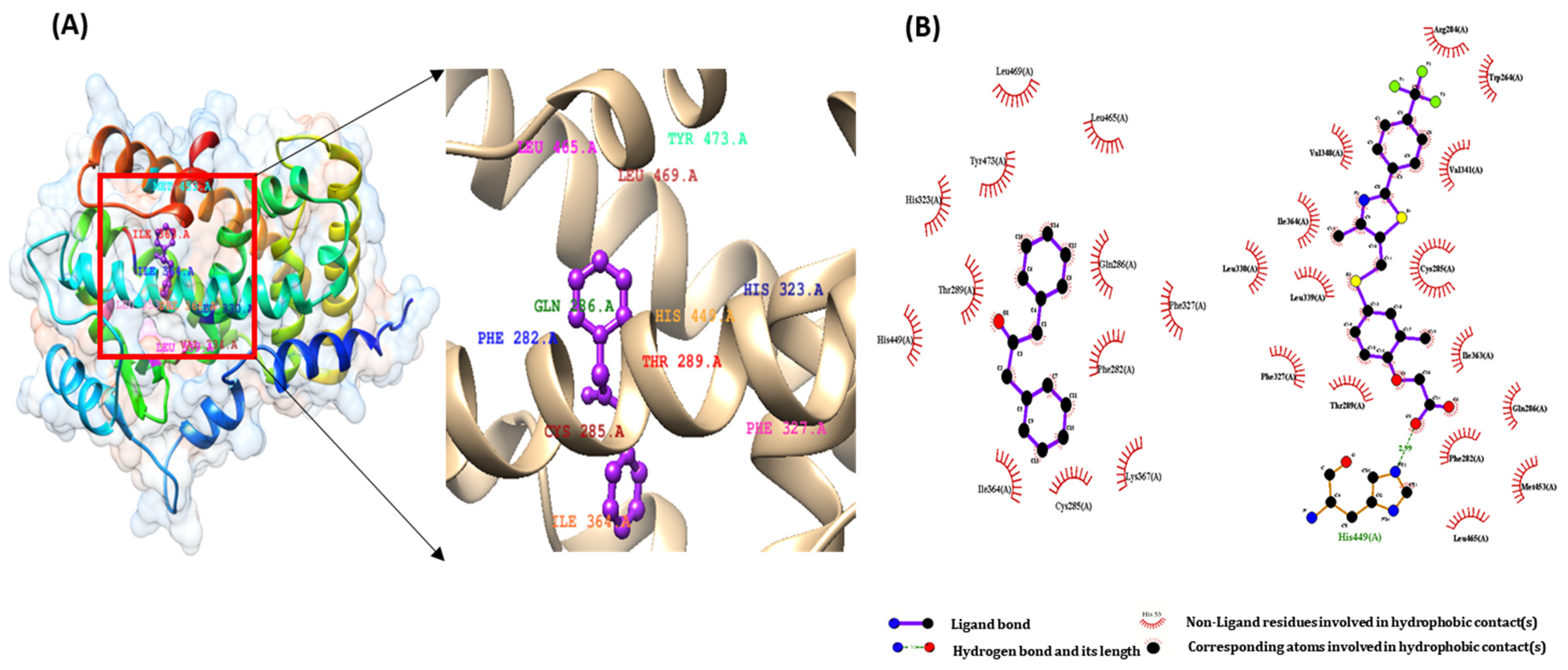
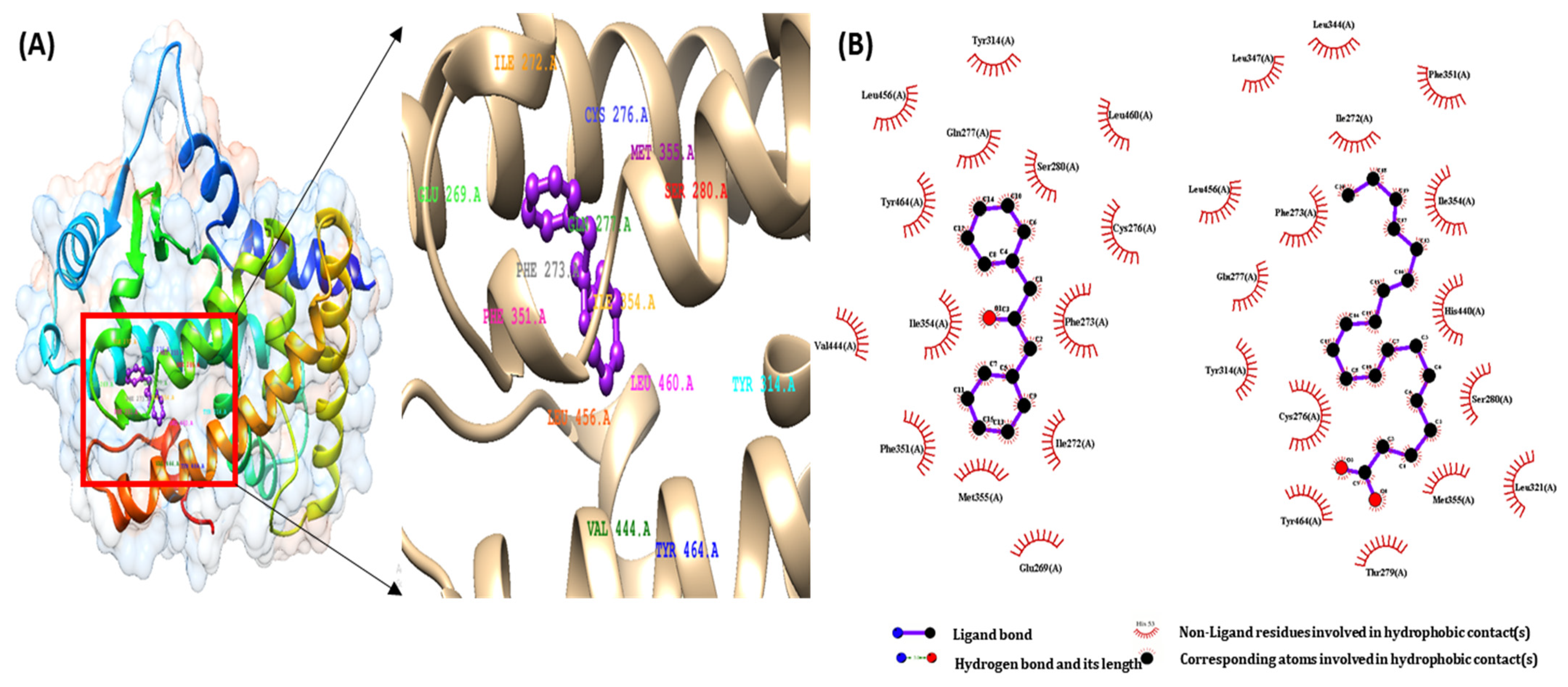
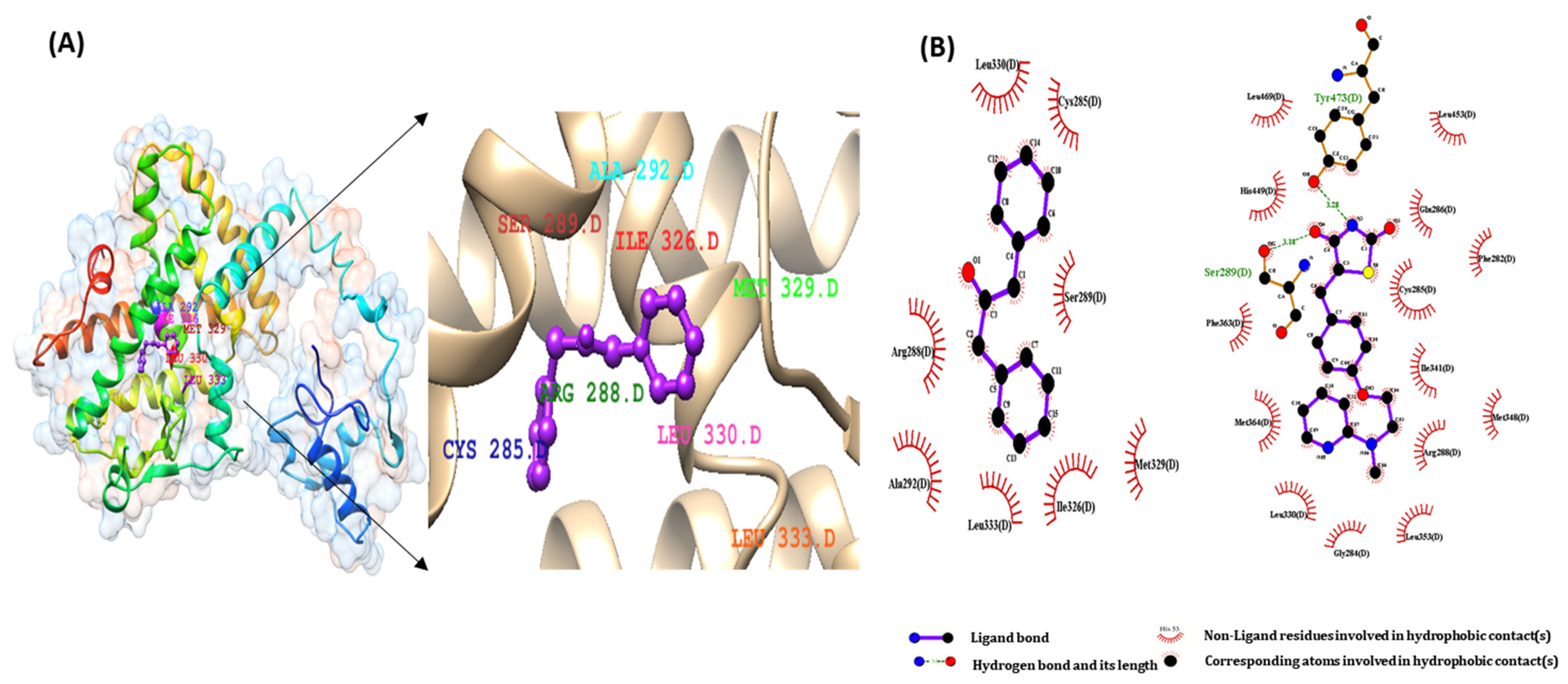
2.2. Molecular Docking Study of DPP as a PPARs Agonist
2.3. Pharmacophore Validation of DPP
2.4. In Silico Toxicity Study of DPP
2.5. ADMET Prediction of DPP
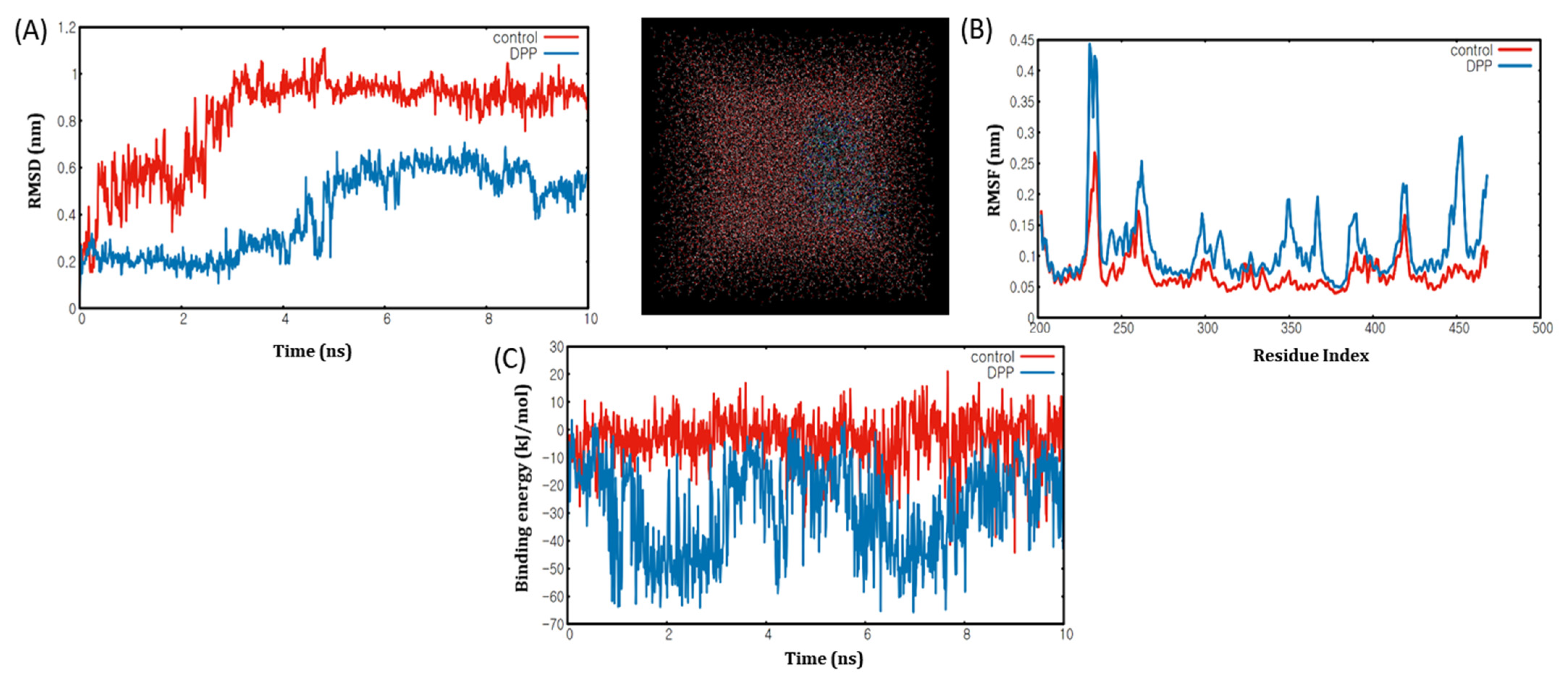
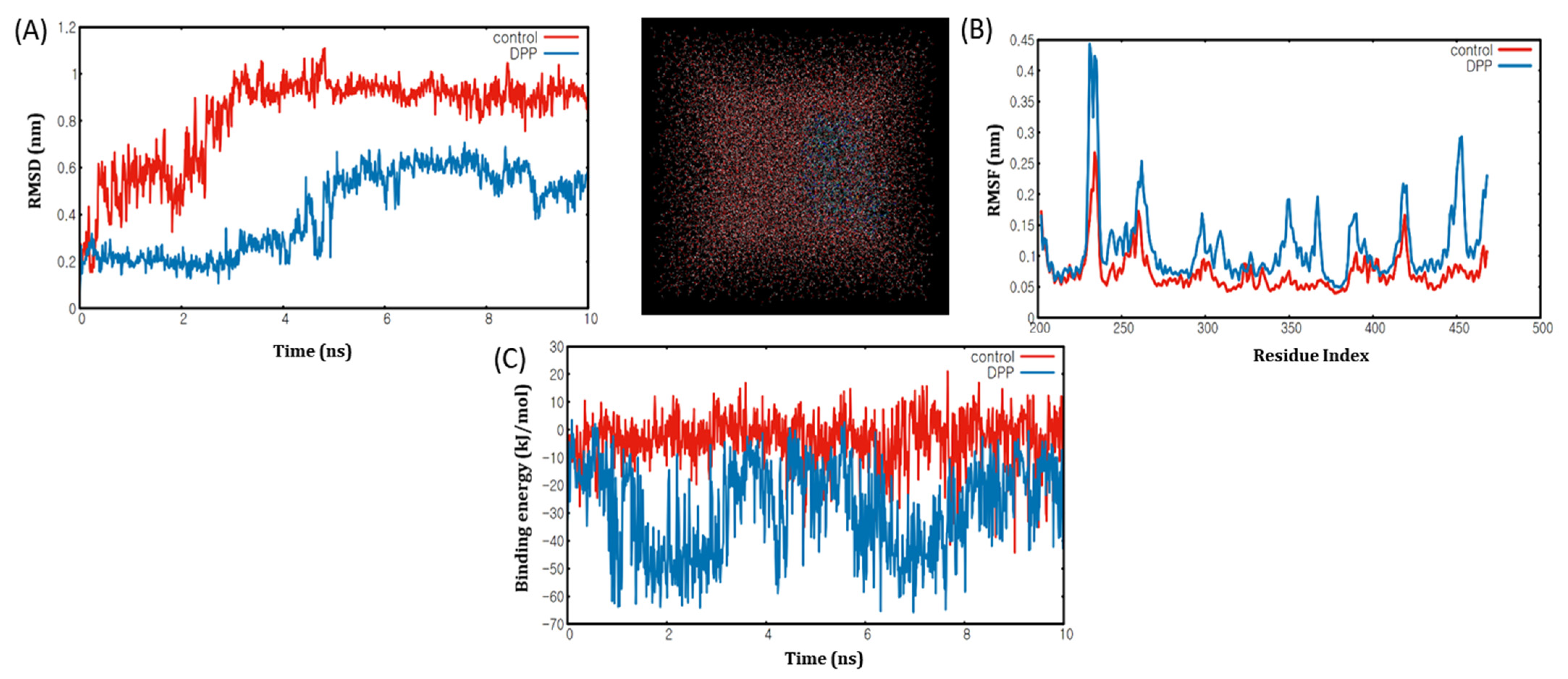
2.6. Molecular Dynamics Simulation Analyses of DPP with PPARα/γ
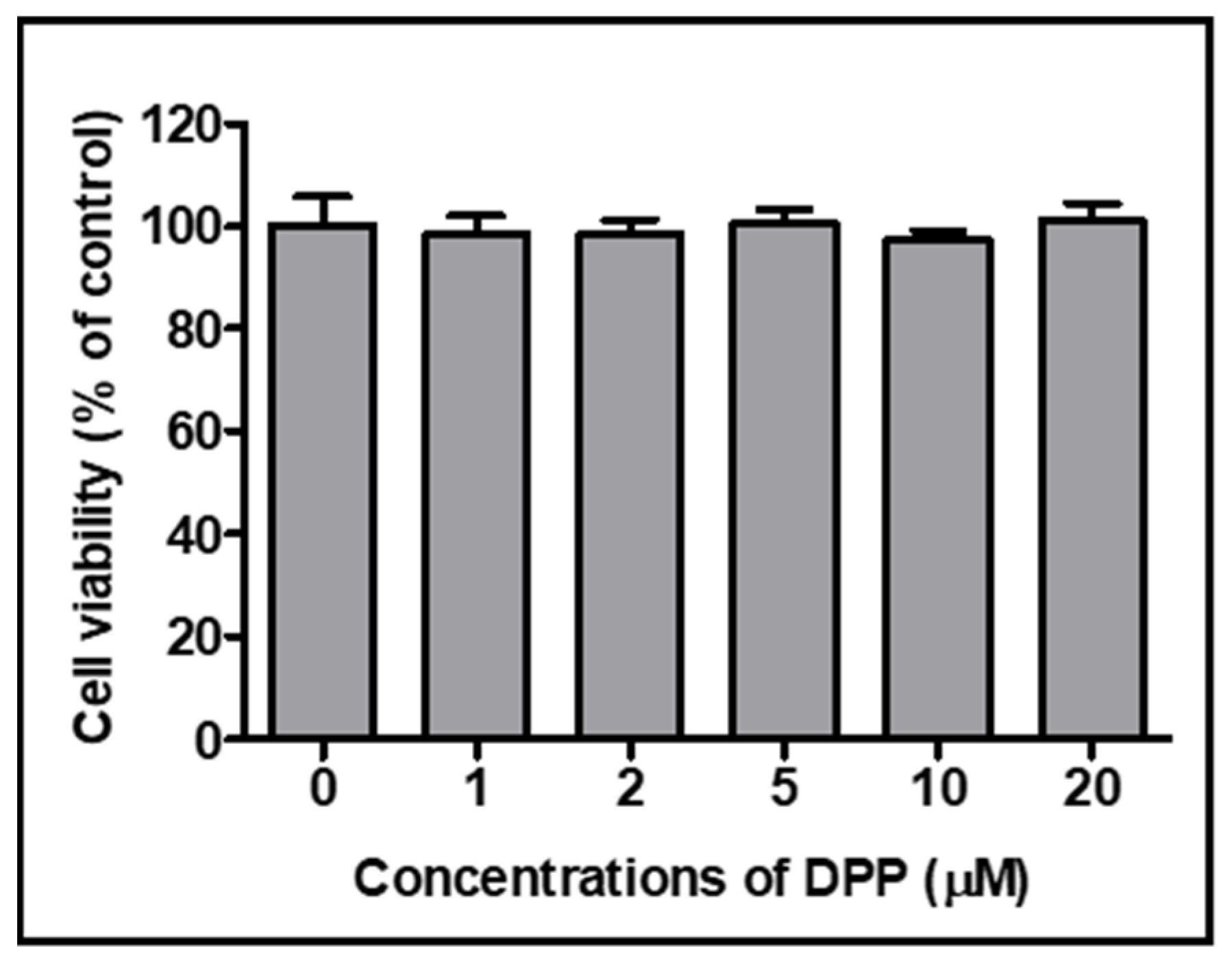
2.7. Evaluation of Toxicity of Volatiles by an In Vitro Analysis
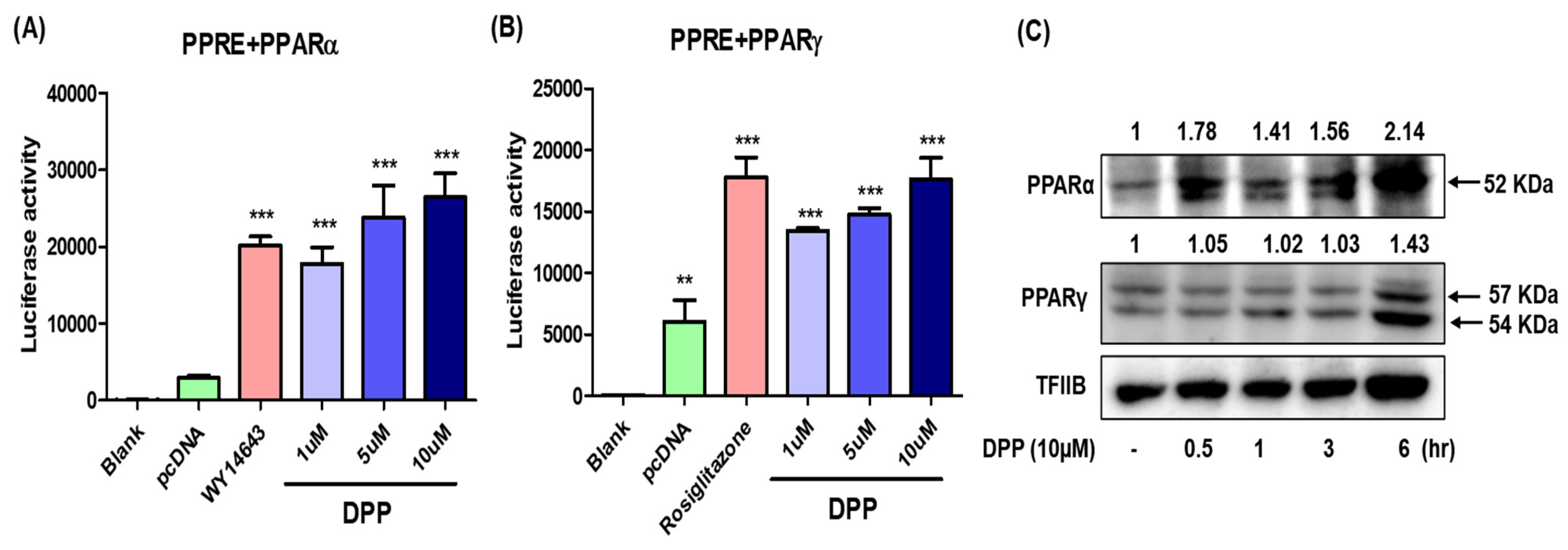
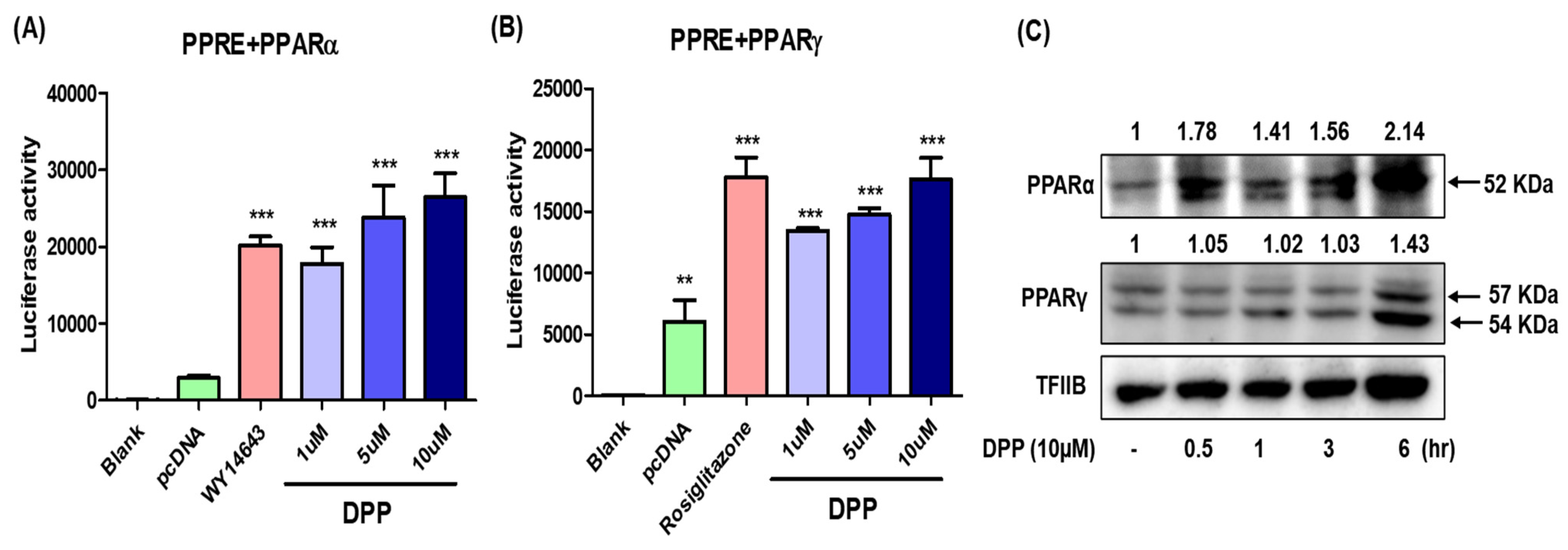
2.8. In Vitro Study of DPP as a PPARα/γ Dual Agonist
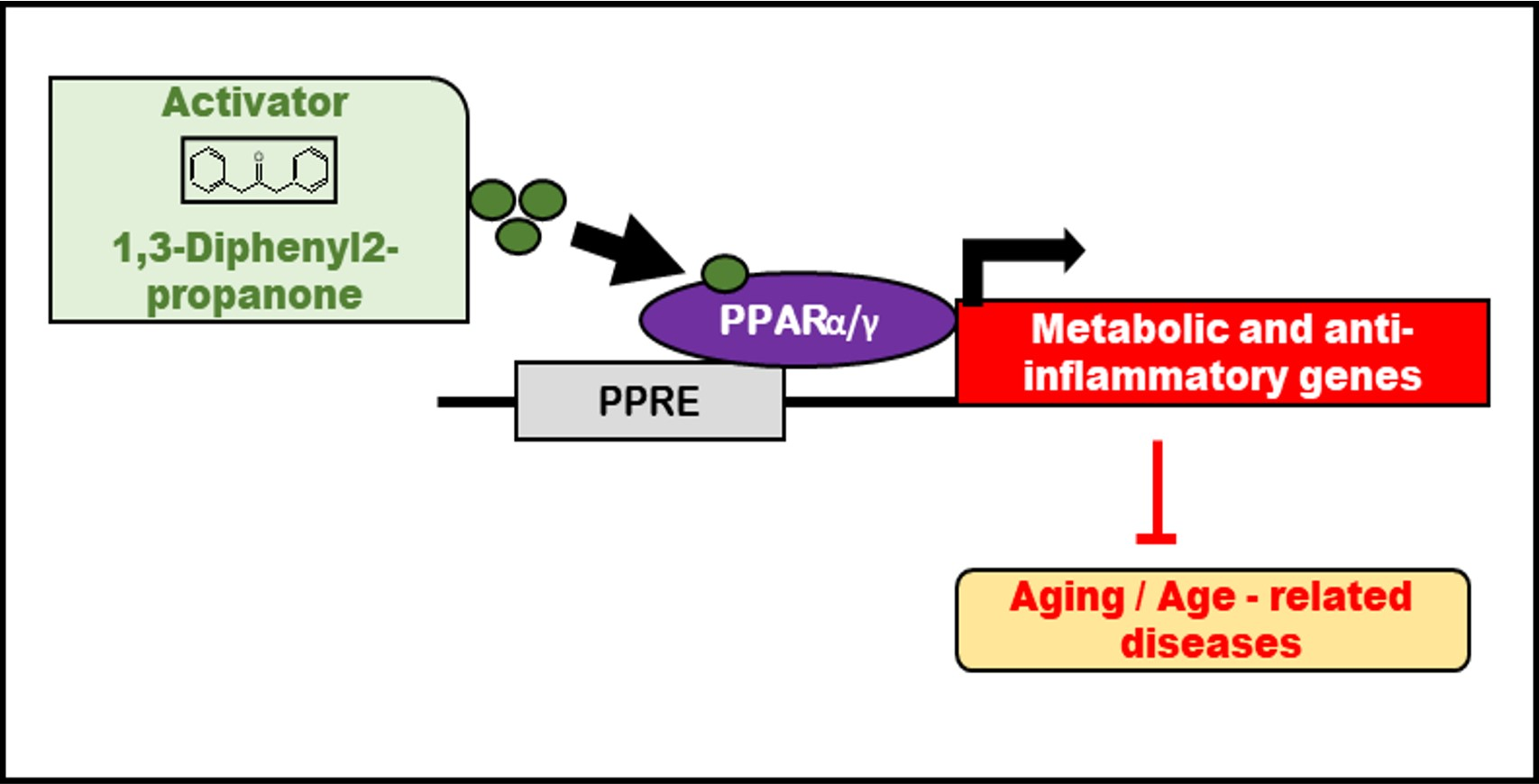
3. Discussion
4. Materials and Methods
4.1. Tools
4.2. Materials
4.3. Protein and Ligand Preparation
4.4. Docking Protocol Validation
4.5. Molecular Docking
4.6. AutoDock 4.2.6
4.7. AutoDock Vina
4.8. Dock6
4.9. Docking Visualization
4.10. Toxicity Screening
4.11. ADMET Prediction
4.12. Molecular Dynamics Simulation
4.13. Chemicals and Reagents
4.14. Cell Culture and Cell Viability Assay
4.15. Transfection and Luciferase Assay
4.16. Western Blot Analysis
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Natarajan, S.; Luthria, D.; Bae, H.; Lakshman, D.; Mitra, A. Transgenic soybeans and soybean protein analysis: An overview. J. Agric. Food Chem. 2013, 61, 11736–11743. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.M.; Lim, H.-J.; Kim, M.-S.; Kim, D.S.; Hwang, C.E.; Nam, S.H.; Joo, O.S.; Lee, B.W.; Kim, J.K.; Shin, E.-C. Time course effects of fermentation on fatty acid and volatile compound profiles of Cheonggukjang using new soybean cultivars. J. Food Drug Anal. 2016, 25, 637–653. [Google Scholar] [CrossRef] [Green Version]
- Baek, J.G.; Shim, S.-M.; Kwon, D.Y.; Choi, H.-K.; Lee, C.H.; Kim, Y.-S. Metabolite profiling of Cheonggukjang, a fermented soybean paste, inoculated with various Bacillus strains during fermentation. Biosci. Biotechnol. Biochem. 2010, 74, 1860–1868. [Google Scholar] [CrossRef]
- Cho, C.-w.; Han, C.-J.; Rhee, Y.K.; Lee, Y.-C.; Shin, K.-S.; Shin, J.-S.; Lee, K.-T.; Hong, H.-D. Cheonggukjang polysaccharides enhance immune activities and prevent cyclophosphamide-induced immunosuppression. Int. J. Biol. Macromol. 2015, 72, 519–525. [Google Scholar] [CrossRef]
- Bae, M.-J.; Shin, H.S.; See, H.-J.; Chai, O.H.; Shon, D.-H. Cheonggukjang ethanol extracts inhibit a murine allergic asthma via. suppression of mast cell-dependent anaphylactic reactions. J. Med. Food 2014, 17, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Choi, J.N.; Choi, J.H.; Cha, Y.S.; Muthaiya, M.J.; Lee, C.H. Effect of fermented soybean product (Cheonggukjang) intake on metabolic parameters in mice fed a high-fat diet. Mol. Nutr. Food Res. 2013, 57, 1886–1891. [Google Scholar] [CrossRef]
- Go, J.; Kim, J.E.; Kwak, M.H.; Koh, E.K.; Song, S.H.; Sung, J.E.; Kim, D.S.; Hong, J.T.; Hwang, D.Y. Neuroprotective effects of fermented soybean products (Cheonggukjang) manufactured by mixed culture of Bacillus subtilis MC31 and Lactobacillus sakei 383 on trimethyltin-induced cognitive defects mice. Nutr. Neurosci. 2016, 19, 247–259. [Google Scholar] [CrossRef]
- Belfiore, A.; Genua, M.; Malaguarnera, R. PPAR-gamma agonists and their effects on IGF-I receptor signaling: Implications for cancer. PPAR Res. 2009, 2009, 830501. [Google Scholar] [CrossRef] [Green Version]
- Tenenbaum, A.; Fisman, E.Z.; Motro, M. Metabolic syndrome and type 2 diabetes mellitus: Focus on peroxisome proliferator activated receptors (PPAR). Cardiovasc. Diabetol. 2003, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Kota, B.P.; Huang, T.H.-W.; Roufogalis, B.D. An overview on biological mechanisms of PPARs. Pharmacol. Res. 2005, 51, 85–94. [Google Scholar] [CrossRef]
- Fruchart, J.-C.; Staels, B.; Duriez, P. The role of fibric acids in atherosclerosis. Curr. Atheroscler. Rep. 2001, 3, 83–92. [Google Scholar] [CrossRef]
- Desvergne, B.; Michalik, L.; Wahli, W. Be fit or be sick: Peroxisome proliferator-activated receptors are down the road. Mol. Endocrinol. 2004, 18, 1321–1332. [Google Scholar] [CrossRef] [Green Version]
- Auwerx, J. PPARγ, the ultimate thrifty gene. Diabetologia 1999, 42, 1033–1049. [Google Scholar] [CrossRef] [Green Version]
- Burdick, A.D.; Kim, D.J.; Peraza, M.A.; Gonzalez, F.J.; Peters, J.M. The role of peroxisome proliferator-activated receptor-beta/delta in epithelial cell growth and differentiation. Cell Signal 2006, 18, 9–20. [Google Scholar] [CrossRef]
- Al-Salman, J.; Arjomand, H.; Kemp, D.G.; Mittal, M. Hepatocellular injury in a patient receiving rosiglitazone. A case report. Ann. Intern. Med. 2000, 132, 121–124. [Google Scholar] [CrossRef]
- Forman, L.M.; Simmons, D.A.; Diamond, R.H. Hepatic failure in a patient taking rosiglitazone. Ann. Intern. Med. 2000, 132, 118–121. [Google Scholar] [CrossRef]
- Cariou, B.; Charbonnel, B.; Staels, B. Thiazolidinediones and PPAR gamma agonists: Time for a reassessment. Trends Endocrinol. Metab. 2012, 23, 205–215. [Google Scholar] [CrossRef]
- Bongartz, T.; Coras, B.; Vogt, T.; Scholmerich, J.; Muller-Ladner, U. Treatment of active psoriatic arthritis with the PPAR gamma ligand pioglitazone: An open-label pilot study. Rheumatology 2005, 44, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [Green Version]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Tacke, F.; Weiskirchen, R. Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)-related liver fibrosis: Mechanisms, treatment and prevention. Ann. Transl. Med. 2021, 9, 729. [Google Scholar] [CrossRef]
- Chukeatirote, E.; Eungwanichayapant, P.; Kanghae, A. Determination of volatile components in fermented soybean prepared by a co-culture of Bacillus subtilis and Rhizopus oligosporus. Food Res. 2017, 1, 225–233. [Google Scholar] [CrossRef]
- Kastritis, P.; Bonvin, A.M.J.J. On the binding affinity of macromolecular interactions: Daring to ask why proteins interact. J. R. Soc. Interface 2013, 10, 20120835. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.R.; Woolfson, D.N.; Muskett, F.W.; Stoneman, R.G.; Urbaniak, M.D.; Caddick, S. Protein-small molecule interactions in neocarzinostatin, the prototypical enediyne chromoprotein antibiotic. Chembiochem 2007, 8, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Tassa, C.; Duffner, J.L.; Lewis, T.; Weissleder, R.; Schreiber, S.L.; Koehler, A.N.; Shaw, S.Y. Binding affinity and kinetic analysis of targeted small molecule-modified nanoparticles. Bioconjugate Chem. 2010, 21, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, K.S.; Rao, A.L.; Rao, M.B. Design, synthesis, biological evaluation and molecular docking studies of novel 3-substituted-5-[(indol-3-yl)methylene]-thiazolidine-2,4-dione derivatives. Heliyon 2018, 4, e00807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balavignesh, V.; Srinivasan, E.; Ramesh Babu, N.G.; Saravanan, N. Molecular docking study ON NS5B polymerase of hepatitis c virus by screening of volatile compounds from Acacia concinna and ADMET prediction. Int. J. Pharm. Life Sci. 2013, 4, 2548–2558. [Google Scholar]
- Khalid, S.; Hanif, R.; Jabeen, I.; Mansoor, Q.; Ismail, M. Pharmacophore modeling for identification of anti-IGF-1R drugs and in-vitro validation of fulvestrant as a potential inhibitor. PLoS ONE 2018, 13, e0196312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, L.; Lv, M.; Pei, R.; Li, P.; Pei, Z.; Wang, Y.; Su, W.; Xie, X.-Q. AlzPlatform: An Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J. Chem. Inf. Model. 2014, 54, 1050–1060. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.C.; Yih, H.; Shwu, J.L.; Hsuan, L.L. Discovery of Novel N-Glycoside and Non-Glycoside hSGLT2 Inhibitors for the Treatment of Type 2 Diabetes Mellitus. J. Diabetes Mellit. 2019, 9, 77–104. [Google Scholar] [CrossRef] [Green Version]
- Van Raalte, D.H.; Li, M.; Pritchard, P.H.; Wasan, K.M. Peroxisome proliferator-activated receptor (PPAR)-α: A pharmacological target with a promising future. Pharm. Res. 2004, 21, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Seo, A.Y.; Chung, S.W.; Kim, M.K.; Leeuwenburgh, C.; Yu, B.P.; Chung, H.Y. Molecular mechanism of PPAR in the regulation of age-related inflammation. Ageing Res. Rev. 2008, 7, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Howroyd, P.; Swanson, C.; Dunn, C.; Cattley, R.C.; Corton, J.C. Decreased longevity and enhancement of age-dependent lesions in mice lacking the nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha). Toxicol. Pathol. 2004, 32, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, B.; Park, S.; Yu, B.P.; Chung, H.Y. Modulation of PPAR in aging, inflammation, and calorie restriction. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2004, 59, B997–B1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iemitsu, M.; Miyauchi, T.; Maeda, S.; Tanabe, T.; Takanashi, M.; Irukayama-Tomobe, Y.; Sakai, S.; Ohmori, H.; Matsuda, M.; Yamaguchi, I. Aging-induced decrease in the PPAR-α level in hearts is improved by exercise training. Am. J. Physiol. Circ. Physiol. 2002, 283, H1750–H1760. [Google Scholar] [CrossRef]
- Sanguino, E.; Roglans, N.; Alegret, M.; Sanchez, R.M.; Vazquez-Carrera, M.; Laguna, J.C. Atorvastatin reverses age-related reduction in rat hepatic PPAR alpha and HNF-4. Br. J. Pharmacol. 2005, 145, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Montagner, A.; Polizzi, A.; Fouche, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Regnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPAR alpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPAR alpha and the fatty acid oxidation pathway aggravates renal fibrosis during aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237. [Google Scholar] [CrossRef] [Green Version]
- Chiarelli, F.; Di Marzio, D. Peroxisome proliferator-activated receptor-gamma agonists and diabetes: Current evidence and future perspectives. Vasc. Health Risk Manag. 2008, 4, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Shiomi, Y.; Yamauchi, T.; Iwabu, M.; Okada-Iwabu, M.; Nakayama, R.; Orikawa, Y.; Yoshioka, Y.; Tanaka, K.; Ueki, K.; Kadowaki, T. A novel peroxisome proliferator-activated receptor (PPAR) alpha agonist and PPAR gamma antagonist, Z-551, ameliorates high-fat diet-induced obesity and metabolic disorders in mice. J. Biol. Chem. 2015, 290, 14567–14581. [Google Scholar] [CrossRef] [Green Version]
- Khuchua, Z.; Glukhov, A.I.; Strauss, A.W.; Javadov, S. Elucidating the beneficial role of PPAR agonists in cardiac diseases. Int. J. Mol. Sci. 2018, 19, 3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.-P.; Neumann, D. A new lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-score—a comprehensive scoring function for evaluation of chemical drug-likeness. Med. Chem. Commmun. 2019, 10, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. A molecular dynamics approach to explore the intramolecular signal transduction of PPAR-α. Int. J. Mol. Sci. 2019, 20, 1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]

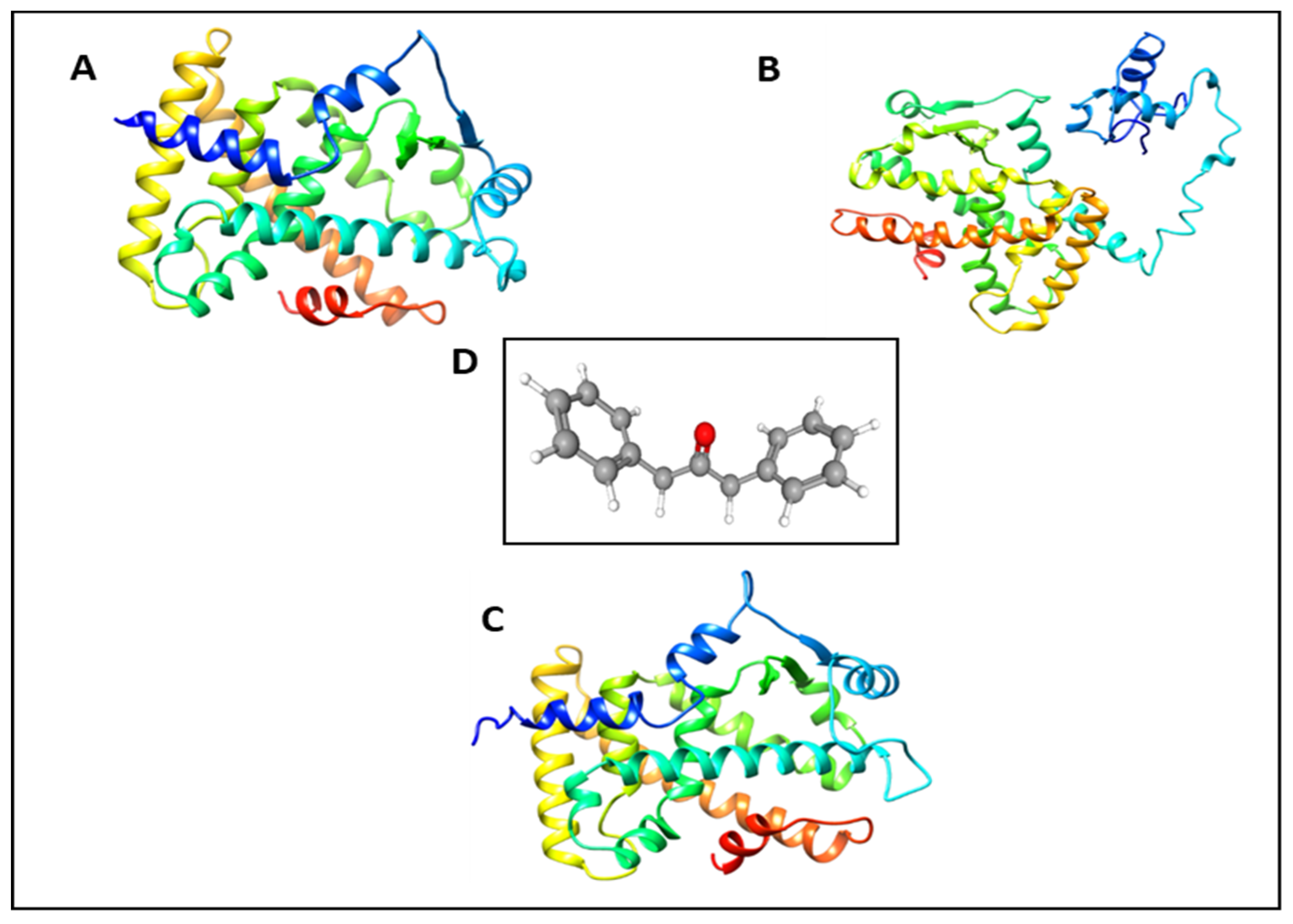




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial No | Volatile Compounds | NF | F |
|---|---|---|---|
| Ketones | |||
| 1–181 | 1,3-Diphenyl-2-propanone | − | + |
| 2 | 2,3-Butandione | − | + |
| 3 | 2,6-Dihydroxyacetophenone | − | + |
| 4 | 3-Penten-2-one | − | + |
| 5 | Pyrovalerone | − | + |
| Alcohols | |||
| 6 | 1-Dodecanol | − | + |
| 7 | 1-Octanol | − | + |
| 8 | 2,3-Butanediol | − | + |
| 9 | 2,5-Dimethyl-3-hexanol | − | + |
| 10 | 3-Methyl-2-butanol | − | + |
| 11 | Benzyl alcohol | − | + |
| 12 | Ethanol | − | + |
| Acids and esters | |||
| 13 | 2-Methyl-decanoic acid | − | + |
| 14 | 2-Methyl-hexanoic acid | − | + |
| 15 | 3-Methyl-butanoic acid | − | + |
| 16 | 3-Methyl-pentanoic acid | − | + |
| 17 | Butanoic acid, 3-hydroxy-ethyl ester | − | + |
| 18 | Formic acid, 1-methylpropyl ester | − | + |
| 19 | 9,12-Octadecadienoic acid methyl ester | − | + |
| 20 | Benzeneacetic acid, ethyl ester | − | + |
| 21 | Octanoic acid ethyl ester | − | + |
| Miscellaneous | |||
| 22 | 2,4-Di-tert-butylphenol | − | + |
| 23 | 2-Methoxy-4-(2-propenyl)phenol | − | + |
| 24 | 3,4-Dihydroxyphenylglycol | − | + |
| 25 | trans-Calamenene | − | + |
| Pyrazine | |||
| 26 | 2,5-Dimethyl-pyrazine | − | + |
| 27 | 2-Butyl-3,5-dimethyl-pyrazine | − | + |
| 28 | 3-Ethyl-2,5-dimethyl-pyrazine | − | + |
| 29 | Tetramethyl-pyrazine | − | + |
| Aldehyde | |||
| 30 | Acetaldehyde | − | + |
| 31 | Alpha-ethylidene-benzeneacetaldehyde | − | + |
| 32 | Nonanal | − | + |
| 33 | Piperonal | − | + |
| 34 | Benzeneacetaldehyde | − | + |
| Aromatic compounds | |||
| 35 | 4-(Nonafluoro-tert-butyl) nitrobenzene | − | + |
| Component | PPARα | PPARβ/δ | PPARγ | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Vina | AD4 | Dock6 | Vina | AD4 | Dock6 | Vina | AD4 | Dock6 | |
| Control | −6.7 | −7.81 | −35.45 | −7.8 | −7.44 | −41.006 | −6.6 | −7.89 | −32.666 |
| 1,3-Diphenyl-2-propanone | −8.8 | −8.14 | −30.4204 | −7.5 | −7.36 | −34.8391 | −7.0 | −7.53 | −36.0086 |
| 2,4-Di-tert-butylphenol | −6.8 | −7.17 | −28.6838 | −7 | −7.03 | −27.4858 | −5.6 | −6.74 | −28.516 |
| 4-(Nonafluoro-tert-butyl) nitrobenzene | −6.1 | −5.25 | −25.5059 | −7.7 | −5 | −24.3869 | −6.1 | −4.86 | −28.3937 |
| 9,12-Octadecadienoic acid methyl ester | −6.3 | −8.18 | −26.8669 | −6.9 | −7.34 | −28.9908 | −6.3 | −7.83 | −27.3687 |
| Pyrovalerone | −7.5 | −8.41 | −124.603 | −7.6 | −7.75 | −129.638 | −6.7 | −7.78 | −130.539 |
| trans-Calamenene | −6.8 | −8.02 | −29.2324 | −7.6 | −7.8 | −28.4842 | −6.5 | −7.71 | −27.7687 |
| Component | AMPK | LKB1 | PAR2 | |||||
|---|---|---|---|---|---|---|---|---|
| Vina | AD4 | Dock6 | Vina | AD4 | Vina | AD4 | Dock6 | |
| Control | −5.7 | −10.81 | −132.631 | −7.4 | −7.22 | −4.8 | −8.19 | −105.242 |
| 1,3-Diphenyl-2-propanone | −7.2 | −10.73 | −37.2376 | −7.7 | −8.81 | −5 | −7.07 | −29.2367 |
| 2,4-Di-tert-butylphenol | −7 | −6.41 | −25.7078 | −7.1 | −6.06 | −4.6 | −5.49 | −21.2142 |
| 4-(Nonafluoro-tert-butyl) nitrobenzene | −7.7 | −9.65 | −29.9102 | −7.3 | −8.48 | −5.2 | −6.93 | −26.5166 |
| 9,12-Octadecadienoic acid methyl ester | −7.2 | −3.51 | −28.1999 | −7.3 | −4.15 | −4.4 | −2.89 | −22.5024 |
| Pyrovalerone | −6.4 | −6.57 | −25.6285 | −5 | −5.46 | −3.4 | −3.85 | −21.3088 |
| trans-Calamenene | −6.8 | −7.19 | −133.185 | −6 | −7.09 | −4.4 | −6.06 | −99.2638 |
| Component | SIRT1 | SIRT2 | SIRT3 | SIRT6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Vina | AD4 | Dock6 | Vina | AD4 | Dock6 | Vina | AD4 | Dock6 | Vina | AD4 | |
| Control | −7.9 | −9.23 | −38.382 | −11.4 | −12.01 | −53.2537 | −5.6 | −11.44 | −231.781 | −9.9 | −8.5 |
| 1,3-Diphenyl-2-propanone | −6.9 | −9.67 | −34.439 | −9.1 | −11.58 | −37.1523 | −9.4 | −10.59 | −42.1555 | 5.1 | 0.63 |
| 2,4-Di-tert-butylphenol | −8.7 | −7.63 | −29.9781 | −8.1 | −7.22 | −29.2715 | −6.9 | −6.37 | −24.8486 | −7.1 | −7.27 |
| 4-(Nonafluoro-tert-butyl) nitrobenzene | −8.4 | −11.65 | −34.5245 | −8.9 | −10.87 | −33.2557 | −8.5 | −9.31 | −37.0224 | −5.6 | −10.36 |
| 9,12-Octadecadienoic acid methyl ester | −9 | −5.63 | −26.8674 | −8.5 | −4.71 | −23.4719 | −7.5 | −4.14 | −23.8888 | −5.4 | −5.15 |
| Pyrovalerone | −7.5 | −8.4 | −33.1864 | −7.7 | −8.18 | −29.2381 | −6.7 | −6.74 | −26.5441 | −6.8 | −8.32 |
| trans-Calamenene | −8.5 | −8.78 | −126.213 | −8.5 | −8.34 | −123.2 | −7.3 | −7.92 | −126.672 | −7.3 | −9.27 |
| Compound | Binding Energy (kcal/mol) | Inhibition Constant Ki (μM) | Intermolecular Energy (kcal/mol) | Bonded Residues |
|---|---|---|---|---|
| 1,3-Diphenyl-2-propanone | −8.14 | 1.09 | −9.33 | CYS276, GLN 277, SER 280, TYR 314, MET 355, LEU 456, and TYR 464 |
| Compound | Binding Energy (kcal/mol) | Inhibition Constant Ki (μM) | Intermolecular Energy (kcal/mol) | Bonded Residues |
|---|---|---|---|---|
| 1,3-Diphenyl-2-propanone | −7.36 | 4.05 | −8.37 | PHE 282, CYS 285, THR 289, ILE 364, and HIS 449 |
| Compound | Binding Energy (kcal/mol) | Inhibition Constant Ki (μM) | Intermolecular Energy (kcal/mol) | Bonded Residues |
|---|---|---|---|---|
| 1,3-Diphenyl-2-propanone | −7.35 | 3.05 | −8.18 | CYS 285, ARG 288, SER 289, and LEU 330 |
| Serial No | Volatile Compounds | Toxicity LD50 (mg/Kg) |
|---|---|---|
| 1 | 1,3-Diphenyl-2-propanone | 2000 |
| 2 | 2,4-Di-tert-butylphenol | 700 |
| 3 | 4-(Nonafluoro-tert-butyl) nitrobenzene | 1000 |
| 4 | 9,12-Octadecadienoic acid methyl ester | 20,000 |
| 5 | Pyrovalerone | 350 |
| 6 | trans-Calamenene | 6700 |
| Rules | Eicosapentaenoic Acid (PPARα) | Rosiglitazone (PPARγ) | 1,3-Diphenyl-2-propanone | 2,4-Di-tert-butylphenol | 4-(Nonafluoro-tert-butyl) Nitrobenzene | 9,12-Octadecadienoic Acid Methyl Ester | Pyrovalerone | trans-Calamenene | b Required Parameters |
|---|---|---|---|---|---|---|---|---|---|
| a MW | 302.5 | 357.4 | 210.27 | 206.32 | 341.13 | 294.5 | 245.36 | 202.33 | ≤500 |
| a HBD | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | ≤5 |
| a HBA | 2 | 6 | 1 | 1 | 11 | 2 | 2 | 0 | ≤10 |
| a LogP | 5.6 | 3.1 | 3.1 | 4.9 | 5.6 | 6.9 | 3.8 | 5.1 | 2–5 |
| a Rot bonds | 13 | 7 | 4 | 2 | 1 | 15 | 5 | 1 | ≤10 |
| Compounds | Absorption | Distribution | ||||
|---|---|---|---|---|---|---|
| Human Intestinal Absorption (HIA %) | Caco-2 Cell Permeability (nm s−1) | MDCK Cell Permeability (nm s−1) | Skin Permeability (logKp, cm h−1) | Plasma Protein Binding (%) | Blood–Brain Barrier Penetration (Cbrain/Cblood) | |
| Eicosapentaenoic acid (PPARα) | 97.940242 | 30.0843 | 76.2528 | −0.566312 | 100 | 6.59907 |
| Rosiglitazone (PPARγ) | 97.451401 | 28.616 | 2.07646 | −3.44324 | 91.095751 | 0.012398 |
| 1,3-Diphenyl-2-propanone | 100 | 54.5905 | 209.282 | −1.6951 | 90.912956 | 1.75109 |
| 2,4-Di-tert-butylphenol | 100 | 44.8684 | 116.431 | −0.739126 | 100 | 10.0918 |
| 4-(Nonafluoro-tert-butyl) nitrobenzene | 96.809262 | 22.6752 | 3.91751 | −0.758863 | 100 | 1.01857 |
| 9,12-Octadecadienoic acid methyl ester | 100 | 47.1151 | 67.0846 | −0.538746 | 100 | 13.8625 |
| Pyrovalerone | 100 | 57.4649 | 157.355 | −1.55534 | 76.382571 | 1.46433 |
| trans-Calamenene | 100 | 23.4586 | 60.7588 | −0.761116 | 100 | 11.9795 |
| Compounds | CYP2C19 Inhibition | CYP2C9 Inhibition | CYP2D6 Inhibition | CYP2D6 Substrate | CYP3A4 Inhibition | CYP3A4 Substrate |
|---|---|---|---|---|---|---|
| Eicosapentaenoic acid (PPARα) | Inhibitor | Inhibitor | Non | Non | Inhibitor | Non |
| Rosiglitazone (PPARγ) | Non | Inhibitor | Non | Non | Non | Weakly |
| 1,3-Diphenyl-2-propanone | Inhibitor | Inhibitor | Non | Non | Inhibitor | Weakly |
| 2,4-Di-tert-butylphenol | Non | Inhibitor | Non | Non | Inhibitor | Substrate |
| 4-(Nonafluoro-tert-butyl) nitrobenzene | Non | Inhibitor | Non | Non | Inhibitor | Substrate |
| 9,12-Octadecadienoic acid methyl ester | Inhibitor | Inhibitor | Non | Non | Inhibitor | Non |
| Pyrovalerone | Non | Non | Inhibitor | Weakly | Non | Substrate |
| trans-Calamenene | Inhibitor | Inhibitor | Non | Non | Inhibitor | Substrate |
| Donor | Hydrogen | Acceptor |
|---|---|---|
| GLN277 NE2 | GLN277 HE21 | 469 O1 |
| HIS440 NE2 | HIS440 HE2 | 469 O1 |
| Serial No | Compound Name | IUPAC Name | Molecular Weight (Da) | Uses |
|---|---|---|---|---|
| 1 | 1,3-Diphenylacetone | 1,3-diphenylpropan-2-one | 210.27 | Food additive, flavor |
| 2 | 2,4-Di-tert-butylphenol | 2,4-ditert-butylphenol | 206.32 | Making chemicals |
| 3 | 4-(Nonafluoro-tert-butyl) nitrobenzene | 1-[1,1,1,3,3,3-hexafluoro-2-(trifluoromethyl)propan-2-yl]-4-nitrobenzene | 341.13 | Industrial application |
| 4 | 9,12-Octadecadienoic acid methyl ester | methyl octadeca-9,12-dienoate | 294.5 | Personal care, cosmetics |
| 5 | Pyrovalerone | 1-(4-methylphenyl)-2-pyrrolidin-1-ylpentan-1-one | 245.36 | manufacturing of drugs |
| 6 | trans-Calamenene | (1S,4R)-1,6-dimethyl-4-propan-2-yl-1,2,3,4-tetrahydronaphthalene | 202.33 | Preservative, Insecticidal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arulkumar, R.; Jung, H.-J.; Noh, S.-G.; Park, D.; Chung, H.-Y. Cheonggukjang-Specific Component 1,3-Diphenyl-2-Propanone as a Novel PPARα/γ Dual Agonist: An In Vitro and In Silico Study. Int. J. Mol. Sci. 2021, 22, 10884. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910884
Arulkumar R, Jung H-J, Noh S-G, Park D, Chung H-Y. Cheonggukjang-Specific Component 1,3-Diphenyl-2-Propanone as a Novel PPARα/γ Dual Agonist: An In Vitro and In Silico Study. International Journal of Molecular Sciences. 2021; 22(19):10884. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910884
Chicago/Turabian StyleArulkumar, Radha, Hee-Jin Jung, Sang-Gyun Noh, Daeui Park, and Hae-Young Chung. 2021. "Cheonggukjang-Specific Component 1,3-Diphenyl-2-Propanone as a Novel PPARα/γ Dual Agonist: An In Vitro and In Silico Study" International Journal of Molecular Sciences 22, no. 19: 10884. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910884